

Abstract
Previous studies have demonstrated that stress may increase prodynorphin gene expression, and κ opioid agonists suppress drug reward. Therefore, we tested the hypothesis that stress-induced release of endogenous dynorphin may mediate behavioral responses to stress and oppose the rewarding effects of cocaine. C57Bl/6 mice subjected to repeated forced swim testing (FST) using a modified Porsolt procedure at 30°C showed a characteristic stress-induced immobility response and a stress-induced analgesia observed with a tail withdrawal latency assay. Pretreatment with the κ opioid receptor antagonist nor-binaltorphimine (nor-BNI; 10 mg/kg, i.p.) blocked the stress-induced analgesia and significantly reduced the stress-induced immobility. The nor-BNI sensitivity of the behavioral responses suggests an activation of the κ opioid receptor by a stress-induced release of dynorphin peptides. Supporting this hypothesis, transgenic mice possessing a disrupted prodynorphin gene showed no increase in immobility or stress-induced analgesia after exposure to repeated FST. Because both stress and the κ opioid system can modulate the response to drugs of abuse, we tested the effects of forced swim stress on cocaine-conditioned place preference (CPP). FST-exposed mice conditioned with cocaine (15 mg/kg, s.c.) showed significant potentiation of place preference for the drug-paired chamber over the responses of unstressed mice. Surprisingly, nor-BNI pretreatment blocked stress-induced potentiation of cocaine CPP. Consistent with this result, mice lacking the prodynorphin gene did not show a stress-induced potentiation of cocaine CPP, whereas wild-type littermates did. The findings suggest that chronic swim stress may activate the κ opioid system to produce analgesia, immobility, and potentiation of the acute rewarding properties of cocaine in C57Bl/6 mice.
Keywords: κ, opioid, dynorphin, stress, depression, cocaine, conditioned place preference
Introduction
Stress is a normal element of life, serving to evoke adaptive responses of an organism to the environment. This response to stress may be of survival value, as demonstrated by the phenomena of stress-induced analgesia (SIA; Maier et al., 1980). However, persistent and inescapable stressors that overwhelm an organism's adaptive abilities can be harmful, leading to detrimental behaviors such as drug abuse (Rhoads, 1983; Kosten et al., 1986; Najavits et al., 1998). Animal studies correlate exposure to an inescapable stressor with increases in both the rewarding properties and self-administration of drugs of abuse (Piazza et al., 1990; Shaham and Stewart, 1994; Will et al., 1998; Kosten et al., 2000). The mechanisms mediating this stress-induced enhancement are unclear. In one possible mechanism, stress-induced activation of the hypothalamo-pituitary-adrenal axis may subsequently activate the endogenous κ opioid system (Przewlocki et al., 1987; Nabeshima et al., 1992; Watanabe et al., 1995) presumably thereby modulating drug reward and self-administration (Mello and Mendelson, 1997; Carlezon et al., 1998; Kreek and Koob, 1998). However, Schenk et al. (1999) have suggested that the κ system actually opposes drug-rewarding effects, because administration of the κ agonist U69,593 both decreases cocaine self-administration and blocks cocaine sensitization. Further understanding of the role of the κ opioid system in mediating the response to stress would likely provide new insights into the issues of stress adaptation and drug abuse.
Endogenous opioid systems have been implicated in multiple stress-induced behavioral responses, making them logical candidates for study. For example, SIA induced after a forced swim test (FST) stress was absent in mice lacking β-endorphin (Rubinstein et al., 1996). Moreover, δ opioid receptors have been associated with stress–response behaviors, because δ agonists reduced the immobility of rats in a forced swim test (Broom et al., 2002).
