

Abstract
SUMMARY
Despite their known transforming properties, the effects of leukemogenic FLT3-ITD mutations on hematopoietic stem and multipotent progenitor cells and on hematopoietic differentiation are not well understood. We report a mouse model harboring an ITD in the murine Flt3 locus that develops myeloproliferative disease resembling CMML and further identified FLT3-ITD mutations in a subset of human CMML. These findings correlated with an increase in number, cell cycling and survival of multipotent stem and progenitor cells in an ITD dose-dependent manner in animals that exhibited alterations within their myeloid progenitor compartments and a block in normal B-cell development. This model provides insights into the consequences of constitutive signaling by an oncogenic tyrosine kinase on hematopoietic progenitor quiescence, function, and cell fate.
SIGNIFICANCE
Activating FLT3 mutations are among the most common genetic events in AML and confer a poor clinical prognosis. Essential to our understanding of how these lesions contribute to myeloid leukemia is the development of a Flt3-ITD ‘knock-in’ murine model that has allowed examination of the consequences of constitutive FLT3 signaling on primitive hematopoietic progenitors when expressed at appropriate physiologic levels. These animals informed us to the existence of FLT3-ITD-positive human CMML, which has clinical importance given the availability of FLT3 small molecule inhibitors. This model will not only serve as a powerful biological tool to identify mutations that cooperate with FLT3 in leukemogenesis, but also to assess molecular therapies that target either FLT3 or components of its signaling pathways.
INTRODUCTION
The FMS-like tyrosine kinase 3 (FLT3) gene encodes a class III receptor tyrosine kinase (RTK) that shares strong sequence and structural similarities to other family members including FLT1, FMS, PDGF βR, and c-KIT (Rosnet and Birnbaum, 1993). FLT3 plays a critical role in normal hematopoiesis [for reviews see (Gilliland and Griffin, 2002; Stirewalt and Radich, 2003)] and within the hematopoietic system, its expression occurs primarily in immature myeloid and lymphoid progenitors, including CD34+ cells with high levels of CD117 (c-KIT) expression (Rasko et al., 1995; Rosnet et al., 1996), but not in erythroid cells (Gabbianelli et al., 1995), megakaryocytes (Ratajczak et al., 1996), or mast cells (Hjertson et al., 1996). Although targeted disruption of Flt3 results in healthy adult mice with normal mature hematopoietic populations, these animals demonstrate deficiencies in primitive pro-B and pre-B cell lymphoid compartments (Mackarehtschian et al., 1995). Moreover, bone marrow reconstitution experiments revealed a reduced ability of stem cells lacking Flt3 to reconstitute both T cells and myeloid cells (Mackarehtschian et al., 1995), together indicating an important role for FLT3 in the development of multipotent hematopoietic stem cells and lymphoid cells.
High levels of wild type FLT3 expression have been detected in a number of hematologic malignancies including the vast majority of patients with AML (70->90%) (Carow et al., 1996; Rosnet et al., 1996) and a large proportion of precursor B-cell acute lymphoblastic leukemia (ALL) including a subset of ALL carrying a chromosomal translocation involving the 11q23 locus (Armstrong et al., 2002; Drexler, 1996; Rosnet et al., 1996). Congruent with these findings, FLT3 is also expressed at high levels in both leukemia and lymphoma cell lines (DaSilva et al., 1994; Meierhoff et al., 1995) including pre-B, myeloid, and monocytic cell lines.
Internal tandem duplications (ITD) within the juxtamembrane (JM) domain of FLT3 in patients with AML were first reported in 1996 (Nakao et al., 1996) and occur in approximately 25% of patients, making it one of the most single common mutations in adult AML (Frohling et al., 2002; Kiyoi et al., 1999; Kottaridis et al., 2001; Schnittger et al., 2002; Whitman et al., 2001). FLT3-ITD mutations result in ligand-independent receptor dimerization (Kiyoi et al., 1998), constitutive FLT3 signaling, and activation of the STAT5, RAS/MAPK, and PI3K pathways and confer factor-independent growth to 32D and Ba/F3 cells (Hayakawa et al., 2000; Mizuki et al., 2000). Another major class of FLT3 mutations that also cause constitutive FLT3 activation and induce autonomous proliferation of cytokine-dependent cell lines, occurs within the activation loop (AL) of the second kinase domain (Yamamoto et al., 2001). This group of mutations is comprised of substitutions, small deletions, or insertions most commonly involving codons 835 and 836 and is detected in approximately 5-10% of patients with AML. More recently, AL mutations in FLT3 have also been described in cases of acute lymphoblastic leukemias (ALL) that harbor rearrangements of the Mixed Lineage Leukemia (MLL) gene on chromosome 11q23 (Armstrong et al., 2003) as well as AL and ITD mutations in a small subset of T-ALL (Paietta et al., 2004) implicating FLT3 in the pathogenesis of both lymphoid and myeloid disease. Although both FLT3-AL and FLT3-ITD mutations cause constitutive activation of the receptor tyrosine kinase, their signal transduction properties and transforming abilities appear to differ considerably from one another (Choudhary et al., 2005; Grundler et al., 2005) arguing for differential roles of these classes of mutations in AML pathogenesis. Lastly, a group of point mutations within the JM domain have also recently been described in approximately 1% of AML cases involving several amino acids including residues 579, 590, 591, 592, and 594 (Reindl et al., 2006; Stirewalt et al., 2004).
Gain of function mutations in FLT3, in particular FLT3-ITD mutations, are of significant clinical consequence, and a number of studies have shown that FLT3-ITD mutations may be associated with disease progression (Horiike et al., 1997; Ishii et al., 1999) and are associated with an increased risk of relapse and shorter overall survival (Frohling et al., 2002; Kiyoi et al., 1999; Kottaridis et al., 2001; Schnittger et al., 2002; Thiede et al., 2002; Whitman et al., 2001). Moreover, patients with low or absent levels of wild-type FLT3, consistent with homozygosity for the FLT3-ITD allele appear to have a particularly dismal outcome (Thiede et al., 2002; Whitman et al., 2001), suggesting FLT3-ITD gene dosage has biologic and prognostic significance.
Given their frequency and relevance to AML disease pathogenesis and prognosis, there is a compelling basis for understanding the role of activating FLT3 mutations in leukemogenesis and critical to this advancement is the development of accurate leukemogenic FLT3 animal models. In this study, we have generated a murine model whereby we have introduced an internal tandem duplication (ITD) mutation in the endogenous murine Flt3 locus. This model has distinct advantages over prior retroviral transduction and transgenic platforms employing heterologous promoters where differences in expression levels of activated FLT3 may affect disease phenotype and avoids potential cooperating mutations introduced by retroviral integration (Baldwin et al., 2007; Kelly et al., 2002b; Lee et al., 2005). Employing this model, we have explored the biological impact of constitutive FLT3 signaling on primitive hematopoietic stem and progenitor populations, providing significant mechanistic insights into how FLT3-ITD mutations contribute to myeloid leukemogenesis and demonstrate that FLT3-ITD expression results in the development of a fully penetrant myeloproliferative disorder within these animals. The murine phenotype closely resembles human chronic myelomonocytic leukemia (CMML) and genomic analysis of a large series of primary patient samples led to the identification of a subset of FLT3-ITD positive CMML. This mouse model provides an accurate framework for understanding the biological mechanisms governing the genesis and progression of FLT3 associated myeloid disease. Moreover, not only can it serve as a powerful system to identify cooperating mutations with FLT3 in leukemogenesis, but also allows the ability to assess the therapeutic efficacy of molecular agents that target FLT3 or downstream signaling pathways activated by the FLT3-ITD allele in these cooperative models of acute leukemia.
RESULTS
Generation of a FLT3-ITD ‘knock-in’ murine model
To better understand the role of FLT3-ITD mutations in disease pathogenesis when expressed physiologically from its endogenous promoter, we engineered a human ITD (W51) (Kelly et al., 2002b) into exon 14 of the murine Flt3 locus by homologous recombination in embryonic stem (ES) cells (Figure 1A-1C). Correctly targeted ES cells were used to produce viable mice carrying mutant ITD alleles. Heterozygous (Flt3+/ITD) mice were intercrossed to produce homozygous Flt3ITD/ITD animals that were viable and fertile with predicted Mendelian ratios (data not shown). Both heterozygous and homozygous animals expressed comparable levels of Flt3 protein to wild-type (wt) littermates (Figure 1D and 1E). Compared to wt BM and spleen cells, Flt3ITD/ITD mutant mice also demonstrated a consistent increase in the levels of phospho-Flt3 and the known downstream signaling intermediate, phospho-Stat5 (Rocnik et al., 2006). This was also observed to a lesser degree in Flt3+/ITD animals (Figure 1F).
