

Abstract
A new method is presented that makes it possible to inject electrons rapidly into redox-active proteins by means of a short light flash. Reduced carboxymethylated cytochrome c (CmCyt c) with carbon monoxide bound to the heme iron is mixed with the oxidized acceptor protein. Upon rapid photodissociation of CO the apparent redox potential of CmCyt c drops, resulting in electron transfer to the electron acceptor. In this study we have used mitochondrial cytochrome c oxidase as the acceptor protein, but the method also can be used to investigate electron transfer to other proteins that can interact with cytochrome c. In principle, it can be used with any redox protein into which a CO binding site at the heme iron can be engineered.
Keywords: electron transfer, carboxymethylated cytochrome c, cytochrome c oxidase, flash photolysis
Many key reactions in biological energy conversion, e.g. in photosynthesis and respiration, involve long-range electron-transfer reactions within and between proteins. In many cases these reactions are rapid and cannot be resolved in time using conventional techniques. In this study, a new method is presented that makes it possible to inject an electron rapidly into redox-active proteins by means of a short laser flash. This method relies on the fact that carbon monoxide has a high affinity for ferrous heme proteins but does not bind to the ferric heme counterparts. The effect of CO is therefore to increase the apparent redox potential of any heme protein that has a vacant sixth coordination position and is thus able to bind CO. The magnitude of this effect on the apparent redox potential is CO-concentration dependent.
Generally, CO complexes of ferrous heme proteins are light sensitive, such that a brief actinic light pulse, e.g. from a laser, will dissociate the CO from the heme group, leaving this as a five-coordinate high-spin ferrous species. The quantum yields for such reactions are generally high so that complete photodissociation is easily achieved (see e.g. ref. 1). Five-coordinate heme proteins invariably have a lower redox potential than their hexaccordinate forms and thus photodissociation of CO has the effect of rapidly (within a few nanoseconds, if a laser is used) switching the redox potential of the protein rendering a good electron acceptor (the CO adduct) into a potent electron donor (five-coordinate ferrous). We have exploited these properties to provide a way to inject electrons into redox proteins. In this study we have used mitochondrial cytochrome c oxidase as the acceptor protein and a chemically modified form of its natural substrate, carboxymethyl-cytochrome c (CmCyt c) (2–7), as electron donor. In CmCyt c the iron is pentacoordinated (8) because methionine-80 is carboxymethylated so that it can no longer act as a ligand for the heme iron. This allows other ligands, such as CO, to bind at the coordination site normally occupied by the sulfur atom of the methionine residue. The method can be used to investigate redox reactions in any protein that can interact with cytochrome c (e.g., cytochrome c peroxidase, flavocytochrome b2, NO2 reductase, N2O reductase, NO reductase, plastocyanin, cellobiose oxidase, etc.) In principle, it is applicable to any redox protein into which a CO binding site at the heme iron can be engineered (e.g., cytochrome b5).
Cytochrome c oxidase is a membrane-bound, redox-driven proton pump, which catalyzes oxidation of cytochrome c by dioxygen (for a recent review, see ref. 9). The enzyme comprises four redox-active metal sites; two copper sites, CuA and CuB, and two heme-bound irons, Fea (heme a) and Fea3 (heme a3). During turnover, an electron is first transferred from cytochrome c to CuA and then consecutively to Fea and the binuclear center, Fea3-CuB, where O2 is reduced to water. Upon electron injection into the fully oxidized enzyme equilibration with Fea is rapid (≈104 s−1), whereas electron transfer to Fea3-CuB is several orders of magnitude slower (reviewed in refs. 9 and 10; see also ref. 11). Electron injection into cytochrome c oxidase has been studied previously using the stopped-flow technique (reviewed in ref. 10), but the time resolution of this technique is limited to about 1 ms. Rapid light-induced electron injection from, e.g., Ru-modified cytochrome c has been studied with a much better time resolution (refs. 12 and 13; see also ref. 10), but with a very small yield. The method described here is both rapid and provides high yield.