The involvement of the endogenous κ opioid system in the behavioral response to stress is less clear. Although κ agonists produce a dysphoria similar to that noted in depression and chronic stress (Pfeiffer et al., 1986), κ opioid receptor (KOR) knock-out mice demonstrated responses similar to those of wild-type mice in a single brief trial of the FST, prompting the authors to suggest that KOR is not involved (Filliol et al., 2000). However, stress-induced analgesia was blocked by peripheral administration of the KOR antagonist nor-binaltorphimine (nor-BNI) in multiple studies with both rats and mice (Takahashi et al., 1990; Watkins et al., 1992; Menendez et al., 1993). These results are supported by the finding that intracerebroventricular administration of dynorphin A (1–17) or a stable analog, E2078, potentiates the immobility response to a stressor, an effect blocked by opioid antagonists (Katoh et al., 1990). Moreover, subanalgesic doses of dynorphin A (1–13) and (1–10) prolonged SIA in forced swim-stressed mice but not unstressed controls (Starec et al., 1996). Finally, expression of herpes simplex virus-cAMP response element-binding protein (CREB) in rat brain elevated CREB levels to produce an increase in immobility in the FST that was reduced by expression of a dominant negative mutant of CREB and prevented by nor-BNI treatment, thereby suggesting that CREB-mediated induction of prodynorphin gene expression may have mediated the stress-induced immobility (Pliakas et al., 2001). Overall, these reports suggest that activation of the endogenous κ opioid systems may potentiate the immobility and analgesic responses to a stressor.
In this study, we tested the hypothesis that repeated exposure to stress activates the endogenous κ opioid system to produce changes in behavioral immobility, pain threshold, and drug reward. Supporting this, we report that mice exposed to forced swimming stress demonstrated a nor-BNI-sensitive, dynorphin-mediated increase in pain threshold and behavioral immobility as well as a subsequent potentiation of cocaine conditioned place preference.
Materials and Methods
Animals and housing. Male C57Bl/6 mice (Charles River Laboratories, Wilmington, MA) weighing 23–33 gm were used in these experiments. All mice used were adult males, ranging from 12 to 16 weeks of age. Breeding pairs of heterozygous prodynorphin knock-out (KO) mice (Sharifi et al., 2001) backcrossed 12 generations onto the C57Bl/6 background were used to generate KO and wild-type (WT) littermate controls for this study. The C57Bl/6 mouse strain was chosen for this work because it has been demonstrated to produce robust immobility in the FST (Dalvi and Lucki, 1999; Lucki et al., 2001) and is a well characterized background strain for transgenic studies (Banbury Conference, 1997). The prodynorphin gene-disrupted animals show no discernible differences from wild-type littermates in growth, life span, overt behavior, or locomotor activity (Sharifi et al., 2001). All mice used were group-housed, four to a cage, in self-standing plastic cages (28 cm long × 16 cm wide × 13 cm high) using Bed-A-Cob for home bedding within the Animal Core Facility at the University of Washington, and maintained in a specific pathogen-free housing unit. All mice tested were transferred 1 week before use into a colony room adjacent to the testing room to acclimate to environment. All housing rooms were illuminated on a 12 hr light/dark cycle with lights on at 7 A.M. Food pellets and water were available ad libitum. All procedures with mice were approved by the institutional Animal Care and Use Committee in accordance with the 1996 National Institutes of Health Guide for the Care and Use of Laboratory Animals, and mice were inspected regularly by veterinary staff to ensure compliance.
Genotyping of prodynorphin wild-type and knock-out mice by PCR. The presence or absence of the prodynorphin gene was confirmed in genomic DNA isolated from tail tissue samples taken from each mouse using PCR analysis and a procedure described previously (Sharifi et al., 2001). Briefly, a fraction of isolated DNA was used in PCR assays using the following primers: dynorphin, exon 3, 5′-GTGCAGTGAGGATTCAGGATGGG; and neomycin, 5′-ATCCAGGAAACCAGCAGCGGCTAT. PCR products were resolved on a 1.5% agarose gel with ethidium bromide and then photographed for analysis. Homologous prodynorphin KO animals produce PCR products with the neomycin but not dynorphin primer, whereas homologous prodynorphin WT mice produce PCR products with the dynorphin but not neomycin primer. Heterologous prodynorphin mice yield both PCR products, thereby allowing their exclusion from the study.