Figure 1. ‘Knock-in’ of an internal tandem duplication (ITD) mutation in the murine Flt3 gene.
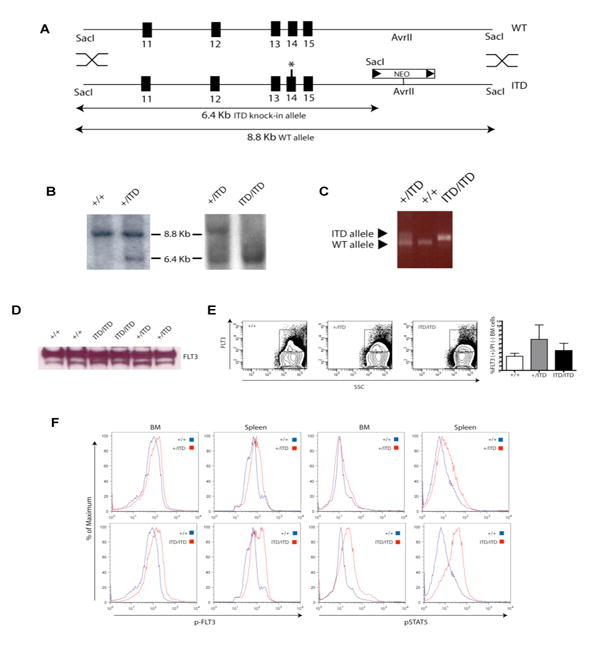
(A) Schematic of the Flt3 locus (WT) and targeting vector (ITD) comprised of murine genomic sequences encompassing exons 11-15 and an in-frame human W51 ITD mutation (*) within exon 14 and a neomycin resistance cassette flanked by two loxP sites (arrowheads) downsteam of exon 15. (B) Southern blot analysis with the 5′ probe of SacI-digested DNA from Flt3+/+, Flt3+/ITD, and Flt3ITD/ITD mice. (C) PCR analysis of mouse tail DNA from Flt3+/+, Flt3+/ITD, and Flt3ITD/ITD animals. (D) Immunoblot of Flt3 expression in primary spleen cells from mutant and wt animals. Equivalent amounts of protein were loaded from spleen protein lysates from two independent animals with the indicated genotype and blotted with anti-Flt3 antibody. (E) Representative flow cytometric analysis of Flt3 expression in total, live (PI negative) BM cells in Flt3+/+, Flt3+/ITD, and Flt3ITD/ITD mice. Data in bar graph represent the percentage of Flt3 positive in total live BM cells (mean +/- S.E.M.) Flt3+/+ (n=3), Flt3+/ITD (n=4), Flt3ITD/ITD (n=5) (p=NS), although the cell numbers which express FLT3 are increased FLT3-ITD mice, the mean fluorescence intensity range which correlates with FLT3 protein expression is similar for all 3 genotypes. (F) Flow cytometric analysis of levels of phospho-Flt3 (p-FLT3) and phospho-Stat5 (pSTAT5) in whole BM and spleen cells from mutant mice and wt littermate controls. Data are representative of two independent experiments.
Flt3+/ITD and Flt3ITD/ITD animals develop a myeloproliferative disease with monocytic features
Comparative analysis of Flt3+/ITD and Flt3ITD/ITD with age-matched wt littermates demonstrated dose-dependent development of progressive splenomegaly (Figure 2A and Supplementary Figure 1A). Mutant animals showed mildly decreased liver size, hemoglobin levels, and platelet counts (Supplementary Figure 1A-1D), and Flt3ITD/ITD animals exhibited significant leukocytosis and monocytosis with a reversal of neutrophil to lymphocyte ratios seen in both hetero- and homozygous animals compared to Flt3+/+ wt controls (Figure 2B and 2C). Morphologic and flow cytometric analysis of mutant animals further confirmed the expanded granulocyte (Gr1+/Mac1+/FSChi/SSChi) and monocyte (Gr1-/lo/Mac1+/FSCint/SSCint) populations in their peripheral blood (PB) (Figure 2D). Histopathology demonstrated an expansion of splenic red pulp in mutant animals comprised mainly of mature myeloid and admixed erythroid elements as well as a marked white pulp expansion by cells resembling maturing monocytes. These cells were also present in mutant BM and livers in a background of increased numbers of maturing myeloid forms (Figure 3A) and exhibited variably positive non-specific esterase (NSE), but negative sudan black B (SBB) staining confirming their monocytic nature (Figure 3D). Flow analysis of mutant BM and spleens demonstrated significantly increased mature myeloid (Gr1+/Mac1+) and monocytic (Gr1-/lo/Mac1+/F4-80+) populations as well as immature (Mac1+/-/ckit+) cells versus wt controls (Figure 3B and data not shown), which were accompanied by a concomitant decrease in the percentage of B and T lymphoid cells (Figure 3B). These findings were supported by B220, CD3, and Mac1 immunohistochemistry of mutant and wt spleens, that also demonstrated an absence of staining for MPO, CD34, and Mac3 within the expanded splenic white pulp (Figure 3C and data not shown). Additional analysis of the B cell population indicated that there was a significant loss of mature B cells (B220+IgM+; data not shown) in both the BM and the spleen from mutant animals and further examination indicated a specific block of B cell development at the pre-B stage (B220+CD43-; Figure 3E).
Figure 2. Myelomonocytic expansion in Flt3+/ITD (+/ITD) and Flt3ITD/ITD (ITD/ITD) animals versus wild type Flt3+/+ (+/+) mice.
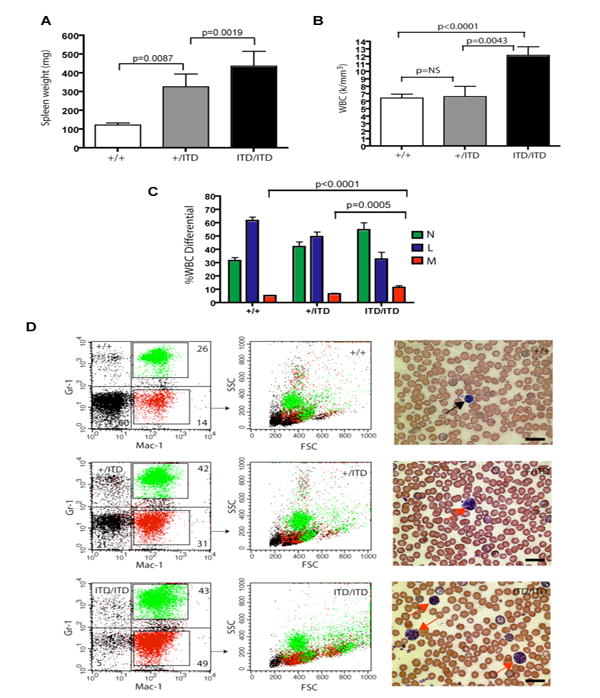
(A) composite data from age-matched littermates of indicated genotypes in the C57BL6 background (mean +/- S.E.M.; Flt3+/+, n=20; Flt3+/ITD, n=21; Flt3ITD/ITD, n=18) demonstrating ITD dose-dependent splenomegaly. (B) Total white blood cell (WBC) counts and (C) WBC differential values from age-matched littermates of mutant and wt animals (mean +/- S.E.M.). (D) Representative flow cytometric analysis of peripheral blood from Flt3+/+, Flt3+/ITD, Flt3ITD/ITD mice for Gr1 and Mac1 highlights expanded granulocytic (green) and monocytic (red) populations, the former exhibiting high forward (FSChi) and side (SSChi) scatter properties and the latter exhibiting characteristic intermediate forward (FSCint) and side (SSCint) scatter properties. Peripheral blood images illustrate an unremarkable smear and lymphocyte (black arrow) in a wt animal, but progressively increased numbers of monocytes (red arrows) in mutant animals (Wright-Giemsa; scale bars, 20 μm).
Figure 3. Flt3+/ITD and Flt3ITD/ITD mice develop a chronic myeloproliferative disease with a prominent monocytic component.