MATERIALS AND METHODS
CmCyt c was prepared as described previously (7) from horse heart cytochrome c (Sigma, type VI). Cytochrome c oxidase from bovine heart was purified essentially as described (14). It was extracted from mitochondrial membranes using Triton X-100, followed by hydroxyapatite chromatography and exchange of Triton X-100 for 0.1% (wt/vol) dodecyl β-d maltoside (Sigma). The enzyme was stored under liquid nitrogen until used. Reduced carboxy-CmCyt c was prepared by repeated evacuation of the protein solution on a vacuum line and replacement of air by CO (99.5%), followed by an anaerobic addition of a 2-fold molar excess of dithionite (from a 5 mM solution in 100 mM Hepes buffer at pH 7.2). An anaerobic solution of cytochrome c oxidase was prepared as described above, but it was flushed with N2 instead of CO.
The solutions were mixed using a locally modified stopped-flow apparatus (Applied Photophysics, DX-17MV). The output signal from the photomultiplier was fed into a 3-MHz current-to-voltage converter and a preamplifier with a variable RC filter (Tektronix, model AM 502). The output voltage was recorded using a digital transient recorder (Nicolet, model 490). The stop-syringe switch was connected to a Nd-YAG laser (Spectra Physics), which was triggered about 100 ms after mixing. The photomultiplier was protected from the laser light using various interference filters, depending on wavelength. The cuvette path length was 1.00 cm.
RESULTS AND DISCUSSION
The binding properties of the CO ligand to CmCyt c have been investigated in detail previously, and it has been shown that CO binds strongly exclusively to the reduced form with a binding constant, KCO, of about 106 mM−1 at pH 5.9 (5). Thus the apparent redox potential of the CmCyt c-CO complex, Eapp (in mV), increases with increasing CO concentration:
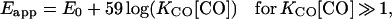 |
1 |
where E0 is the redox potential (in mV) in the absence of CO and [CO] is the CO concentration (in mM). Consequently, upon dissociation of CO the apparent redox potential of CmCyt c drops by approximately 350 mV at 1 mM CO, a property that is used to inject electrons rapidly into an acceptor bound to CmCyt c.
The optical absorption difference spectra of the reduced-minus-oxidized and the carboxy-minus-reduced CmCyt c are shown in Fig. 1A. Illumination of the CmCyt c-CO complex with a laser flash results in rapid (>107 s−1) dissociation of CO followed by recombination with a rate constant of about 2⋅103 s−1 at 1 mM CO (not shown), consistent with the previously determined second-order recombination rate constant of 1.6·106 M−1·s−1 (5).
Figure 1.

Absorption difference spectra of (A) CmCyt c; ferro minus ferri (solid line) and carboxy-ferro minus ferro (dashed line). (B) Heme a of cytochrome c oxidase measured as the difference between reduced and fully oxidized enzyme with cyanide bound to Fea3 (CuB).The spectra were recorded with 6.3 μM and 2.4 μM for CmCyt c and cytochrome c oxidase, respectively, but in the figure they are scaled to a 1 μM concentration. Note the different absorbance scales in the right and left panels. Conditions: 10 mM Hepes, pH 7.1, 22 ± 1°C.
Recombination of CO with CmCyt c competes with electron transfer from CmCyt c to cytochrome c oxidase. If necessary, the recombination reaction can be slowed by reducing the CO concentration. However, a lower CO concentration also results in a smaller increase in the apparent redox potential of CmCyt c upon CO binding (see Eq. 1). Therefore, the CO concentration must be adjusted so that CO recombination is much slower than electron transfer, yet the apparent redox potential of the complex is higher than that of the electron acceptor. In this study we used a CO concentration of about 0.1 mM. Fig. 2 illustrates absorbance changes associated with flash photolysis of CO followed by recombination. At 0.1 mM CO the recombination rate constant was approximately 100 s−1.
Figure 2.

Absorbance changes at 412 nm after flash photolysis of the CmCyt c-CO complex. Conditions: 1 μM CmCyt c, 0.1 mM CO, 10 mM Hepes, pH 7.1, 22 ± 1°C.