FST. To induce stress, C57Bl/6 mice were exposed to a modified forced swim test. Testing was performed on the basis of methods previously described by Porsolt et al. (1977a,b). The modified Porsolt forced swim test paradigm used was a 2 d procedure in which mice swam without the opportunity to escape. In all trials, mice were placed in an opaque 5 l beaker (40 cm tall × 25 cm in diameter) filled with 3.5 l of 30°C water. After the trial, mice were removed, dried with towels, and returned to their home cages for at least 7 min before further testing. On day 1, animals were placed in water to swim for a single trial of 15 min. The time spent immobile in the last 4 min of the trial was recorded. On day 2, animals were again placed in water to swim but through a series of four trials, each 6 min long; trials were separated by a 7–12 min return to a home cage. Multiple trials were used to determine the effects of extended exposure to the inescapable stressor. The second day of FST was typically marked by a facilitated immobility characterized by a rapid and prolonged adoption of the immobile posture. Immobility is defined as the time at which the mice initiate and maintain a stationary posture. In this characteristic posture, the mouse's forelimbs are motionless and directed forward, the tail is directed outward, and the hind legs are in limited motion. To qualify as immobility, this posture must be clearly visible and sustained for a minimum of 2 sec. Difficulty in swimming or staying afloat were criteria for exclusion; however, no mice met these criteria in this study. When mice were pretreated with a drug, experiments were performed with the experimenter blind to the pretreatment.
Measurement of mouse body temperature. Reports elsewhere show that mice exposed to forced swimming in 20°C water demonstrate opioid-sensitive stress-induced analgesia coinciding with a significant decrease in body temperature, an example of hypothermia (Mogil et al., 1996). To reduce possible complicating effects of hypothermia in this study, the water used in all forced swim trials was maintained at 30°C. The effect of forced swimming under these conditions on body temperature was further monitored using temperature transponders implanted subcutaneously (above the shoulder blades) in mice 1 d before swim testing (IPTT-200; Biomedic Data Systems Inc., Seaford, DE). Body temperature was read remotely (DAS5007 pocket scanner; Biomedic Data Systems Inc.) before, immediately after the last 6 min swim in the 2 d protocol, and 10 min after forced swim. Before forced swimming, mice had an average body temperature of 36.7 ± 0.15°C. These values were not significantly changed by vehicle or nor-BNI pretreatment alone. Immediately after forced swim, mice showed a transient decrease in body temperature to 33.8 ± 0.39°C but recovered within 10 min to 36.1 ± 0.61°C, a value not statistically different from the baseline (preswim) temperature. Mice receiving nor-BNI 60 min before the forced swim also showed a transient drop in body temperature (34.3 ± 0.43°C), which was not significantly different from that of vehicle-treated mice. In the conditioned place preference (CPP) training protocol used (see below), mice repeatedly exposed to forced swimming were next put into the cocaine-conditioning chamber. Body temperatures were measured immediately before and then 15 and 30 min after administration of cocaine or vehicle, and no significant differences between groups were found. For example, at the end of the 30 min conditioning session, mice not exposed to forced swimming that did not receive cocaine were at 38.4 ± 0.14°C; mice that had not swum but had received cocaine (15 mg/kg, s.c.) were at 38.0 ± 0.23°C; mice that had swum but had not received cocaine were at 37.4 ± 0.18°C; mice that had swum and had received cocaine were at 37.4 ± 0.63°C; mice receiving nor-BNI (10 mg/kg, i.p.) before swimming but not cocaine were at 37.8 ± 0.25°C; and mice receiving nor-BNI before swimming and cocaine were at 37.8 ± 0.25°C. In these tests, no treatment produced a statistically significant difference in body temperature compared with untreated, unstressed mice (F(5,3,15) = 1.54; p > 0.05).