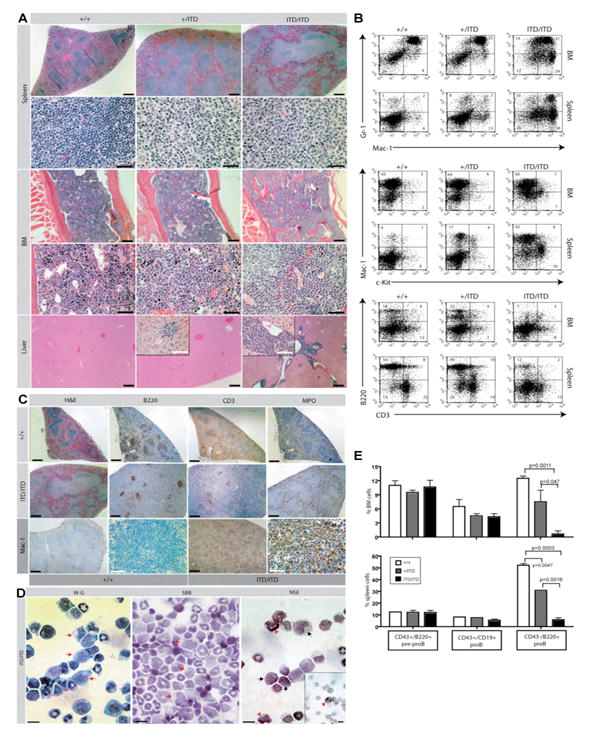
(A) Histopathologic sections of spleen, bone marrow (BM), and liver from representative Flt3+/+ (+/+), Flt3+/ITD (+/ITD), and Flt3ITD/ITD (ITD/ITD) animals in the C57BL6 background. Splenic red pulp is expanded largely by maturing myeloid and erythroid elements in both hetero- and homozygous animals which also display a prominent white pulp expansion comprised largely of intermediate sized cells with moderate amounts of pale cytoplasm (monocytes) compared to the normal small lymphocyte populations seen in wt control white pulp [spleen: first row (H&E; scale bars, 500 μm), second row (H&E; scale bars, 50 μm)]. Clusters of morphologically similar pale cells are observed in BM and liver of Flt3ITD/ITD mice in a background of significant maturing myeloid hyperplasia (also seen in heterozygous animals to a lesser degree) when compared with unremarkable BM from wt controls displaying normal maturing trilineage hematopoiesis [BM: first row (H&E; scale bars, 200 μm), second row (H&E; scale bars, 50 μm); Liver (H&E; scale bars, 200 μm; insets, 60 μm)]. (B) Flow cytometric analysis of single cell suspensions of BM and spleen of representative Flt3+/+ (+/+), Flt3+/ITD (+/ITD), and Flt3ITD/ITD (ITD/ITD) mice demonstrates a progressive increase in both mature granulocytic (Gr1+/Mac1+) and monocytic (Gr1-/lo/Mac1+) populations as well as immature myelomonocytic (Mac1+/-/ckit+) cells with a corresponding decrease in the amount of B (B220+) and T (CD3+) lymphoid cell populations. (C) Immunohistochemistry staining of paraffin-embedded spleen sections from Flt3+/+ and Flt3ITD/ITD mice for B220, CD3, and Mac1 illustrate similar reductions in the B and T lymphoid populations and an expanded Mac1 population in mutant versus wt control respectively for both Flt3+/+ and Flt3ITD/ITD mice. Scale bars; 500 μm (black); 50 μm (white). Stains for myeloperoxidase (MPO) highlight increased myeloid populations confined within splenic red pulp of homozygous mutant animals. (D) Cytospins of BM cells from Flt3ITD/ITD mice demonstrates variably weak (black arrows; third panel) to strong (red arrowheads; third panel) staining of the expanded monocyte population [first panel: red arrows, Wright-Giemsa (W-G)] for napthol AS-D chloroacetate esterase [third panel: NSE), but negative staining for sudan black B [middle panel: red arrows, SBB]. Scale bars, 20 μm. (E) Flow cytometric analysis of percentage (mean +/- S.E.M.) of pre-proB (CD43+/B220+), proB (CD43+/CD19+), and preB (CD43-/B220+) cells in BM and spleen from Flt3+/+, Flt3+/ITD and Flt3ITD/ITD mice indicates an ITD allelic dose-dependent block at the preB stage of development.
Flt3-ITD myeloproliferative disease including this block in B cell development was transplantable into secondary recipients as determined by spleen weight, histopathologic examination and flow cytometric studies performed on lethally irradiated (2 × 550 rad) wild type recipient animals, receiving 1×106 BM cells from either Flt3+/+ or Flt3ITD/ITD animals (Supplemental Figure 2), at experimental endpoint, supporting the notion that the phenotypic effects of the ITD allele were cell autonomous.
Cell biological and developmental impact of FLT3-ITD expression in the hematopoietic stem and progenitor compartment
When plated in methylcellulose cultures in the presence of growth factors, BM cells from Flt3ITD/ITD mice demonstrated increased colony numbers overall, with proportionately greater numbers of monocyte colony forming units (CFU-M; Figure 4A and 4B) as well as fewer erythroid burst forming units (BFU-E) and granulocyte-erythroid-monocyte-megakarocyte units (CFU-GEMM) (Figure 4A) compared to Flt3+/ITD and Flt3+/+ littermates. In addition, colonies derived from Flt3+/ITD or Flt3ITD/ITD BM did not show any enhanced serial replating activity and consistently gave rise to fewer numbers of colonies in the second round of replating (Figure 4C) demonstrating that the ITD allele does not confer properties of self-renewal to hematopoietic progenitors in vitro.
Figure 4. Colony forming assays, hematopoietic stem cell (HSC) and progenitor analysis of mutant Flt3 animals.
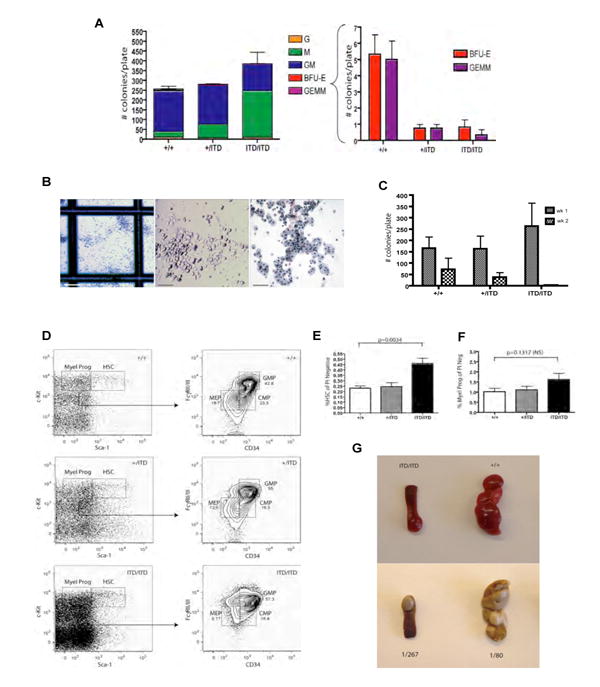
(A) BM cells from Flt3+/+, Flt3+/ITD, Flt3ITD/ITD mice were plated on M3434 methylcellulose medium (containing SCF, IL-3, IL-6, EPO) and scored for colony formation 7-10 days later (G=granulocyte, M=macrophage, GM=granulocyte macrophage, BFU-E=burst-forming uniterythroid, GEMM=granulocyte, erythroid, macrophage, megakaryocyte). Results are the average of three independent experiments performed in duplicate (mean +/- S.E.M. are shown). (B) Morphology of increased numbers of CFU-M colonies [left panel, scale bar, 500 μm; middle panel; scale bar, 200 μm, right panel; scale bar, 100 μm (Wright-Giemsa)]. (C) Bone marrow cells harboring the mutant Flt3-ITD allele do not demonstrate serial replating capability and exhibit a dose-dependent decreased replating potential capacity. Results presented are the mean +/- S.E.M. of two independent experiments performed in duplicate. (D-F) Multiparameter flow cytomery demonstrates significantly increased numbers of Lin-Sca1+ckit+ cells and a trend towards increased myeloid progenitors in Flt3ITD/ITD mice over heterozygous and wt animals with a progressive increase in the relative proportions of granulocyte-monocyte progenitors (GMP) in these mice. Plotted in (E) and (F) are the values from two independent experiments (mean +/- S.E.M.; Flt3+/+, n=5; Flt3+/ITD, n=6; Flt3ITD/ITD, n=6). (G) Spleen colony forming unit (CFU-S) ability is diminished in Flt3ITD/ITD HSC (Lin-Sca1+ckit+) cells. Duplicate wt C57BL6 recipient mice were transplanted with 500 Lin-Sca1+ckit+ cells sorted from two independent Flt3+/+ or Flt3ITD/ITD donor mice and spleens from representative recipient animals are shown. Frequency of CFU-S is calculated as 1/(# cells transplanted/# colonies observed).