To investigate electron transfer from CmCyt c to cytochrome c oxidase, the CmCyt c-CO complex was mixed with the oxidized enzyme in the absence of oxygen. A low ionic strength was used to ensure complex formation between CmCyt c and the oxidase. In the dark the CO-off rate is ≈10−3 s−1, and no electron transfer was observed during the first ≈10 s after mixing, which indicates that the CmCyt c-CO complex has a higher redox potential than heme a/CuA (see also discussion below). A short time (≈100 ms) after mixing, CO was photodissociated, resulting in a drop of the apparent CmCyt c redox potential and electron transfer to the oxidase. The reaction was followed spectrophotometrically at a number of wavelengths in the region 400–450 nm and at 605 nm. Fig. 3 A and B shows absorbance changes at 445 nm and 605 nm, where heme a has the largest contribution to the reduced-minus-oxidized changes in the Soret and alpha regions of the spectrum, respectively, whereas the contribution from CmCyt c is <5% of the total absorbance change (see Fig. 1 A and B). A rapid, unresolved increase in absorbance associated with CO dissociation was followed by a slower increase with a rate constant of 1.0⋅104 s−1, consistent with reduction of Fea. This absorbance increase vanished upon addition of 300 mM KCl (Fig. 3A), which prevents formation of the electrostatic CmCyt c-cytochrome c oxidase complex. Similarly, when fully reduced cytochrome c oxidase was substituted for the oxidized enzyme the 104 s−1 change was not observed (not shown).
Figure 3.

(A) Absorbance changes after pulsed illumination of the ferro CmCyt c-CO complex about 100 ms after mixing with an anaerobic solution of cytochrome c oxidase (COX) (upper trace). The rapid initial increase in absorbance is due to CO dissociation. The following slower increase is associated with oxidation of CmCyt c and reduction of Fea. It was fit with an exponential function with a rate constant of 9,700 s−1 (solid noise-free line). The lower trace was recorded after addition of KCl to a concentration of ≈300 mM to the CmCyt c solution before the experiment. (B) Same experiment as upper trace in A but recorded at 605 nm. At this wavelength there is essentially no contribution from oxidation of ferro-CmCyt c (see Fig. 1). A laser artifact at t = 0 has been truncated. The ratio of the absorbance changes at 445 nm and 605 nm is consistent with reduction of heme a during the 9,700 s−1 phase. Conditions after mixing: 0.55 μM photoactive CmCyt c, 2.5 μM cytochrome c oxidase, 0.1 mM CO, 0.1% dodecyl maltoside, 10 mM Hepes, pH 7.1, 22 ± 1°C.
The CO-recombination rate in our experiments was about two orders of magnitude slower (≈102 s−1) than that of electron transfer (≈104 s−1). Consequently, <5% of CmCyt c recombined with CO during the time of the electron transfer.
Fig. 4A shows a kinetic difference spectrum (i.e., the change in absorbance as a function of wavelength) of the initial very rapid phase assigned to CO photodissociation. This spectrum fits well to the static CmCyt c2+-minus-CmCyt c2+(CO) spectrum (cf. Fig. 1A), confirming the assignment. From the fit, the concentration of photoactive CmCyt c(CO) was determined to be 0.55 μM. The kinetic difference spectrum of the 104 s−1 absorbance change, ΔAET, is shown in Fig. 4B. It was consistent with oxidation of CmCyt c and a fractional reduction of heme a, i.e.,
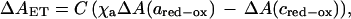 |
2 |
where C and χaC are the concentrations (in μM) oxidized CmCyt c and reduced heme a, respectively, and ΔA(ared-ox) and ΔA(cred-ox) are the absorption difference spectra of reduced-minus-oxidized heme a and CmCyt c, respectively, normalized to 1 μM.
Figure 4.

(A) Kinetic difference spectrum of the initial rapid absorbance changes following immediately after the flash (•) (see Fig. 3). The solid line is the difference spectrum of ferro- minus carboxy-ferro-CmCyt c (cf. Fig. 1A), scaled to a concentration of 0.55 μM. (B) Kinetic difference spectrum of the 9,700 s−1 absorbance changes (•) (see Fig. 3). The solid line represents the absorbance calculated from the reduced-minus-oxidized heme a and reduced-minus-oxidized CmCyt c spectra (see Eq. 2). In both A and B a positive absorbance change is defined as an increase in absorbance after the flash. Error bars (SD) are based on 1–3 measurements on three different samples at each wavelength.
