

Abstract
Glial cell line-derived neurotrophic factor (GDNF)-dependent activation of the tyrosine kinase receptor RET is necessary for kidney and enteric neuron development, and mutations in RET are associated with human diseases. Activation of RET by GDNF has been shown to require an accessory component, GDNFR-α (RETL1). We report the isolation and characterization of rat and human cDNAs for a novel cell-surface associated accessory protein, RETL2, that shares 49% identity with RETL1. Both RETL1 and RETL2 can mediate GDNF dependent phosphorylation of RET, but they exhibit different patterns of expression in fetal and adult tissues. The most striking differences in expression observed were in the adult central and peripheral nervous systems. In addition, the mechanisms by which the two accessory proteins facilitate the activation of RET by GDNF are quite distinct. In vitro binding experiments with soluble forms of RET, RETL1 and RETL2 demonstrate that while RETL1 binds GDNF tightly to form a membrane-associated complex which can then interact with RET, RETL2 only forms a high affinity complex with GDNF in the presence of RET. This strong RET dependence of the binding of RETL2 to GDNF was confirmed by FACS analysis on RETL1 and RETL2 expressing cells. Together with the recent discovery of a GDNF related protein, neurturin, these data raise the possibility that RETL1 and RETL2 have distinctive roles during development and in the nervous system of the adult. RETL1 and RETL2 represent new candidate susceptibility genes and/or modifier loci for RET-associated diseases.
The RET protooncogene encodes a receptor tyrosine kinase that is expressed in a variety of tissues during development including the peripheral and central nervous systems and the kidney. Analysis of ret null mice has defined ret as critical for the migration and innervation of enteric neurons to the hindgut and for proliferation and branching of the ureteric bud epithelium during kidney development (1). In humans, mutations in RET can engender at least four different disease phenotypes (2–4). Somatic rearrangements of RET which result in receptor activation are associated with papillary thyroid carcinoma and germline activating mutations of RET are linked to the cancer syndromes multiple endocrine neoplasia type 2A and 2B (MEN2A and MEN2B). Familial Hirshsprung disease (HSCR), which is characterized by a lack of enteric nerve innervation to the hindgut, can arise from mutations in the endothelin pathway or in RET.
The search for a key component of the RET signaling pathway, the RET ligand, has been an area of intensive research. Recently, it has been shown that mice null for the gene encoding glial cell line-derived neurotrophic factor (GDNF) manifest a phenotype similar to that of mice null for ret (5–7). Both GDNF and ret null mice exhibit renal agenesis or severe dysgenesis and lack enteric neurons. The latter phenotype is reminiscent of human HSCR (1, 5–7). The similarity in the mutant phenotypes implied that GDNF and ret act in the same pathway. Initial experiments confirmed that GDNF could activate RET on cells, but a direct physical interaction between GDNF and RET was not demonstrated (8–10). Recent studies have shown that GDNF-dependent RET signaling requires a cell-surface-associated accessory protein, GDNFR-α. GDNFR-α binds GDNF to form a stable complex that can activate RET (11, 12). No binding of GDNFR-α to RET was seen in the absence of GDNF.
Using a direct expression cloning strategy, we have isolated a cDNA for GDNFR-α (RETL1) by its ability to interact with the extracellular domain of RET, demonstrating for the first time that a direct interaction between GDNFR-α and RET can be observed. We also report the identification of a novel cell surface protein, RETL2, which can mediate GDNF-dependent phosphorylation of RET. Human RETL2 shares 49% identity with human RETL1 but functions in a mechanistically distinct way. In contrast to RETL1, RETL2 can only bind GDNF with high affinity in the presence of RET. RETL1 and RETL2 display different expression patterns that could account for some of the tissue-specific phenotypic differences observed in human disease patients carrying RET mutations. RETL1 and RETL2 may represent new candidate susceptibility or modifying genes for RET-associated diseases.
MATERIALS AND METHODS
RET Fusion Proteins.
A cDNA encoding the extracellular domain of rat c-ret was isolated using the reverse transcription–PCR method. Poly(A) selected RNA from the day 14 embryonic rat kidney was converted to cDNA using avian myeloblastosis virus reverse transcriptase and amplified using Taq polymerase in a standard PCR with oligomers kid-013 (nucleotides 150–169 of GenBank sequence X15262X15262; human c-ret) and kid-015 (complement of nucleotides 1894–1914 of GenBank sequence X67812X67812; murine c-ret). The resulting PCR fragment was cloned and sequenced and found to encode the extracellular domain of rat RET, which exhibits 92% identity to murine RET (data not shown). To generate a rat RET–Ig fusion protein, a DNA fragment encoding amino acids 1–637 of rat RET was ligated to a fragment containing the Fc domain of human IgG1 and cloned into the Biogen expression vector pMDR901 to generate plasmid pJC022. Plasmid pJC022 was transfected into Chinese hamster ovary cells to generate a stable cell line producing the fusion protein. Clarified conditioned media from the cell line was loaded by gravity directly onto Protein A Sepharose (Pharmacia). The column was washed with five column volumes each of PBS, PBS containing 0.5 M NaCl, and 25 mM sodium phosphate, 100 mM NaCl (pH 5.0). The bound protein was eluted with 25 mM NaH2PO4, 100 mM NaCl (pH 2.8) and immediately neutralized with 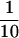 fraction volume of 0.5 M Na2HPO4 (pH 8.6). A plasmid encoding a fusion protein between the extracellular domain of rat RET and placental alkaline phosphatase (AP) was constructed as described (13) in the CH269 expression vector (described below) and expressed in 293-Epstein–Barr virus-encoded nuclear antigen (EBNA) cells. Conditioned medium was produced and used without further purification. SDS/PAGE analysis of the rat RET–AP fusion protein indicated a size consistent with its predicted molecular weight and gel filtration analysis indicated that it was produced as a dimer.