Evaluation of the Lin-Sca1+ckit+ (LSK) compartment (containing long-term and short-term HSC, as well as multipotent progenitors cells) and myeloid progenitor (Lin-Sca1-ckit+) populations of Flt3+/ITD and Flt3ITD/ITD BM demonstrated a ∼2 fold increase in the number of LSK cells (p=0.0034) and a mild trend of increased myeloid progenitors in Flt3ITD/ITD mice over heterozygous and wt littermates (Figure 4D). Although total numbers of common lymphoid progenitors (CLP) and total myeloid progenitors were not significantly different between genotypes (Supplementary Figure 3 and Figure 4F), the ITD allele affected subpopulations within the myeloid progenitor compartment, with an expansion in the relative percentage of immunophenotypic granulocyte-monocyte progenitors (GMP), and a corresponding decrease in megakaryocyte-erythroid progenitors (MEP) (Figure 4D), consistent with the myeloproliferative phenotype observed in the mice and enhanced CFU-M and reduced BFU-E. We also observed significantly reduced size and numbers of CFU-S in mice transplanted with Flt3ITD/ITD LSK compared with wt LSK (Figure 4G). As CFU-S activity typically reflects primitive multipotent progenitors or ST-HSCs with considerable megakaryocyte-erythroid potential, these observations further support the erythroid potential of ST-HSCs being negatively affected in Flt3ITD/ITD mice.
Analysis of the effects of the Flt3-ITD allele on cell cycle of LSK and myeloid progenitor compartments demonstrated an ITD dose-dependent increase in the proportion of LSK cells in S/G2/M with a corresponding decrease in the proportion in G0/G1 (Figure 5A and 5B). This effect appeared specific to LSK cells as no discernable differences in cell cycle distributions were detected in myeloid progenitors from wt and mutant animals (Figure 5A and 5C). In contrast, both LSK and myeloid progenitor populations demonstrated an ITD dose-dependent decrease in the number of cells undergoing apoptosis (Figure 5D-5F), suggesting that constitutive FLT3 signaling confers differential proliferative and anti-apoptotic effects within these two compartments. Similar results in cell cycle, survival, stem cell and progenitor analyses were observed in a younger cohort of animals (∼1 month) (Supplemental Figure 4), further supporting the cell intrinsic nature of the effects conferred by the ITD allele in these animals.
Figure 5. Effects of activated Flt3 expression on cell cycle distribution and survival within multipotent hematopoietic stem and progenitor cell compartments.
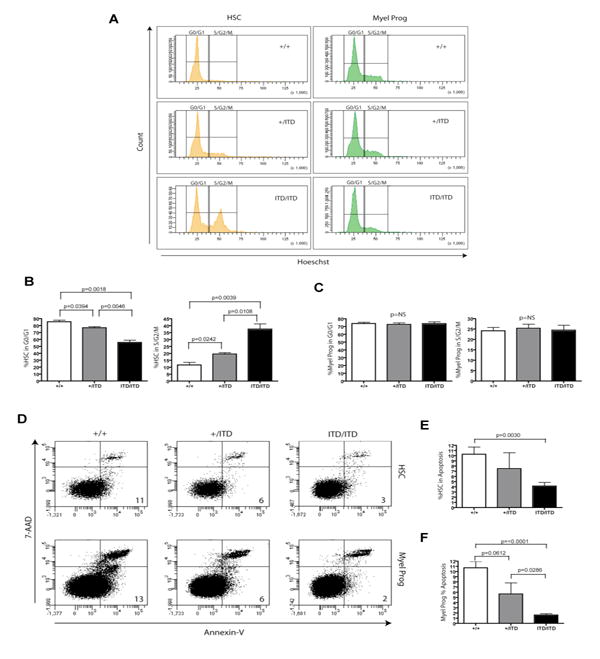
(A-C) Multiparameter flow cytometry of primitive stem and progenitor (Lin-Sca1+ckit+) and myeloid progenitor (Lin-Sca1-ckit+) populations demonstrate an LSK-specific increase of cells in S/G2/M and proportional decrease in G0/G1 in an ITD dose-dependent manner. (B-C) Representative cell cycle data from one of two independent experiments (mean +/- S.E.M.; Flt3+/+, n=3; Flt3+/ITD, n=3; Flt3ITD/ITD, n=3; differences were statistically significant in both independent experiments, student's t-test). (D-F) Survival analysis of HSC (Lin-Sca1+ckit+) and myeloid progenitor (Lin-Sca1-ckit+) cells show a dose-dependent decrease in the percentage of apoptotic cells (7-AAD-/Annexin-V+) in both compartments. (E-F) Representative apoptosis data from one of two independent experiments (mean +/- S.E.M.; Flt3+/+, n=3; Flt3+/ITD, n=3; Flt3ITD/ITD, n=3; differences were statistically significant in both independent experiments, student's t-test).
FLT3-ITD mutations are present in a subset of human CMML
FLT3 mutations have been primarily found in patients with AML. However, the hematopoietic phenotype resembling human CMML that was observed in our murine model, the fact that approximately 20% of CMML patients develop secondary AML (Fenaux et al., 1988; Onida et al., 2002), and the high prevalence of FLT3 mutations in myelomonocytic and monocytic variants of de novo AML [subtypes M4 and M5 according to the French-American-British (FAB) classification] (Kottaridis et al., 2001; Schnittger et al., 2002; Thiede et al., 2002) led us to hypothesize that activating FLT3 mutations may also be present in patients with CMML. We therefore analyzed BM samples from 194 patients with newly diagnosed CMML for the presence of FLT3 ITD mutations and FLT3 AL mutations involving codons D835 and I836. This analysis identified FLT3-ITD mutations in 6 (3.1%) of 194 cases (Table 1).
Table 1.
Mutations identified in CMML patients
total number of patients analyzed (n=194)
ITD, internal tandem duplication
AL, activation loop
FLT3-ITD and N-RAS/K-RAS mutations identified were mutually exclusive
Of the 6 FLT3 mutant patients, 4 presented with “proliferative CMML”, as defined by the FAB classification, that was associated with constitutional symptoms, involvement of extramedullary organs (hepatomegaly, splenomegaly, pleural effusion; Table 2), anemia requiring transfusions, and thrombocytopenia. Furthermore, 5 of the 6 FLT3 mutant cases had a short (less than 3 months) history of an antecedent hematologic disorder. Kaplan-Meier plots indicated that the median survival of CMML patients with FLT3-ITD (8.5 mos.) was shorter than that of CMML patients with FLT3 wildtype (18.2 mos.) (p=0.09, log-rank test; Figure 6). Univariate Cox proportional hazards models confirmed a significant association of FLT3-ITD with survival (p=0.021, hazard ratio (HR) = 2.66, 95% confidence interval (CI) 1.56-6.13), however such an association was not significant in a multivariate Cox model, presumably due to limited cohort size.
Table 2.
Characteristics of CMML patients with FLT3-ITD mutations
| UPIN | WHO | MDAPS | Karyotype | N,K-RAS | Organ Involve. | Cause of Death |
|---|---|---|---|---|---|---|
| 5886 | CMML-2 | Int-2 | 45,XX,-13[1]* | wt | Pleura liver | Progressive disease |
| 4536 | CMML-1 | Int-1 | 46,XY | wt | spleen | Progressive disease |
| 1883 | CMML-1 | Int-1 | 46,XX | wt | none | AML, sepsis |
| 7054 | CMML-1 | Int-2 | 46,XY | wt | Liver spleen | Cardiac, post chemo |
| 1339 | CMML-1 | Int-2 | 46,XY | wt | liver spleen | Progressive Disease |
| 0729 | CMML-1 | Int-1 | -7 | wt | none | AML, sepsis |
42-46,XX, +1,-3,-7,-8,del(1)(q21q42),t(6;11)(q21;q23),del(7)(p22),+mar[20] UPIN (unique patient identifier number); WHO (World Health Organization classification; CMML-1 or 2); MDAPS [MD Anderson Prognostic Scoring System based upon a 4-scale system of Low, Intermediate-1, (Int-1), Intermediate-2 (Int-2), or High (Beran et al., 2007; Onida et al., 2002)]; N-,K-RAS – genotypic status.