Antinociceptive testing with the 55°C warm water tail withdrawal assay. The 55°C water is a commonly used nociceptive stimulus for opioid analgesia testing (Vaught and Takemori, 1979). Latency to withdraw the tail was taken as the end point, with the duration of tail immersion (or latency of response) measured by stopwatch. After determining baseline latencies, mice received a single intraperitoneal dose of vehicle (saline, 0.9%) or the κ opioid receptor antagonist nor-BNI before forced swim testing (detailed above). The antinociceptive effect of the forced swim stressor was measured 5–9 min after conclusion of the forced swim testing for that day. If the mouse failed to remove its tail within 15 sec, it was removed, and the animal was assigned a maximal antinociceptive score. Note that none of the animals used in this study had a baseline tail withdrawal latency longer than 5 sec, thereby requiring exclusion.
CPP. C57Bl/6 wild-type or prodynorphin knock-out mice were used in place-conditioning studies using a three-compartment box, similar to methods used by Carlezon et al. (1998). The compartmentalized box was divided into two equal-sized outer sections (25 × 25 × 25 cm) joined by a small central compartment (8.5 × 25 × 25 cm) accessed through a single doorway (3 × 3 cm). The compartments differed in wall striping (vertical vs horizontal alternating black and white lines, 1.5 cm in width), floor texture (wood chips vs Bed-A-Cob), and lighting intensity. Mice were tested on the morning of day 1 before any treatment to establish preconditioning responses and any possible box bias. Testing involved placing individual animals in the small central compartment and allowing them to freely explore the entire apparatus for 30 min. The time each mouse spent in the two outer compartments was recorded by stopwatch for the full 30 min period, using the placement of the front and rear paws as the determining factor. Animals that demonstrated a baseline preference >18 min spent in a particular compartment on the first day were rejected from the study. (Note that only one animal was rejected.)
Mice on average showed a slight preconditioning preference between the two chambers that was statistically significant (94 ± 33 sec; n = 51; F(1,100) = 12.4; p < 0.05) for the chamber with horizontal stripes and Bed-A-Cob flooring. A “biased” approach was used wherein individual animals were given cocaine in the less preferred compartment identified in the preconditioning test. This method has been shown to produce reliable conditioned place preference responses comparable with other experimental designs (for discussion, see Bardo et al., 1995). On day 2, mice were injected subcutaneously with 15 mg/kg cocaine, a dose demonstrated previously to produce a measurable but not maximal conditioned place preference in C57Bl/6 mice (Miner, 1997; Romieu et al., 2002; Zhang et al., 2002), and immediately confined for 30 min to the drug-paired outer compartment. Four hours later, mice received saline conditioning by administration of saline (0.3 ml/30 gm of body weight) and immediate confinement for 30 min to the opposite, vehicle-paired, outer compartment. On day 3, mice again were conditioned first with cocaine, followed by saline 4 hr later, in the appropriate compartments. On day 4, testing of conditioned preference was conducted in the morning to conclude the experiment. Mice were placed in the preference apparatus for 30 min, and the time spent in each compartment was measured.
To examine the effect of forced swim stress on cocaine CPP, mice were exposed to 2 d of vehicle or nor-BNI (10 mg · kg –1 · d –1, i.p.) pretreatment and forced swim testing after preconditioning preference testing as described above. Ten minutes after the second day forced swim session, mice were injected with cocaine (15 mg/kg, s.c.) and placed in the non-preferred compartment for 30 min to begin conditioning as described above. The experiment concluded on day 4 with testing of conditioned place preference.