The electron-transfer reaction is modeled as follows:
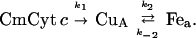 |
3 |
The fraction reduced Fea, χa, is related to the equilibrium constant K2 = k2/k−2 by:
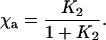 |
4 |
A fit of the model (Eqs. 2 and 3) to the data in Fig. 4B gives C = 0.40 μM and χa = 60 ± 10%, which using Eq. 4 gives K2 = 1.6, which is consistent with other studies (15, 16). Because the net photoactive CmCyt c concentration is 0.55 μM, the fraction CmCyt c oxidized by cytochrome c oxidase is 0.40 μM/0.55 μM = 73%.
The electron-transfer rate from Ru-modified cytochrome c to cytochrome c oxidase previously has been shown to depend on the position of the Ru-binding site (12, 13). A rate of ≈104 s−1 was observed when Lys-25, on the periphery of the heme crevice domain, was modified, whereas a rate of ≈105 s−1 was observed when Ru was attached at residue 39 opposite to the cytochrome c oxidase-binding site. It is likely that in CmCyt c the Lys-25 residue is also modified, which may account for the observed electron-transfer rate.
The redox potential of CmCyt c in aqueous solution has been reported to be −218 mV (17), i.e. about 0.5 V lower than that of the native cytochrome c (18). The dramatic decrease of the redox potential in the modified protein has been attributed to a larger degree of water exposure (17), which stabilizes the ferric form (19). Upon formation of a complex of CmCyt c and cytochrome c oxidase (or other acceptor proteins) water is likely to be released from the contact surface, which results in less water around the heme, thus increasing its redox potential (19).
After mixing the CmCyt c-CO complex with the oxidase in the absence of O2, no electron transfer was observed in the dark (≤10% in 10 s). The redox potential of CuA is below 300 mV (20, 21) whereas that of heme a in the fully oxidized enzyme is about 380 mV (22). Because these two centers are in rapid equilibrium, the absence of electron transfer implies that the redox potential of the CmCyt c-CO complex bound to the enzyme is at least about 400 mV. Consequently, after photolysis of CO the redox potential of the oxidase-bound CmCyt c would be at least 100 mV (300 mV lower than that of the CO complex, c.f. Eq. 1 with [CO] = 0.1 mM), which is similar to that of myoglobin (≈50 mV), a high-spin heme protein with a cavity protected from water exposure (23).
Fig. 5 shows a schematic energy-level diagram that models the reactions after photodissociation of CO from CmCyt c-CO complex bound to an electron acceptor. Reduced CmCyt c is mixed with the acceptor protein. The CO ligand is flashed off, resulting in a decrease of the apparent redox potential. This is then followed by electron transfer from CmCyt c to the acceptor.
Figure 5.

Energy-level diagram. After mixing of CmCyt c-CO (Fec2+-CO) and the electron acceptor (A) a complex is formed. Flash photolysis of CO results in a drop of the redox potential (increase in energy) by about 360 mV at 1 mM CO. The CO-recombination rate is ≈103 s−1 (at 1 mM CO). After dissociation of CO, electron transfer from CmCyt c to A proceeds with a rate constant kET. This is followed by slow re-reduction of CmCyt c by A− and CO recombination. Alternatively, if a reductant is present in solution (as in the experiments in this work), it may re-reduce CmCyt c directly.
The method we report here is, we believe, a very useful addition to the armory of techniques that can be used to study electron transfer. It has wide applicability and can be refined by substituting the more discriminating and specific methods of site-directed mutagenesis for the chemical modification we have used here to create a CO binding site in cytochrome c. We hope it also will provide a powerful way in which to study the role (if any) of specific amino acids on putative connectivity pathways linking the redox centers. To fully realize the potential of this method it will be important to determine more accurately the driving force for electron transfer. This will require determination of the redox potential of cytochrome c while in the electrostatic complex with its redox partner.
Acknowledgments
This study was supported by grants from the British Council, the Biotechnology and Biological Sciences Research Council of the United Kingdom, the Swedish Natural Science Research Council, and Carl Trygger’s Foundation.