Figure 6. Kaplan-Meier survival plots of FLT3-ITD positive versus FLT3 wildtype (WT) patients with CMML.
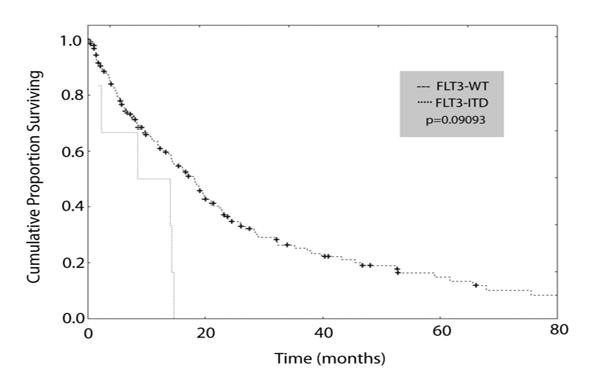
Survival data available from 193 newly diagnosed patients with CMML at the MD Anderson Cancer Center (MDACC) were plotted, including FLT3-ITD positive (n=6) and FLT3-ITD negative (wildtype FLT3; n=187) patients. FLT3-ITD positive patients exhibited a median survival time of 8.5 months compared to 18.2 months for FLT3 wildtype CMML patients (p=0.09093; log-rank analysis). Median survival times and statistical analyses were calculated using STATISTICA data analysis software.
Demographic information and comprehensive laboratory data were available for 168 of the 194 patients (Supplemental Table 1). While no appreciable differences were observed in the majority of parameters, we noted that median WBC counts and serum LDH levels were modestly higher in FLT3 mutant patients than in patients with FLT3 wildtype. Although small sample size precluded attainment of statistically significant WBC and LDH parameters available for FLT3-ITD (n=6) with those of FLT3 wildtype (n=185), comparison between these two groups displayed trends for increased frequency of features associated with proliferative type CMML (FAB classification) (including an abnormal elevated LDH >700 U/L, WBC count >13.0 × 109/L, and the presence of organomegaly) in FLT3-ITD positive versus FLT3 wildtype patients (data not shown).
FLT3-ITD associated CMML displayed no evidence of eosinophilia in both human cases and in our murine model (Supplemental Table 2) unlike CMML associated with PDGFRβ fusion proteins. Chromosome banding analysis revealed normal cytogenetics in 4 of the 6 FLT3 mutant cases, whereas the remaining 2 cases exhibited a complex karyotype and isolated monosomy 7, respectively. Of note, none of the 6 FLT3 mutant cases carried a mutation in K-RAS or N-RAS, as compared to 38 (20.2%) of the 188 cases of the FLT3 wildtype patients (Table 1).
DISCUSSION
Murine models of activating tyrosine kinase mutations and fusion proteins implicated in leukemia provide instructive platforms to examine their effects in vivo. While informative, the use of retroviral transduction or transgenic systems employing exogenous promoters in many of these models precludes accurate physiologic expression levels of these oncogenes and may ultimately affect the resultant biological and phenotypic outcome (Ren, 2004). This concept is supported in results from prior bone marrow transplant studies (Kelly et al., 2002b) and transgenic animals modeling activated FLT3 mutations including our vav FLT3-ITD mice (Lee et al., 2005) and a recently reported Tel-FLT3 model (Baldwin et al., 2007) wherein the expression of a constitutively activated FLT3 in the form of a Tel-FLT3 fusion protein is driven by an exogenous cytomegalovirus (CMV) promoter. While all of these models have provided valuable insights into FLT3-mediated leukemogenesis, the variable phenotypes observed among these aforementioned systems support the notion that promoter choice (e.g., FLT3-ITD: vav and LTR; Tel-FLT3: CMV) plays an important role in both the nature and severity of disease phenotype.
To more precisely assess the effects of activated FLT3 expression at more appropriate physiologic levels, we have generated a murine model in which expression of a constitutively activated form of the FLT3 tyrosine kinase is under the control of the endogenous murine Flt3 promoter, representing to our knowledge, one of the few ‘knock-in’ leukemogenic tyrosine kinase models.
Examination of these animals has revealed insights into the in vivo effects on immature hematopoietic stem and progenitor cell populations of the FLT3-ITD, one of the most common activating tyrosine kinase mutations implicated in leukemia. Our murine data demonstrate that the leukemogenic properties conferred by FLT3-ITD are due, in part, to increased multipotent stem and progenitor cell cycling and enhanced survival properties within these compartments. These findings are supported by recent lentiviral-based FLT3-ITD expression studies in human hematopoietic stem and progenitor cells that reported increased survival and proliferation in FLT3-ITD transduced human cell populations (Li et al., 2007).
While enhanced cell survival was observed in both FLT3-ITD positive myeloid progenitor (Lin-Sca1-ckit+) and more primitive LSK (Lin-Sca1+ckit+) populations containing long-term and short-term HSC, as well as multipotent progenitors cells, increased cell cycling appeared specific to the LSK population (Supplemental Figure 5). Interestingly, this LSK-specific cell cycle aberrancy is nearly identical to that observed in triply null FoxO1, FoxO3, FoxO4 animals when conditionally deleted in the hematopoietic system (Tothova et al., 2007). Prior reports have demonstrated that activated FLT3 receptor signaling induces phosphorylation of FoxO family members (FoxO3a) in Ba/F3 cells, with resultant nuclear exclusion of FoxO proteins and transcriptional repression of FoxO-target genes critical to normal cell survival and proliferation (Brandts et al., 2005; Scheijen et al., 2004). Taken together, our data suggests that enhanced cell proliferation within the LSK compartment of our mutant FLT3 mice is mediated, at least in part, through inactivation of the FoxO transcription factor family, although we have not formally examined if there are alterations among FoxO family members within these mice.
Our studies also illustrate that constitutive signaling by FLT3-ITD at physiologic levels directs hematopoietic differentiation towards the myeloid/monocytic lineage with concomitant suppression of megakaryocyte-erythroid development (Supplemental Figure 5). Moreover, while the levels of common lymphoid progenitors (CLP) appear largely indiscernible between mutant FLT3 animals and their wild-type littermates, constitutive FLT3 signaling appears to induce a block in normal B cell development, which is consistent with the prevalence of FLT3-ITD mutations associated with acute myeloid versus acute lymphoid leukemia. This finding is also intriguing given the fact that absence of Flt3 also leads to deficiencies in normal B cell development, indicating that constitutive FLT3-ITD signaling likely interferes with normal FLT3 gene function (Mackarehtschian et al., 1995).
The phenotypic consequences of the aforementioned alterations in hematopoietic stem and progenitor cell populations results in a chronic MPD in Flt3+/ITD and Flt3ITD/ITD animals resembling human CMML which prompted us to look for the presence of activating FLT3 mutations in patient samples. Our findings along with previous reports (Lin et al., 2006; Pardanani et al., 2003), indicate that although the incidence of FLT3 mutations is relatively uncommon in chronic MPD, in the instances where isolated FLT3 lesions in chronic MPD are detected, these patients may frequently present with CMML. Although it would be premature to conclude that CMML with FLT3-ITD is a “unique” subtype of this disease that is associated with particular clinical and laboratory features at the current time, our data suggest that this molecular feature may represent an independent negative prognostic factor in CMML. Clearly, analysis of larger cohorts will need to be examined to see whether this notion bears out in multivariate analysis models. Importantly, however our findings support that molecular assessment for the presence of FLT3 mutations should be performed in individuals with CMML as their survival may be improved by inhibition of FLT3-ITD with available small molecule compounds, which have shown efficacy in AML (Smith et al., 2004; Stone et al., 2005; Wadleigh et al., 2005). As imatinib mesylate has demonstrated considerable clinical efficacy in the small subset of CMML associated with PDGFRβ rearrangements involving chromosome 5q31-q33 (Apperley et al., 2002; David et al., 2007), similar effects might be predicted with the use of available FLT3 small molecule inhibitors in patients with FLT3-ITD associated CMML.