Because the duration of stress-induced potentiation affecting the conditioning might be transient, the acquisition of cocaine CPP was monitored with an altered CPP protocol. Preconditioning testing and forced swim exposure (when used) were performed during days 1 and 2 as described above. Cocaine place preference conditioning began 10 min after the last FST trial on day 2. Instead of receiving a second training session with saline, animals were returned to their home cage to separate the saline treatment from stress. On the morning of day 3 and each subsequent morning to the conclusion of the experiment, mice were tested for conditioned preference in the preference apparatus for 30 min, with the time spent in each compartment measured. On the afternoon of day 3, saline conditioning was performed by administration of saline (0.3 ml/30 gm of body weight) and immediate confinement to the opposite, vehicle-paired, outer compartment for 30 min. Mice were tested again for conditioned preference on day 4 and subsequently conditioned with cocaine that afternoon in the same compartment used on day 2. On day 5, mice were tested again for conditioned preference and subsequently conditioned with saline that afternoon in the same compartment used on day 3. On day 6, final testing of conditioned preference was conducted in the morning to conclude the experiment. Note that all results are plotted as the difference in the times spent on the drug-paired side versus the vehicle-paired side. Therefore, a positive value demonstrates the animals' preference for the drug-paired side.
Data analysis. All data were analyzed by repeated measures ANOVA. Significant results demonstrated by ANOVA were further analyzed for significance with Student's t test for significant pair-wise comparisons. Dependent variables were expressed as the time spent immobile during forced swimming in all FST experiments and the latency of time spent before removing the tail in the tail withdrawal tests. Comparisons were analyzed for swim-stressed groups receiving nor-BNI or vehicle pretreatment, with the additional factor of wild-type or prodynorphin gene disruption. All CPP experiments express the dependent variable as the difference in time spent in the drug- and saline-paired compartments. Data for all CPP groups were analyzed for cocaine or vehicle conditioning, with the additional factors of swim-stressed versus unstressed groups, nor-BNI or vehicle pretreatment, and wild-type or prodynorphin gene disruption. Analysis compared differences in time spent in the (eventual) drug- and saline-paired compartments before and after FST exposure. Seconds spent in the drug-paired, saline-paired, and neutral zone compartments were additionally analyzed separately. Body temperature readings collected in triplicate were averaged together by mouse and then treatment group, with the dependent variable expressed as the body temperature (in degrees Celsius). Data for all temperature groups were analyzed for swimming or no swimming effects with the additional factor of nor-BNI or vehicle pretreatment before swimming. Moreover, the effects of cocaine or vehicle administration on body temperature in the conditioning apparatus were analyzed, with the additional factors of unstressed versus swim-stressed groups and nor-BNI or vehicle pretreatment. All data are presented as means ± SEM of the animal treatment group, with significance set at p < 0.05.
Results
Stress-induced analgesia is mediated by dynorphin peptides. C57Bl/6 wild-type mice were treated with vehicle and exposed to the forced swim test over 2 d. On both days, tail withdrawal latency in the 55°C warm-water tail withdrawal assay was significantly increased (day 1, F(1,50) = 28.49; p < 0.001; day 2, F(1,47) = 91.90; p < 0.001) threefold after forced swim testing (Fig. 1A,B, left). The SIA induced by FST exposure was blocked by pretreatment with the KOR antagonist nor-BNI before FST exposure (Fig. 1A,B, center; day 1, F(1,41) = 2.50; p > 0.05; day 2, F(1,39) = 0.57; p > 0.05). Notably, nor-BNI alone had no acute effect on baseline tail withdrawal response in mice not exposed to forced swimming (Fig. 1A, center left; F(1,12) = 0.04; p > 0.05). These results suggest that endogenous opioids were released by exposure to the forced swim stressor to produce the change in tail withdrawal latency. To test this, C57Bl/6 mice lacking prodynorphin gene products and their wild-type littermates were administered vehicle and exposed to forced swim testing. Consistent with previous results, swim-stressed wild-type mice demonstrated a significant increase in tail withdrawal latency at the end of testing on day 1(from 2.4 ± 0.31 to 5.6 ± 0.7 sec; n = 9; F(1,16) = 18.43; p < 0.001) and day 2 (2.4 ± 0.3 to 4.6 ± 0.4 sec; F(1,16) = 18.16; p < 0.001). In contrast, prodynorphin knock-out mice demonstrated no significant change (day 1, F(1,16) = 0.19; p > 0.05; day 2, F(1,16) = 0.14; p > 0.05) in tail withdrawal latency after forced swimming on either day (Fig. 1A,B, right). For wild-type mice, the stress-induced increase in tail flick latency on day 2 was not significantly different from the increase on day 1 (F(1,48) = 1.77; p > 0.05), suggesting that there was comparable activation of the nor-BNI-sensitive endogenous opioid system on both days.