fraction volume of 0.5 M Na2HPO4 (pH 8.6). A plasmid encoding a fusion protein between the extracellular domain of rat RET and placental alkaline phosphatase (AP) was constructed as described (13) in the CH269 expression vector (described below) and expressed in 293-Epstein–Barr virus-encoded nuclear antigen (EBNA) cells. Conditioned medium was produced and used without further purification. SDS/PAGE analysis of the rat RET–AP fusion protein indicated a size consistent with its predicted molecular weight and gel filtration analysis indicated that it was produced as a dimer.
Expression Cloning.
A cDNA library was prepared from Wistar rat day 18 embryonic kidney mRNA in vector CH269 (derived from the Invitrogen vector, pCEP4, by excising the EBNA-1 gene), containing approximately 1 × 106 clones with an average insert size of 1.5 kb. Pools of 5000 colonies from the library were generated; part of the culture was used to make glycerol stocks that were stored and the rest of the culture was used to make DNA. DNA was purified using Qiagen (Chatsworth, CA) Qiafilter cartridges and Qiagen plasmid midi kits. Screening of the library was performed essentially as described by Cheng and Flanagan (13), except for modifications described below. DNAs from 256 pools were individually transfected into 293-EBNA cells (8 × 105 cells on a 60-mm Biocoat plate from Collaborative Biomedical Products, Bedford, MA) using lipofectamine (Bethesda Research Laboratories). After 48 hr, the cells were washed with 0.5 mg/ml BSA, 20 mM Hepes (pH 7.0) and 0.1% NaN3 and incubated with 20 μg/ml rat RET–Ig in Tris-buffered saline plus 1 mM MgCl2 and CaCl2 for 60–90 min at room temperature. Following this incubation, the cells were washed four times with 0.5 mg/ml BSA, 20 mM Hepes (pH 7.0), and 0.1% NaN3 and then fixed with 60% acetone/3% formaldehyde/20 mM Hepes (pH 7.0) for 30 sec. Following two washes with HBS buffer (150 mM NaCl/20 mM Hepes, pH 7.0), the cells were incubated with an AP-coupled secondary antibody [goat F(ab′)2 anti-human IgG Fc-γ-specific; Jackson ImmunoResearch; 1:5,000 dilution in Tris-buffered saline plus 1 mM MgCl2 and 1 mM CaCl2] for 60 min at room temperature. The cells were then washed twice with HBS buffer and twice with AP substrate buffer (100 mM NaCl/5 mM MgCl2/1.5 mM Lavamisole/100 mM Tris⋅HCl, pH 9.5). The last wash was left for 15 min. The AP substrates nitroblue tetrazolium chloride (0.33 mg/ml) and 5-bromo-4-chloro-3-indoylphosphate p-toluidine salt (BCIP) (0.17 mg/ml) were then added in AP substrate buffer containing 1.25 mM Lavamisole and incubated with the cells for 5–20 min. The plates were then washed with water and inspected under a dissecting microscope for the presence of a purple precipitate on cells as described (13). From an analysis of 256 pools, 17 positive pools were identified in the primary screen. DNA from each positive pool was retransfected into 293-EBNA cells and the above procedure repeated. Ten out of the 17 positive pools showed staining only with RET–Ig and not with a control IgG fusion protein. Two of the pools were broken down into smaller subpools until individual cDNA clones, 86–17 and OG-1, were identified. DNA sequence analysis of both clones revealed the same open reading frame encoding a protein of 468 amino acids (referred to as RETL1). A human RETL1 clone, GJ107, was isolated from a human embryonic kidney library obtained from Clontech, using a probe from the rat RETL1 clone, 86–17. The human RETL2 clone, DSW240, was isolated from a human fetal liver cDNA library obtained from Clontech, using two oligonucleotide probes corresponding to nucleotides 38–67 and 156–175 of GenBank sequence H12981H12981.
RET Phosphorylation Assay.