The disease phenotype findings in prior FLT3 murine models (Baldwin et al., 2007; Kelly et al., 2002a; Kelly et al., 2002b; Lee et al., 2005) and our current ‘knock-in’ model suggest that in patients with isolated FLT3 mutations, progression to AML is unlikely in the absence of pre-existing or the subsequent development of additional genetic lesions. These data also suggest that subsequent mutations must complement constitutive FLT3 activation to ensure full transformation of hematopoietic cells and that certain combinations of mutations are not only cooperative but also interdependent in the pathogenesis of AML (Supplemental Figure 5). Our model will serve as a valuable biological tool to explore the individual contributions of these different classes of leukemogenic mutations in the development of AML and molecular therapeutic regimens that target them.
EXPERIMENTAL PROCEDURES
Transgenic Mice
Gene targeting and chimera formation were used to generate an ITD ‘knock-in’ allele in exon 14 of the murine Flt3 locus. A high fidelity PCR based cloning strategy was employed to construct a targeting vector encompassing intronic murine genomic sequences upstream of exon 11 and downstream of exon 15 and harboring previously described human internal tandem duplication (ITD) mutation W51(Kelly et al., 2002b) in exon 14 and a neomycin (NEO) selection cassette flanked by two loxP sites downstream of exon 15 at the AvrII restriction site (Figure 1A). In brief, W51 results in duplication of human FLT3 amino acids 596-602 (R-E-Y-E-Y-D-L). Human exon 14 DNA sequences harboring the W51 mutation were cloned from pGEM-FLT3 W51(Kelly et al., 2002b) by PCR as a BstXI-AvaI fragment that was subsequently subcloned into murine exon 14 between the BstXI and AvaI restriction sites. All intronic sequences (including splice acceptor and donor sites), exons, and the W51 ITD mutation were verified by DNA sequencing. Correctly targeted ES cells were confirmed by Southern blot using probes lying beyond the regions of homology contained within the targeting vector and chimeric mice were subsequently generated by standard methods (Brigham and Women's Hospital Transgenic Core Facility). Germline transmission of the ITD allele was achieved by crossing highly chimeric mice to wild-type C57BL/6 or Balb/c mice. Analyses were performed on animals in both the C57BL/6 and Balb/c background which showed similar phenotypes overall (data not shown). All data displayed in this report are from animals in the C57BL/6 background. Flt3+/ITD and Flt3ITD/ITD and wt littermate mice were genotyped by PCR with primers Flt314F (5′-AGGTACGAGAGTCAGCTGCAGATG-3′) and Flt314R (5′–TGTAAAGATGGAGTAAGTGCGGGT-3′) using the following parameters: 95°C for 2 min, followed by 35 cycles of 95°C for 1 min, 60°C for 1 min, and 72°C for 50 s. Animals were monitored 2-3x/week for the presence of disease by general inspection and palpation. Peripheral blood was collected from the retro-orbital cavity using an EDTA-treated glass capillary and automated total and differential blood cell counts were determined using Hemavet 950 (Drew Scientific). Collected blood was also used to prepare blood smears, which were stained with Wright and Giemsa. Following sacrifice, mice were examined for the presence of tumors or other abnormalities, and organs were collected for further cell and histopathologic analysis. Single cell suspensions were made from hematopoietic organs, washed; red blood cells lysed (Puregene) and were frozen in 10% dimethylsulfoxide/90% fetal bovine serum (FBS) until analysis. All mice were housed in a pathogen-free animal facility in microisolator cages and experiments were conducted with the ethical approval of the Harvard Medical Area Standing Committee on Animals.
Flow cytometry and expression analysis
For multiparameter flow cytometry, bone marrow mononuclear cells were isolated from age-matched littermates between 1-4 months of age backcrossed to C57BL/6 at least 6 generations and were flushed from hind leg bones with RPMI (Cambrex, Biowhittaker)+10%FCS (Gibco)+Penicillin/Streptomycin (Cambrex, Biowhittaker), lysed on ice with red blood cell lysis solution (Puregene) and washed in PBS (Gibco) +2%FBS. HSC (LSK), CMP, GMP, MEP and CLP populations were analyzed and sorted using a FACSAria instrument (Becton Dickinson, Mountain View, CA) as previously reported (Akashi et al., 2000; Kondo et al., 1997). Apoptosis was determined by staining freshly harvested bone marrow mononuclear cells with lineage, stem and progenitor markers, followed by Annexin-V and 7-AAD staining. Cell cycle analysis was carried out as previously reported (Cheng et al., 2000). Additional analyses were carried out on a 4-color FACSCalibur cytometer (Becton Dickinson, Mountain View, CA) using samples that were washed with PBS and 0.1% bovine serum albumin (BSA, Sigma), blocked with Fc-block (BD-Pharmingen) for 10 minutes and stained with the following monoclonal antibodies that were conjugated to either PE, FITC, PerCP, or APC: Gr-1, Mac-1, c-kit, F4/80, CD3, B220, IgM, CD45.1, CD45.2 in PBS+0.1% BSA for 30 minutes. Viable cells were assessed with staining for 7-AAD. Antibodies for phospho-FLT3 (pFLT3) and phospho-Stat5 were obtained from Cell Signaling and flow cytometry carried out on permeabilized and fixed murine bone marrow and spleen cells as previously described (Wernig et al., 2006). A minimum of 10,000 events was acquired and analyzed using CellQuest and FlowJo software. Immunoblot expression analysis for murine Flt3 protein on primary bone marrow and spleen cells was carried out as previously described (Lee et al., 2005) using anti-Flt3 rabbit polyclonal antibody (M20) (Santa Cruz Biotechnologies).
Colony forming assays
Myeloid colony-plating assays were performed in methylcellulose-based medium (M3434) containing 3U/mL erythropoietin, 10ng/mL recombinant murine interleukin-3 (rmIL-3), 10 ng/mL rmIL-6 and 50 ng/ml rmSCF (Stem Cell Technologies, Vancouver, BC Canada). For primary methylcellulose cultures, 1×104 or 1×105 cells were seeded in duplicate and scored for colony formation 7-10 days later. Serial replating was carried out as previously described (Deguchi et al., 2003) in three independent experiments with 104 cells replated in duplicate for each round of replating and colony counts performed on day 7. For CFU-S assays, 500 HSC (Lin-Sca1+ckit+) cells were sorted from BM harvested on a FACSAria instrument (Becton Dickinson, Mountain View, CA) from two Flt3+/+ or Flt3ITD/ITD littermate mice each and injected into lateral tail veins of lethally irradiated wt C57/BL6 recipient mice in duplicate. Day 12 CFU-S assay was carried out as previously described (Morrison and Weissman, 1994).
Histopathology, immunohistochemistry and cytochemistry analysis
Tissues were fixed for at least 72 hours in 10% neutral buffered formalin (Sigma), dehydrated in alcohol, cleared in xylene, and infiltrated with paraffin on an automated processor (Leica Bannockburn, IL, USA). Tissue sections (4μm thick) were placed on charged slides, deparaffinized in xylene, rehydrated through graded alcohol solutions, and stained with hematoxylin and eosin (H&E). Immunohistochemistry was performed on paraffin-embedded tissue sections using primary antibodies to B220 (Pharmingen), myeloperoxidase (MPO; Dako), CD3 (Dako), and Mac1 (AbCam). Cytochemical staining for Napthol AS-D Chloroacetate Esterase (NSE) (Sigma-Aldrich) and Sudan Black B (SBB) (Sigma-Aldrich) were performed on cytospin preparations from bone marrow cells according to manufacturer's protocols.
Sample collection and DNA sequence analysis
Bone marrow samples from 194 patients with chronic myelomonocytic leukemia (CMML) were collected from the MD Anderson Cancer Center between 1999 and 2005 and analyzed for both internal tandem duplication (ITD) and activation loop (AL) mutations. All patients provided informed consent. PCR amplification and DNA sequencing of exons 14 and 20 of human FLT3 was performed using M13-tailed primer as previously described (Levine et al., 2005). Sequence analysis of bidirectional sequence traces was performed using Mutation Surveyor version 2.28 (SoftGenetics, State College, Pennsylvania, USA). Candidate mutations were reamplified and sequenced from original DNA for independent verification. Studies involving patient samples were approved by the M.D. Anderson Ethics Committee and all subjects gave informed consent in accordance with the Declaration of Helsinki.