Figure 1.
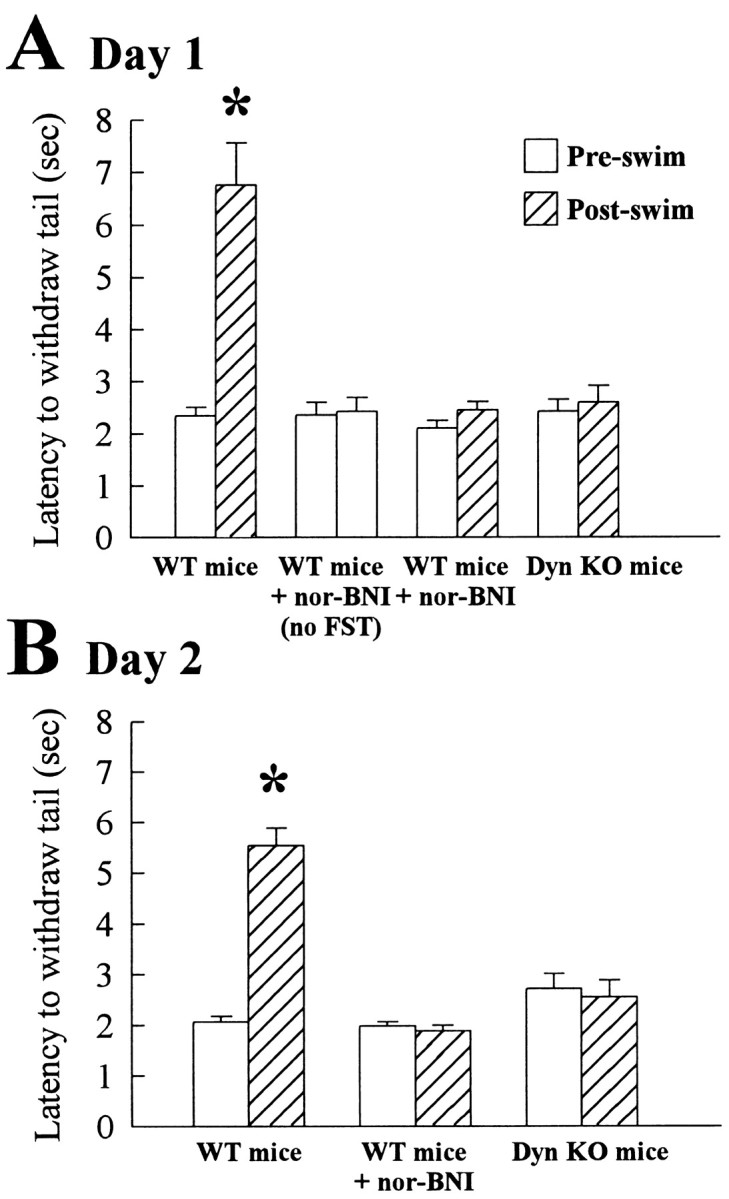
Forced swim stress-induced analgesia is blocked by pretreatment with nor-BNI or prodynorphin (Dyn) knock-out. Tail withdrawal latencies presented were obtained 5–9 min after the forced swimming on the first (A) or second (B) day. Mice were tested in the 55°C warm water tail withdrawal assay before (open bars) and after (hatched bars) exposure to the forced swim test, as described in Materials and Methods. On either day, C57Bl/6 mice pretreated with vehicle (0.3 ml/30 gm of body weight) demonstrated a tail withdrawal response that was increased nearly threefold after forced swim stress (A, B, left pair). Pretreatment 60 min before FST with the κ-selective antagonist nor-BNI (10 mg/kg, i.p.) did not significantly change baseline tail withdrawal latencies (A, left center pair) but blocked the increase in stress-induced analgesia produced by forced swimming (A, B, right center pair). Likewise, disruption of the prodynorphin gene prevented forced swim stress-induced analgesia (A, B, right pair). Wild-type littermates of the prodynorphin knock-out mice displayed SIA not significantly different from that of vehicle-treated C57Bl/6 mice (data in Results). *Significantly different from matching preswim latencies; p < 0.05, as determined by ANOVA followed by Student's t test. Bars represent n = 20–24 wild-type animals or 9 each of prodynorphin KO mice in swim test trials and 7 wild-type animals in tests of nor-BNI effect without FST exposure.
Opioid receptor antagonist selectivity of nor-BNI assessed
Although nor-BNI is reported to be a KOR-selective antagonist (Takemori et al., 1988; Horan et al., 1992), selectivity under these experimental conditions was directly assessed. Nor-BNI (10 mg/kg, i.p., 1 hr before testing) effectively blocked the increase in tail withdrawal latency produced by the KOR agonist. (trans)-3,4-Dichloro-N-methyl-N-[2-(1-pyrrolidinyl)-cyclohexyl]benzeneacetamide (U50,488; 25 mg/kg, i.p.) alone produced a tail withdrawal latency of 11.0 ± 1.61 