NB41A3 cells (1 × 106; ATCC CCL 147) were seeded into 60-mm tissue culture dishes and after 24 hr, transfected with 6 μg of plasmid DNA (full-length cDNA for rat RETL1, human RETL1, or human RETL2 in the CH269 expression vector) using lipofectamine. After 48 hr, cells were washed with DMEM without serum and treated for 10 min at 37°C with 100 ng/ml human GDNF (Promega) in DMEM without serum. Cells were rinsed once with PBS, then lysed for 15 min on ice with 0.5% Nonidet P-40, 0.2% sodium deoxycholate, 10 mM Tris⋅Cl (pH 8.0), 50 mM NaF, and 0.1 mM Na3VO4. The lysates were clarified by centrifugation for 20 min and precleared with Protein A Sepharose for 30 min at 4°C. RET was precipitated from the lysates with a hamster mAb raised against the rat RET–Ig fusion protein, AA.GE7.3, and Protein A Sepharose. The resin was washed twice with lysis buffer and bound proteins eluted with reducing sample buffer. The samples were boiled for 5 min, fractionated by SDS/PAGE (4–20% gel), and transfered to nitrocellulose (Hybond enhanced chemiluminescence, Amersham) for Western blot analysis. Total RET protein was detected using a rabbit polyclonal antibody against a C-terminal peptide of human RET (Santa Cruz Biotechnology) and a biotinylated-donkey anti-rabbit IgG antibody (Jackson ImmunoResearch). Phosphorylated RET was detected using a murine mAb against phosphotyrosine (Upstate Biotechnology, Lake Placid, NY) and a biotinylated-donkey anti-mouse IgG antibody (Jackson ImmunoResearch). Signals were visualized with biotinylated horseradish peroxidase (HRP) and avidin (Pierce) and the enhanced chemiluminescence detection system (Amersham).
Assessment of RETL1 and RETL2 Interactions with GDNF and RET by Direct Binding.
Plasmids encoding the rat RETL1-Ig and human RETL2-Ig fusion proteins were generated by ligating DNA fragments encoding amino acids 1–437 of rat RETL1 and amino acids 1–438 of human RETL2, respectively, with a DNA fragment encoding the Fc domain of human IgG1 in the expression vector CH269. The plasmids were transfected into 293-EBNA cells and stable lines were obtained using hygromycin selection. The fusion proteins were purified from conditioned medium using Protein A Sepharose as described above for the RET–Ig fusion protein.
Microtiter plates, either Titertech Linbro for studies with chromogenic substrate or Dynatech Microlite 1 for those using chemiluminescent detection, were coated with 250 ng/ml rhGDNF (Promega) in 50 mM sodium carbonate, pH 9.6. The plates were blocked for 1 hr at room temperature with 1% BSA in 10 mM Tris⋅HCl, pH 7.5, and 150 mM NaCl, and treated with serial dilutions of RETL1-Ig or RETL2-Ig in the presence or absence of 90 ng/ml RET–AP (1 hr, ambient temperature). Complexes were identified either by probing with a HRP-labeled mouse anti-human IgG antibody from Jackson ImmunoResearch (pH 7.5), and 150 mM NaCl plus 1% BSA and detecting HRP with the chromogenic substrate 2–2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid)diammonium salt (Boehringer Mannheim) or by assaying for alkaline phosphatase directly using the chemiluminescent substrate disodium 3-(4-methoxyspiro{1, 2-dioxetane-3, 2′-(5′-chloro)tricyclo[3.3.1.13,7]decan}-4-yl)phenyl phosphate (Tropix, Bedford, MA). Between steps, the plates were washed three times with TBS containing 0.03% Tween 20.
Fluorescence-Activated Cell Sorter (FACS) Analysis.
Stable 293-EBNA cell lines transfected with expression plasmids containing full-length sequences for human RETL1 and human RETL2 in the expression vector CH269, were established by selection in hygromycin. As a negative control, stable transformants containing only the expression vector were generated. To assess the binding of RET–Ig to these cells, the cells were washed with PBS and removed from the flask with 5.0 mM EDTA in PBS. Cells (2 × 105) were incubated with rat RET–Ig (final concentration of 20.0 μg/ml) in 0.1 ml FACS buffer (PBS/BSA/0.1% NaN3) supplemented with 1 mM CaCl2 and 1 mM MgCl2 for 1 hr at room temperature, with or without 0.25 μg/ml recombinant human GDNF (R & D Systems). The cells were washed once with FACS buffer and incubated with 0.05 ml of a 1:100 dilution of a phycoerythrin-conjugated goat F(ab)2 anti-human IgG, Fc fragment specific antibody (Jackson ImmunoResearch) for 20 min at room temperature. The cells were washed once again with FACS buffer, resuspended in 0.2 ml FACS buffer, and read in the FACSCAN. The binding of GDNF to these cells was assessed using essentially the same procedure but in the absence of RET–Ig. GDNF was detected using a murine mAb to GDNF (Promega; final concentration 20.0 μg/ml) for 30 min at room temperature, followed by treatment with a 1:100 dilution of a phycoerythrin-conjugated donkey F(ab)2 anti-mouse IgG,(H+L) antibody (Jackson ImmunoResearch) for 20 min at room temperature.