Supplementary Material
Acknowledgments
We would like to thank C. Scholl and other members of the Gilliland laboratory for support and helpful discussions and the Dartmouth and Brigham and Women's Hospital Transgenic core facilities for technical assistance with generation of mice. This work supported by grants from the National Institutes of Health CA113434 (B.H.L.), CA66996, and CA105423 (D.G.G.), and the Leukemia and Lymphoma Society (D.G.G.). S.F. was supported by grant FR 2113/1-1 from the Deutsche Forschungsgemeinschaft. B.J.P.H. is a senior clinical fellow of the Medical Research Council (UK). R.L.L. is a Basic Science Fellow of the American Society of Hematology, a Young Investigator Award recipient of the American Society of Clinical Oncology, and a Doris Duke Charitable Foundation Clinical Scientist Development Award recipient. D.G.G. is a Doris Duke Charitable Foundation Distinguished Clinical Scientist and an Investigator of the Howard Hughes Medical Institute. The Lund Stem Cell Center is supported by a Center of Excellence grant from the Swedish Foundation for Strategic Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- Apperley JF, Gardembas M, Melo JV, Russell-Jones R, Bain BJ, Baxter EJ, Chase A, Chessells JM, Colombat M, Dearden CE, et al. Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta. N Engl J Med. 2002;347:481–487. doi: 10.1056/NEJMoa020150. [DOI] [PubMed] [Google Scholar]
- Armstrong SA, Kung AL, Mabon ME, Silverman LB, Stam RW, Den Boer ML, Pieters R, Kersey JH, Sallan SE, Fletcher JA, et al. Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression based classification. Cancer Cell. 2003;3:173–183. doi: 10.1016/s1535-6108(03)00003-5. [DOI] [PubMed] [Google Scholar]
- Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, Sallan SE, Lander ES, Golub TR, Korsmeyer SJ. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30:41–47. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- Baldwin BR, Li L, Tse KF, Small S, Collector M, Whartenby KA, Sharkis SJ, Racke F, Huso D, Small D. Transgenic mice expressing Tel-FLT3, a constitutively activated form of FLT3, develop myeloproliferative disease. Leukemia. 2007;21:764–771. doi: 10.1038/sj.leu.2404532. [DOI] [PubMed] [Google Scholar]
- Beran M, Wen S, Shen Y, Onida F, Jelinek J, Cortes J, Giles F, Kantarjian H. Prognostic factors and risk assessment in chronic myelomonocytic leukemia: Validation study of the M.D. Anderson Prognostic Scoring System. Leuk Lymphoma. 2007;48:1150–1160. doi: 10.1080/10428190701216386. [DOI] [PubMed] [Google Scholar]
- Brandts CH, Sargin B, Rode M, Biermann C, Lindtner B, Schwable J, Buerger H, Muller-Tidow C, Choudhary C, McMahon M, et al. Constitutive activation of Akt by Flt3 internal tandem duplications is necessary for increased survival, proliferation, and myeloid transformation. Cancer Res. 2005;65:9643–9650. doi: 10.1158/0008-5472.CAN-05-0422. [DOI] [PubMed] [Google Scholar]
- Carow CE, Levenstein M, Kaufmann SH, Chen J, Amin S, Rockwell P, Witte L, Borowitz MJ, Civin CI, Small D. Expression of the hematopoietic growth factor receptor FLT3 (STK-1/Flk2) in human leukemias. Blood. 1996;87:1089–1096. [PubMed] [Google Scholar]
- Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Schwable J, Brandts C, Tickenbrock L, Sargin B, Kindler T, Fischer T, Berdel WE, Muller-Tidow C, Serve H. AML-associated Flt3 kinase domain mutations show signal transduction differences compared with Flt3 ITD mutations. Blood. 2005;106:265–273. doi: 10.1182/blood-2004-07-2942. [DOI] [PubMed] [Google Scholar]
- DaSilva N, Hu ZB, Ma W, Rosnet O, Birnbaum D, Drexler HG. Expression of the FLT3 gene in human leukemia-lymphoma cell lines. Leukemia. 1994;8:885–888. [PubMed] [Google Scholar]
- David M, Cross NC, Burgstaller S, Chase A, Curtis C, Dang R, Gardembas M, Goldman JM, Grand F, Hughes G, et al. Durable responses to imatinib in patients with PDGFRB fusion gene-positive and BCR-ABL-negative chronic myeloproliferative disorders. Blood. 2007;109:61–64. doi: 10.1182/blood-2006-05-024828. [DOI] [PubMed] [Google Scholar]
- Deguchi K, Ayton PM, Carapeti M, Kutok JL, Snyder CS, Williams IR, Cross NC, Glass CK, Cleary ML, Gilliland DG. MOZ-TIF2- induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cell. 2003;3:259–271. doi: 10.1016/s1535-6108(03)00051-5. [DOI] [PubMed] [Google Scholar]
- Drexler HG. Expression of FLT3 receptor and response to FLT3 ligand by leukemic cells. Leukemia. 1996;10:588–599. [PubMed] [Google Scholar]
- Fenaux P, Beuscart R, Lai JL, Jouet JP, Bauters F. Prognostic factors in adult chronic myelomonocytic leukemia: an analysis of 107 cases. J Clin Oncol. 1988;6:1417–1424. doi: 10.1200/JCO.1988.6.9.1417. [DOI] [PubMed] [Google Scholar]
- Frohling S, Schlenk RF, Breitruck J, Benner A, Kreitmeier S, Tobis K, Dohner H, Dohner K. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002;100:4372–4380. doi: 10.1182/blood-2002-05-1440. [DOI] [PubMed] [Google Scholar]
- Gabbianelli M, Pelosi E, Montesoro E, Valtieri M, Luchetti L, Samoggia P, Vitelli L, Barberi T, Testa U, Lyman S, et al. Multi-level effects of flt3 ligand on human hematopoiesis: expansion of putative stem cells and proliferation of granulomonocytic progenitors/monocytic precursors. Blood. 1995;86:1661–1670. [PubMed] [Google Scholar]
- Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100:1532–1542. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- Grundler R, Miething C, Thiede C, Peschel C, Duyster J. FLT3-ITD and tyrosine kinase domain mutants induce 2 distinct phenotypes in a murine bone marrow transplantation model. Blood. 2005;105:4792–4799. doi: 10.1182/blood-2004-11-4430. [DOI] [PubMed] [Google Scholar]
- Hayakawa F, Towatari M, Kiyoi H, Tanimoto M, Kitamura T, Saito H, Naoe T. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene. 2000;19:624–631. doi: 10.1038/sj.onc.1203354. [DOI] [PubMed] [Google Scholar]
- Hjertson M, Sundstrom C, Lyman SD, Nilsson K, Nilsson G. Stem cell factor, but not flt3 ligand, induces differentiation and activation of human mast cells. Exp Hematol. 1996;24:748–754. [PubMed] [Google Scholar]
- Horiike S, Yokota S, Nakao M, Iwai T, Sasai Y, Kaneko H, Taniwaki M, Kashima K, Fujii H, Abe T, Misawa S. Tandem duplications of the FLT3 receptor gene are associated with leukemic transformation of myelodysplasia. Leukemia. 1997;11:1442–1446. doi: 10.1038/sj.leu.2400770. [DOI] [PubMed] [Google Scholar]
- Ishii E, Zaitsu M, Ihara K, Hara T, Miyazaki S. High expression but no internal tandem duplication of FLT3 in normal hematopoietic cells. Pediatr Hematol Oncol. 1999;16:437–441. doi: 10.1080/088800199276994. [DOI] [PubMed] [Google Scholar]
- Kelly LM, Kutok JL, Williams IR, Boulton CL, Amaral SM, Curley DP, Ley TJ, Gilliland DG. PML/RARalpha and FLT3-ITD induce an APL-like disease in a mouse model. Proc Natl Acad Sci U S A. 2002a;99:8283–8288. doi: 10.1073/pnas.122233699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002b;99:310–318. doi: 10.1182/blood.v99.1.310. [DOI] [PubMed] [Google Scholar]
- Kiyoi H, Naoe T, Nakano Y, Yokota S, Minami S, Miyawaki S, Asou N, Kuriyama K, Jinnai I, Shimazaki C, et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood. 1999;93:3074–3080. [PubMed] [Google Scholar]
- Kiyoi H, Towatari M, Yokota S, Hamaguchi M, Ohno R, Saito H, Naoe T. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia. 1998;12:1333–1337. doi: 10.1038/sj.leu.2401130. [DOI] [PubMed] [Google Scholar]