ABBREVIATION
- CmCyt c
carboxymethylated cytochrome c
References
- 1.Brunori M, Giacometti G M, Antonini E, Wyman J. Proc Natl Acad Sci USA. 1973;70:3141–3144. doi: 10.1073/pnas.70.11.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schejter A, George P. Nature (London) 1965;206:1150–1151. doi: 10.1038/2061150a0. [DOI] [PubMed] [Google Scholar]
- 3.Stellwagen E. Biochemistry. 1968;7:2496–2501. doi: 10.1021/bi00847a008. [DOI] [PubMed] [Google Scholar]
- 4.Schejter A, Aviram I. J Biol Chem. 1970;245:1552–1557. [PubMed] [Google Scholar]
- 5.Wilson M T, Brunori M, Rotilio G C, Antonini E. J Biol Chem. 1973;248:8162–8169. [PubMed] [Google Scholar]
- 6.Wilson M T, Brunori M, Bonaventura J, Bonaventura C. Biochem J. 1973;131:863–865. doi: 10.1042/bj1310863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunori M, Wilson M T, Antonini E. J Biol Chem. 1972;247:6076–6081. [PubMed] [Google Scholar]
- 8.Keller R M, Aviram I, Schejter A, Wüthrich K. FEBS Lett. 1972;20:90–92. doi: 10.1016/0014-5793(72)80024-3. [DOI] [PubMed] [Google Scholar]
- 9.Ferguson-Miller S, Babcock G T. Chem Rev. 1996;96:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 10.Winkler J R, Malmström B G, Gray H B. Biophys Chem. 1995;54:199–209. doi: 10.1016/0301-4622(94)00156-e. [DOI] [PubMed] [Google Scholar]
- 11.Brzezinski P. Biochemistry. 1996;35:5611–5615. doi: 10.1021/bi960260m. [DOI] [PubMed] [Google Scholar]
- 12.Geren L M, Beasley J R, Fine B R, Saunders A J, Hibdon S, Pielak G J, Durham B, Millett F. J Biol Chem. 1995;270:2466–2472. doi: 10.1074/jbc.270.6.2466. [DOI] [PubMed] [Google Scholar]
- 13.Pan L P, Hibdon S, Liu R Q, Durham B, Millett F. Biochemistry. 1993;32:8492–8498. doi: 10.1021/bi00084a014. [DOI] [PubMed] [Google Scholar]
- 14.Brandt U, Schägger H, von Jagow G. Eur J Biochem. 1989;182:705–711. doi: 10.1111/j.1432-1033.1989.tb14882.x. [DOI] [PubMed] [Google Scholar]
- 15.Morgan J E, Li P M, Jang D-J, El-Sayed M A, Chan S I. Biochemistry. 1989;28:6975–6983. doi: 10.1021/bi00443a030. [DOI] [PubMed] [Google Scholar]
- 16.Ädelroth P, Brzezinski P, Malmström B G. Biochemistry. 1995;34:2844–2849. doi: 10.1021/bi00009a014. [DOI] [PubMed] [Google Scholar]
- 17.Di Marino M, Marassi R, Santucci R, Brunori M, Ascoli F. Bioelectrochem Bioenerg. 1987;17:27–34. [Google Scholar]
- 18.Dutton P L, Wilson D F, Lee C P. Biochemistry. 1970;9:5077–5082. doi: 10.1021/bi00828a006. [DOI] [PubMed] [Google Scholar]
- 19.Stellwagen E. Nature (London) 1978;275:73–74. doi: 10.1038/275073a0. [DOI] [PubMed] [Google Scholar]
- 20.Wang H, Blair D F, Ellis W R, Jr, Gray H B, Chan S I. Biochemistry. 1986;25:167–171. doi: 10.1021/bi00349a024. [DOI] [PubMed] [Google Scholar]
- 21.Babcock G T, Vickery L E, Palmer G. J Biol Chem. 1978;253:2400–2411. [PubMed] [Google Scholar]
- 22.Blair D F, Ellis W R, Jr, Wang H, Gray H B, Chan S I. J Biol Chem. 1986;261:11524–11537. [PubMed] [Google Scholar]
- 23.Brunori M, Saggase U, Rotilio G C, Antonini E, Wyman J. Biochemistry. 1971;10:1604–1609. doi: 10.1021/bi00785a016. [DOI] [PubMed] [Google Scholar]