RESULTS
Cloning of cDNAs for RETL1 and RETL2.
An expression cloning strategy was used to identify proteins that interact with the extracellular domain of RET. This was accomplished using a soluble form of rat RET containing the RET extracellular domain fused to the hinge and CH2 and CH3 regions of the human IgG1 heavy chain. The purified rat RET–Ig fusion protein was active in a fetal kidney organ culture assay (14, 15), where RET is essential for the growth and branching of the ureteric bud epithelium that gives rise to the collecting ducts. A clear reduction in the overall size and the degree of branching of the collecting ducts was seen in RET–Ig treated kidneys (data not shown), indicating that the fusion protein blocked RET signaling. The RET–Ig fusion protein was next used in an expression cloning approach to screen a rat day 18 embryonic kidney cDNA library for potential RET binding proteins (see Materials and Methods). A single gene product of 468 amino acids was identified that we refer to as RETL1 because of its ability to bind RET. Subsequent testing revealed that an essential part of the expression cloning protocol was a fixing step. Without the fixing step, no binding of RET–Ig to the RETL1 transfected cells could be observed, suggesting that the interaction between RET–Ig and RETL1 was of relatively low affinity.
RETL1 contains a signal sequence, 31 cysteines, two potential N-linked glycosylation sites, and a hydrophobic C terminus, indicating that it may be linked to the cell via a phosphatidylinositol (PI) linkage (Fig. 1). Transient expression of the RETL1 cDNA clone, 86–17, in 293-EBNA cells, followed by cleavage with PI-specific phospholipase C, confirmed that RETL1 can be tethered to the cell via a PI linkage (data not shown) (11, 12). A cDNA clone, GJ107, encoding the human homolog of RETL1, was isolated from a human embryonic kidney library. The human protein is 93.3% identical to that of the rat (Fig. 1). Subsequent to our identification of RETL1, two groups reported the isolation of rat cDNAs encoding RETL1 that they obtained as a result of their efforts to clone a receptor for the neurotrophic factor GDNF (11, 12). They named the protein GDNFR-α because it binds to GDNF with high affinity. Jing et al. (11) also report the sequence of the human protein, which differs from our human sequence by two single amino acids and a five-amino acid insertion.
Figure 1.

Comparative analysis of RETL1 and RETL2 protein sequences. Alignment of rat and human RETL1 and RETL2 predicted protein sequences. Residues conserved in all four sequences are boxed. Sequences were aligned using the clustal method (16).
The peptide sequence of rat RETL1 was used to search the GenBank database with the program blast (19) to identify related proteins. Two significant matches were obtained. One was with GenBank accession no. R02249R02249, a 229-bp expressed sequence tag (EST) from a combined human fetal liver and spleen cDNA library, and the other was with GenBank accession no. H12981H12981, a 521-bp EST from a human infant brain cDNA library. The two ESTs share 99% identity in a region of overlap, indicating that they are from the same cDNA. A cDNA clone corresponding to these EST sequences was isolated from a human fetal liver cDNA library and found to contain an ORF encoding a protein of 464 amino acids, which we refer to as RETL2 (Fig. 1). The human RETL2 protein is 49.1% identical to human RETL1. It shares in common with human RETL1 a hydrophobic N terminus indicative of a signal sequence, and a hydrophobic C terminus indicative of a phophatidylinositol glycan linkage motif. In addition, 30 cysteines out of the 31 that are present in each protein are conserved. We have also isolated a cDNA for rat RETL2; the protein is 94.6% identical to human RETL2 (Fig. 1).
Functional Analysis of RETL1 and RETL2.
To evaluate whether RETL1 and RETL2 can facilitate GDNF dependent phosphorylation of RET, NB41A3 cells were transfected with RETL1 and RETL2, exposed to GDNF, and analyzed for tyrosine phosphorylation of RET. As shown in Fig. 2, treatment of vector transfected cells with GDNF results in only a very slight increase in RET phosphorylation. In contrast, cells transfected with either RETL1 or RETL2 show a significant increase in the level of RET tyrosine phosphorylation after treatment with GDNF. Jing et al. (11) and Treanor et al. (12) have provided similar results for RETL1 expressed in a different cell line, Neuro2A. The results shown in Fig. 2 provide the first evidence that, like RETL1, RETL2 can also facilitate GDNF dependent phosphorylation of RET.
Figure 2.

RETL1 and RETL2 can mediate GDNF dependent phosphorylation of RET. NB41A3 cells were transfected with the indicated plasmid and 48 hrs later treated with or without GDNF. Cells were lysed, precipitated with a mAb to RET, and subjected to Western blot analysis with either a polyclonal antibody to RET (Lower) or a mAb against phosphotyrosine (Upper).