- Kondo M, Weissman IL, Akashi K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 1997;91:661–672. doi: 10.1016/s0092-8674(00)80453-5. [DOI] [PubMed] [Google Scholar]
- Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, Walker H, Wheatley K, Bowen DT, Burnett AK, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98:1752–1759. doi: 10.1182/blood.v98.6.1752. [DOI] [PubMed] [Google Scholar]
- Lee BH, Williams IR, Anastasiadou E, Boulton CL, Joseph SW, Amaral SM, Curley DP, Duclos N, Huntly BJ, Fabbro D, et al. FLT3 internal tandem duplication mutations induce myeloproliferative or lymphoid disease in a transgenic mouse model. Oncogene. 2005;24:7882–7892. doi: 10.1038/sj.onc.1208933. [DOI] [PubMed] [Google Scholar]
- Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Li L, Piloto O, Kim KT, Ye Z, Nguyen HB, Yu X, Levis M, Cheng L, Small D. FLT3/ITD expression increases expansion, survival and entry into cell cycle of human haematopoietic stem/progenitor cells. Br J Haematol. 2007;137:64–75. doi: 10.1111/j.1365-2141.2007.06525.x. [DOI] [PubMed] [Google Scholar]
- Lin P, Jones D, Medeiros LJ, Chen W, Vega-Vazquez F, Luthra R. Activating FLT3 mutations are detectable in chronic and blast phase of chronic myeloproliferative disorders other than chronic myeloid leukemia. Am J Clin Pathol. 2006;126:530–533. doi: 10.1309/JT5BE2L1FGG8P8Y6. [DOI] [PubMed] [Google Scholar]
- Mackarehtschian K, Hardin JD, Moore KA, Boast S, Goff SP, Lemischka IR. Targeted disruption of the flk2/flt3 gene leads to deficiencies in primitive hematopoietic progenitors. Immunity. 1995;3:147–161. doi: 10.1016/1074-7613(95)90167-1. [DOI] [PubMed] [Google Scholar]
- Meierhoff G, Dehmel U, Gruss HJ, Rosnet O, Birnbaum D, Quentmeier H, Dirks W, Drexler HG. Expression of FLT3 receptor and FLT3-ligand in human leukemia-lymphoma cell lines. Leukemia. 1995;9:1368–1372. [PubMed] [Google Scholar]
- Mizuki M, Fenski R, Halfter H, Matsumura I, Schmidt R, Muller C, Gruning W, Kratz-Albers K, Serve S, Steur C, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood. 2000;96:3907–3914. [PubMed] [Google Scholar]
- Morrison SJ, Weissman IL. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity. 1994;1:661–673. doi: 10.1016/1074-7613(94)90037-x. [DOI] [PubMed] [Google Scholar]
- Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, Sonoda Y, Fujimoto T, Misawa S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–1918. [PubMed] [Google Scholar]
- Onida F, Kantarjian HM, Smith TL, Ball G, Keating MJ, Estey EH, Glassman AB, Albitar M, Kwari MI, Beran M. Prognostic factors and scoring systems in chronic myelomonocytic leukemia: a retrospective analysis of 213 patients. Blood. 2002;99:840–849. doi: 10.1182/blood.v99.3.840. [DOI] [PubMed] [Google Scholar]
- Paietta E, Ferrando AA, Neuberg D, Bennett JM, Racevskis J, Lazarus H, Dewald G, Rowe JM, Wiernik PH, Tallman MS, Look AT. Activating FLT3 Mutations in CD117/KIT PositiveT-Cell Acute Lymphoblastic Leukemias. Blood. 2004 doi: 10.1182/blood-2004-01-0168. [DOI] [PubMed] [Google Scholar]
- Pardanani A, Reeder TL, Kimlinger TK, Baek JY, Li CY, Butterfield JH, Tefferi A. Flt-3 and c-kit mutation studies in a spectrum of chronic myeloid disorders including systemic mast cell disease. Leuk Res. 2003;27:739–742. doi: 10.1016/s0145-2126(02)00303-x. [DOI] [PubMed] [Google Scholar]
- Rasko JE, Metcalf D, Rossner MT, Begley CG, Nicola NA. The flt3/flk-2 ligand: receptor distribution and action on murine haemopoietic cell survival and proliferation. Leukemia. 1995;9:2058–2066. [PubMed] [Google Scholar]
- Ratajczak MZ, Ratajczak J, Ford J, Kregenow R, Marlicz W, Gewirtz AM. FLT3/FLK-2 (STK-1) Ligand does not stimulate human megakaryopoiesis in vitro. Stem Cells. 1996;14:146–150. doi: 10.1002/stem.140146. [DOI] [PubMed] [Google Scholar]
- Reindl C, Bagrintseva K, Vempati S, Schnittger S, Ellwart JW, Wenig K, Hopfner KP, Hiddemann W, Spiekermann K. Point mutations in the juxtamembrane domain of FLT3 define a new class of activating mutations in AML. Blood. 2006;107:3700–3707. doi: 10.1182/blood-2005-06-2596. [DOI] [PubMed] [Google Scholar]
- Ren R. Modeling the dosage effect of oncogenes in leukemogenesis. Curr Opin Hematol. 2004;11:25–34. doi: 10.1097/00062752-200401000-00005. [DOI] [PubMed] [Google Scholar]
- Rocnik JL, Okabe R, Yu JC, Lee BH, Giese N, Schenkein DP, Gilliland DG. Roles of tyrosine 589 and 591 in STAT5 activation and transformation mediated by FLT3-ITD. Blood. 2006;108:1339–1345. doi: 10.1182/blood-2005-11-011429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosnet O, Birnbaum D. Hematopoietic receptors of class III receptor-type tyrosine kinases. Crit Rev Oncog. 1993;4:595–613. [PubMed] [Google Scholar]
- Rosnet O, Buhring HJ, Marchetto S, Rappold I, Lavagna C, Sainty D, Arnoulet C, Chabannon C, Kanz L, Hannum C, Birnbaum D. Human FLT3/FLK2 receptor tyrosine kinase is expressed at the surface of normal and malignant hematopoietic cells. Leukemia. 1996;10:238–248. [PubMed] [Google Scholar]
- Scheijen B, Ngo HT, Kang H, Griffin JD. FLT3 receptors with internal tandem duplications promote cell viability and proliferation by signaling through Foxo proteins. Oncogene. 2004;23:3338–3349. doi: 10.1038/sj.onc.1207456. [DOI] [PubMed] [Google Scholar]
- Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, Loffler H, Sauerland CM, Serve H, Buchner T, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood. 2002;100:59–66. doi: 10.1182/blood.v100.1.59. [DOI] [PubMed] [Google Scholar]
- Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, Murphy KM, Dauses T, Allebach J, Small D. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103:3669–3676. doi: 10.1182/blood-2003-11-3775. [DOI] [PubMed] [Google Scholar]
- Stirewalt DL, Meshinchi S, Kussick SJ, Sheets KM, Pogosova-Agadjanyan E, Willman CL, Radich JP. Novel FLT3 point mutations within exon 14 found in patients with acute myeloid leukaemia. Br J Haematol. 2004;124:481–484. doi: 10.1111/j.1365-2141.2004.04808.x. [DOI] [PubMed] [Google Scholar]
- Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. 2003;3:650–665. doi: 10.1038/nrc1169. [DOI] [PubMed] [Google Scholar]
- Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, Grandin W, Lebwohl D, Wang Y, Cohen P, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105:54–60. doi: 10.1182/blood-2004-03-0891. [DOI] [PubMed] [Google Scholar]
- Thiede C, Steudel C, Mohr B, Schaich M, Schakel U, Platzbecker U, Wermke M, Bornhauser M, Ritter M, Neubauer A, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–4335. doi: 10.1182/blood.v99.12.4326. [DOI] [PubMed] [Google Scholar]
- Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Wadleigh M, DeAngelo DJ, Griffin JD, Stone RM. After chronic myelogenous leukemia: tyrosine kinase inhibitors in other hematologic malignancies. Blood. 2005;105:22–30. doi: 10.1182/blood-2003-11-3896. [DOI] [PubMed] [Google Scholar]
- Wernig G, Mercher T, Okabe R, Levine RL, Lee BH, Gilliland DG. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood. 2006;107:4274–4281. doi: 10.1182/blood-2005-12-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, Carlson BD, Carroll AJ, Mrozek K, Vardiman JW, George SL, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res. 2001;61:7233–7239. [PubMed] [Google Scholar]
- Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, Asou N, Kuriyama K, Yagasaki F, Shimazaki C, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97:2434–2439. doi: 10.1182/blood.v97.8.2434. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.