Direct binding of RETL1 and RETL2 to GDNF and RET were evaluated using an ELISA type format in which 96-well plates were coated with GDNF and incubated with soluble forms of RET, RETL1, and RETL2. The soluble RET protein used in this experiment was a fusion of the RET extracellular domain with AP (RET–AP); both soluble RETL1 and RETL2 proteins were fusions with the Fc portion of human IgG1 (RETL1–Ig and RETL2–Ig). Fig. 3a shows that RETL1–Ig (detected with an anti-human Fc antibody) binds to GDNF coated plates with an apparent Kd ≈5 nM, whereas very little binding of RETL2–Ig is observed under the same conditions. Fig. 3b shows that, in the presence of RET–AP (90 ng/ml), RETL2–Ig binds tightly to GDNF with an apparent Kd ≈1 nM; the same concentration of RET–AP had no significant effect on the binding of RETL1–Ig. Detection of binding via the enzymatic activity of the RET–AP fusion rather than by the anti-human Fc antibody showed that RET–AP is a component in the complexes of both RETL1–Ig and RETL2–Ig with GDNF (Fig. 3c). In additional experiments the binding of RETL1–Ig was shown to be insensitive to the density of GDNF coating on the plate, whereas the extent of RET–AP-dependent binding of RETL2–Ig was sensitive to variations in GDNF density (data not shown). Under all conditions tested the binding of RETL2–Ig to GDNF was very weak in the absence of RET–AP, and was strongly enhanced by its presence.
Figure 3.

Detection of RETL1 and RETL2 interactions with GDNF and RET by direct binding. GDNF-coated microtiter plates were treated with serial dilutions of RETL1–Ig (▪), RETL2–Ig (•), or a control Fc fusion protein with the extracellular domain of LFA3 (▴) in the presence or absence of RET–AP. Samples were tested for complex formation either directly by measuring AP activity using the chemiluminescence substrate disodium 3-(4-methoxyspiro{1, 2-dioxetane-3, 2′-(5′-chloro)tricyclo[3.3.1.13,7]decan}-4-yl)phenyl phosphate, or indirectly by probing for Fc in the complex using an anti-human Fc specific antibody conjugated with HRP and the chromogenic substrate 2–2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid)diammonium salt (measured at 405 nm). (a) Samples were incubated in the absence of RET–AP and detected through the anti-Fc-HRP conjugate. (b) samples were incubated in the presence of RET–AP and detected through the anti-Fc-HRP conjugate. (c) samples were incubated in the presence of RET–AP and detected using the AP readout.
The strong dependence of the binding of RETL2 to GDNF on the presence of RET was confirmed by FACS experiments in which we measured the ability of GDNF to bind to cells stably expressing RETL1 or RETL2. In Fig. 4, a–c show cells evaluated for binding GDNF, while Fig. 4 d–f show cells evaluated for binding RET–Ig in the presence or absence of GDNF. Although cells transfected with the vector CH269 show some background binding of GDNF (Fig. 4a), cells expressing RETL1 show a significant shift (Fig. 4b) compared with the vector control, indicating that GDNF can bind to RETL1 expressing cells. In contrast, the majority of cells expressing RETL2 show only a minor shift (Fig. 4c) relative to the vector control, indicating that GDNF binds relatively poorly to RETL2 expressing cells. The shoulder to the right of this peak in Fig. 4c may represent a subpopulation of cells that express a high level of RETL2. Fig. 4 e and f show that, in the presence of GDNF, cells transfected with either RETL1 or RETL2 bind RET–Ig equally well indicating that, when RET is present, RETL2 is as effective as RETL1 at supporting the formation of the three-component complex.
Figure 4.

Binding of GDNF and RET–Ig to cells expressing RETL1 and RETL2 evaluated by FACS. 293-EBNA cell lines expressing RETL1 (b and e) and RETL2 (c and f) or transformed with the CH269 vector (a and d) were established. (a–c) The ability of these cell lines to bind GDNF was evaluated using a mAb to GDNF as described in the experimental methods. In each of these panels, curves on the left show cells incubated in the absence of GDNF. (d–f) The ability of these cell lines to bind RET-Ig was evaluated using an antibody against the human Fc region as described in the experimental methods. Curves labeled RET–Ig show cells incubated with RET–Ig in the absence of GDNF; curves labeled RET–Ig + GDNF show cells incubated with RET–Ig in the presence of GDNF. In each of these panels, control curves (which fall on top of the curves labeled RET–Ig) show cells incubated in the absence of both RET–Ig and GDNF.
Expression of RETL1 and RETL2.
Northern blot analyses of various embryonic and adult rat tissues were performed to compare the expression profiles of RETL1 and RETL2 (Fig. 5). Three transcripts of 3.1, 4.0, and 8.8 kb were observed with the RETL1 analysis, while two transcripts of 3.1 and 4.0 kb were observed for RETL2. Both RETL1 and RETL2 are expressed in embryonic brain, lung, kidney, and intestine, with higher levels of RETL1 observed in intestine and kidney and higher levels of RETL2 observed in lung. The 4.0 kb RETL2 transcript is expressed in all the adult tissues analyzed; the highest level of expression of RETL2 is in the lung and placenta. The 4.0 kb RETL1 transcript can be detected in all the adult tissues analyzed, while the 8.8-kb RETL1 transcript is expressed in kidney, heart, and placenta.
Figure 5.

Expression of RETL1 and RETL2 mRNA in rat tissues. Northern blots (17) contained 4 μg per lane of poly(A)+ mRNA from the indicated tissue. RETL1 and RETL2 cDNA probes of 1.3 kb and 1.4 kb, respectively, were generated by random priming (18). Human β-Actin and glyceraldehyde-3-phosphate dehydrogenase (G3PDH) control oligonucleotide probes (CLONTECH) were 5′ end-labeled with 32P using T4 polynucleotide kinase.
To further define the expression profiles of RETL1 and RETL2, a Northern analysis of human tissues within the central and peripheral nervous systems was performed (Fig. 6). One predominant transcript of 9.6 kb and two minor transcripts of 3.8 and 1.9 kb were observed with the RETL1 analysis, while two major transcripts of 3.6 and 4.2 kb and one minor transcript of 6.8 kb were observed for RETL2. The highest level of expression of RETL1 is observed in the substantia nigra, caudate nucleus, and spinal cord. In contrast, the highest level of expression of RETL2 is observed in the occipital, frontal, and temporal lobes and the cerebral cortex. Overall, the Northern analysis shows that while RETL1 and RETL2 are sometimes expressed in the same tissues, their expression profiles differ significantly, implying that they may perform distinct roles in the embryo and in the adult.
Figure 6.

Expression of RETL1 AND RETL2 mRNA in human brain. Northern blots containing mRNA from the indicated neuronal tissues (Clontech) were hybridized at 68°C in ExpressHyb (CLONTECH) and washed in 0.1× standard saline citrate (0.15 M sodium chloride/0.015 M sodium citrate, pH 7.0), 0.1% SDS at 50°C. RETL1 and RETL2 cDNA probes of 1.1 kb and 1.2 kb, respectively, were generated by random priming (18).
DISCUSSION
We report the isolation and characterization of rat and human cDNAs for RETL1 and RETL2. RETL1 and RETL2 encode two cell surface proteins that function as accessory molecules for RET. They are widely expressed throughout development but differ in their tissue distribution. The human RETL1 and RETL2 proteins share 49% identity, while the rat and human homologs share 93.3% (RETL1) and 94.6% (RETL2) identity. Both RETL1 and RETL2 can mediate GDNF-dependent phosphorylation of RET. However, our studies indicate that RETL1 and RETL2 differ in their ability to bind GDNF in a RET independent versus RET dependent manner. The differences in mechanisms and in the expression profiles of RETL1 and RETL2 could translate into functional differences in RET signaling in vivo.
RETL1 and RETL2 are expressed in a wide variety of tissues in the embryo and adult. An examination of RETL1 and RETL2 expression in rat embryonic tissues indicates a high level of RETL1 in fetal intestine. This is consistent with the expression profiles of RET and GDNF and the observation that mice null for GDNF or RET exhibit hindgut defects reminiscent of HSCR (1, 5–7, 12). Clearly, it will be interesting to determine if any of the familial cases of HSCR can be attributed to genetic alterations in RETL1. Expression of RETL1 is also observed in the fetal kidney, again consistent with sites associated with GDNF and RET expression and with an organ known to be dysgenic in GDNF and RET null mice. In adult rat tissues RETL1 is most abundant in heart and kidney, while RETL2 shows strong expression in placenta and lung. The high level of RETL2 expression in lung is intriguing because there are a few reported cases of small cell lung carcinomas being associated with alterations in RET (20). RETL1 and RETL2 exhibit different patterns of expression in human adult central nervous system tissues. Most striking is the high level of expression of RETL1 in the substantia nigra and caudate nucleus. Recent studies (21) indicate that GDNF when injected into the substantia nigra can show efficacy in models of Parkinson disease suggesting that GDNF signaling through RET in these cells is mediated via RETL1. RETL2 in the human brain is more discrete and is expressed in the cerebral cortex, predominantly in the occipital and frontal lobes.
The generation of soluble forms of RETL1, RETL2 and RET allowed us to directly study the binding properties of the components in vitro. Although both RETL1 and RETL2 mediate GDNF-dependent RET phosphorylation through formation of a three component complex, the mechanisms through that the complexes form are quite distinct. While we and others have shown that RETL1 can bind GDNF with high affinity in a RET-independent manner, our in vitro data suggest that RETL2 can only form a high affinity complex with GDNF in the presence of RET. These mechanistic differences could result in functional consequences in RET signaling in vivo. The strong RET-dependence of the GDNF/RETL2 interaction suggests that dissociation of the RETL2, RET, GDNF complex would release GDNF from the cell surface while dissociation of the RETL1, RET, GDNF complex would leave GDNF tightly bound to RETL1, and thus free to reassociate with RET and reform the complex. These differences might cause RETL2 expressing cells to be more sensitive than RETL1 expressing cells to fluctuations in the local concentration of free GDNF, because retention of GDNF on the surface of RETL1 expressing cells would allow RET to remain activated even if the concentration of free GDNF had decreased.
A second implication of our data is that the affinity of RETL2 for RET may be higher than that of RETL1 for RET. This raises the possibility that RETL2 and RET might preassociate on the membrane even in the absence of GDNF. Such an interaction might serve to regulate the proportion of RET that is free to engage in GDNF-dependent complexes with RETL1 and RETL2. We have demonstrated that an interaction can be observed between RET–Ig and RETL1 (this paper) and RETL2 (data not shown) expressed on cells, in the absence of GDNF, provided that a fixing step is included. Experiments are currently in progress to evaluate the relative affinities of RET for RETL1 and RETL2.
The ability of GDNF, a transforming growth factor β family member, to activate RET, a receptor tyrosine kinase was a surprising result since most members of the transforming growth factor β superfamily bind serine-threonine kinase receptors (22, 23). RETL1 and RETL2 may have evolved as adaptor molecules that allowed a receptor tyrosine kinase family member to recognize a new class of ligands (i.e., a transforming growth factor β family member). These accessory molecules could also facilitate the interaction of multiple ligands with RET. In fact, the existence of a new GDNF-like molecule, neurturin, has just been reported (24). It will be interesting to determine if neurturin will bind RETL1 and RETL2 and activate RET.
RETL1 and RETL2 represent two new candidate susceptibility genes for RET associated human diseases. Families with heritable cases of MEN2A, -2B, and HSCR not attributable to RET or GDNF (25) should be screened for alterations in RETL1 or RETL2. The existence of RETL1 and RETL2 may already suggest an explanation for the low penetrance seen in patients with familial HSCR due to loss of function mutations. For example, many different wildtype alleles of RETL1 and RETL2 could be present in the population and some of these alleles may compensate for and/or suppress the RET loss of function mutations associated with HSCR. In a few families, activating RET mutations that result in MEN2A occasionally cosegregate with a HSCR disease phenotype (2, 25–28). This observation is often postulated to be due to a difference in degree of penetrance in various tissues of the RET mutation or, alternatively, to a closely linked modifiers. It will be interesting to determine if RETL1 and RETL2 are linked to RET.
Acknowledgments
We thank Dr. Michael Donovan for preparing the embryonic kidney sections; David Willis for work during initial pilot experiments; Konrad Miatowski, Joseph Amatucci, and Werner Meier for assistance in producing the rat RET–Ig fusion protein; Michele McAuliffe, Christopher Tonkin, and Janice Nelson for DNA sequencing; the Biogen Media Preparation Group for providing excellent technical assistance; and Dr. Charis Eng for critical reading of the manuscript.
ABBREVIATIONS
- GDNF
glial cell line-derived neurotrophic factor
- GDNFR-α
GDNF receptor-α
- HSCR
Hirschsprung disease
- MEN2A and -2B
multiple endocrine neoplasia type 2A and B
- AP
alkaline phophatase, HRP, horseradish peroxidase
- EBNA
Epstein–Barr virus-encoded nuclear antigen
- FACS
fluorescence-activated cell sorter, PI, phosphatidylinositol
Footnotes
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. U97142–U97145U97142U97143U97144U97145).
References
- 1.Schuchardt A, D’Agati V, Larsson-Blomberg L, Costantini F, Pachnis V. Nature (London) 1994;367:380–383. doi: 10.1038/367380a0. [DOI] [PubMed] [Google Scholar]
- 2.Eng C. N Engl J Med. 1996;335:943–951. doi: 10.1056/NEJM199609263351307. [DOI] [PubMed] [Google Scholar]
- 3.van Heyningken V. Nature (London) 1996;367:319–320. [Google Scholar]
- 4.Pasini B, Ceccherini I, Romeo G. Trends Genet. 1996;12:138–144. doi: 10.1016/0168-9525(96)10012-3. [DOI] [PubMed] [Google Scholar]
- 5.Sánchez M P, Silos-Santiago I, Frisén J, He B, Lira S A, Barbacid M. Nature (London) 1996;382:70–73. doi: 10.1038/382070a0. [DOI] [PubMed] [Google Scholar]
- 6.Pichel J G, Shen L, Sheng H Z, Granholm A-C, Drago J, Grinberg A, Lee E J, Huang S P, Saarma M, Hoffer B J, Sariola H, Westphal H. Nature (London) 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- 7.Moore M W, Klein R D, Fariñas I, Sauer H, Armanini M, Phillips H, Reichardt L F, Ryan A M, Carver-Moore K, Rosenthal A. Nature (London) 1996;382:76–79. doi: 10.1038/382076a0. [DOI] [PubMed] [Google Scholar]
- 8.Durbec P, Marcos-Gutierrez C V, Kilkenny C, Grigoriou M, Wartiowaara K, Suvanto P, Smith D, Ponder B, Costantini F, Saarma M, Sariola H, Pachnis V. Nature (London) 1996;381:789–792. doi: 10.1038/381789a0. [DOI] [PubMed] [Google Scholar]
- 9.Trupp M, Arenas E, Fainzilber M, Nilsson A-S, Sieber B-A, Grigoriou M, Kilkenny C, Salazar-Grueso E, Pachnis V, Arumäe U, Sariola H, Saarma M, Ibáñez C F. Nature (London) 1996;381:785–788. doi: 10.1038/381785a0. [DOI] [PubMed] [Google Scholar]
- 10.Vega Q C, Worby C A, Lechner M S, Dixon J E, Dressler G R. Proc Natl Acad Sci USA. 1996;93:10657–10661. doi: 10.1073/pnas.93.20.10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jing S, Wen D, Yu Y, Holst P L, Luo Y, Fang M, Tamir R, Antonio L, Hu Z, Cupples R, Louis J-C, Hu S, Altrock B W, Fox G M. Cell. 1996;85:1113–1124. doi: 10.1016/s0092-8674(00)81311-2. [DOI] [PubMed] [Google Scholar]
- 12.Treanor J J S, Goodman L, de Sauvage F, Stone D M, Poulsen K T, et al. Nature (London) 1996;382:80–83. doi: 10.1038/382080a0. [DOI] [PubMed] [Google Scholar]
- 13.Cheng H-J, Flanagan J G. Cell. 1994;74:157–168. doi: 10.1016/0092-8674(94)90408-1. [DOI] [PubMed] [Google Scholar]
- 14.Saxen L, Lehtonen E. Differentiation (Berlin) 1994;36:2–11. doi: 10.1111/j.1432-0436.1987.tb00176.x. [DOI] [PubMed] [Google Scholar]
- 15.Kreidberg J A, Sariola H, Loring J M, Maeda M, Pelletier J, Housman D, Jaenisch R. Cell. 1993;74:679–691. doi: 10.1016/0092-8674(93)90515-r. [DOI] [PubMed] [Google Scholar]
- 16.Higgins D G, Bleasby A J, Fuchs R. Nature (London) 1992;384:467–470. [Google Scholar]
- 17.Cate R L, Mattaliano R J, Hession C, Tizard R, Farber N M, Cheung A, Ninfa E G, Frey A Z, Gash D J, Chow E P, Fisher R A, Bertonis J M, Torres G, Wallner B P, Ramachandran K L, Ragin R C, Manganaro T F, MacLaughlin D T, Donahoe P K. Cell. 1986;45:685–698. doi: 10.1016/0092-8674(86)90783-x. [DOI] [PubMed] [Google Scholar]
- 18.Feinberg A P, Vogelstein B. Anal Biochem. 1984;137:266–267. doi: 10.1016/0003-2697(84)90381-6. [DOI] [PubMed] [Google Scholar]
- 19.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 20.Futami H, Egawa S, Tsukada T, Maruyama K, Bandoh S, Noguchi M, Yamaguchi K. Jpn J Cancer Res. 1995;86:1127–1130. doi: 10.1111/j.1349-7006.1995.tb03304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gash D M, Zhang Z, Ovadia A, Cass W A, Yi A, Simmerman L, Russell D, Martin D, Lapchak P A, Collins F, Hoffer B J, Gerhardt G A. Nature (London) 1996;380:252–255. doi: 10.1038/380252a0. [DOI] [PubMed] [Google Scholar]
- 22.Massagué J. Nature (London) 1996;382:29–30. doi: 10.1038/382029a0. [DOI] [PubMed] [Google Scholar]
- 23.Lindsay R M, Yancopoulos G D. Neuron. 1996;17:571–574. doi: 10.1016/s0896-6273(00)80189-0. [DOI] [PubMed] [Google Scholar]
- 24.Kotzbauer P T, Lampe P A, Heuckeroth R O, Golden J P, Creedon D J, Johnson E M, Jr, Milbrandt J. Nature (London) 1996;384:467–470. doi: 10.1038/384467a0. [DOI] [PubMed] [Google Scholar]
- 25.Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, et al. J Am Med Assoc. 1996;276:1575–1579. [PubMed] [Google Scholar]
- 26.Angrist M, Bolk S, Thiel B, Puffenberger E G, Hofstra R M, Buys C H, Cass D T, Chakravarti A. Hum Mol Genet. 1995;4:821–830. doi: 10.1093/hmg/4.5.821. [DOI] [PubMed] [Google Scholar]
- 27.Mulligan L M, Eng C, Attié T, Lyonnet S, Marsh D J, Hyland V J, Robinson B G, Frilling A, Verellen-Dumoulin C, Safar A, Venter D J, Munnich A, Ponder B A J. Hum Mol Genet. 1994;3:2163–2167. doi: 10.1093/hmg/3.12.2163. [DOI] [PubMed] [Google Scholar]
- 28.Borst M J, VanCamp J M, Peacock M L, Decker R A. Surgery (St Louis) 1995;117:386–391. doi: 10.1016/s0039-6060(05)80057-1. [DOI] [PubMed] [Google Scholar]