sec, but it was 3.13 ± 0.33 sec in the presence of nor-BNI (not significantly different from baseline latency). In contrast, the μ opioid receptor preferring agonist morphine (10 mg/kg, i.p.) increased tail withdrawal latency to 9.33 ± 1.27 sec alone and to 11.1 ± 2.42 sec in the presence of nor-BNI (not significant; p > 0.05). The δ opioid receptor-selective agonist (+)-4-[(αR)-α-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide (SNC-80; 100 nmol, i.c.v.) increased latency to 13.2 ± 0.7 sec alone and to 11.3 ± 1.82 sec in the presence of nor-BNI (not significant; p > 0.05). Moreover, nor-BNI (10 mg/kg, i.p.) injected twice to duplicate its use in the 2 d forced swim protocol also blocked the increase in tail withdrawal latency induced by U50,488 but not morphine or SNC-80 (Fig. 2). Although the receptor-mediating the effects of the endogenously released opioid by forced swim were not directly identified by this pharmacological assay, these results suggest that the nor-BNI treatments used in this study selectively blocked the κ opioid receptor.
Figure 2.

Demonstration of nor-BNI selective antagonism of the κ opioid receptor. Mice were either untreated or pretreated once daily for 2 d with nor-BNI (10 mg/kg, i.p.). On the second day, 30 min after administration of the second dose of nor-BNI, mice were administered the opioid-selective agonists U50,488 (25 mg/kg, i.p., for the κ receptor), morphine sulfate (10 mg/kg, i.p., for the μ receptor), and SNC-80 (100 nmol, i.c.v., for the δ receptor). Preliminary dose–response curves with morphine and SNC-80 demonstrated that the doses used here produced submaximal analgesia comparable with the magnitude produced by U50,488. Animals were then tested in the 55°C warm water tail withdrawal assay 30 min after agonist administration, with the latency of the mouse to withdraw its tail from the water bath taken as the end point. As a control for the SNC-80 experiment, intracerebroventricular administration of vehicle did not produce a significant change in tail withdrawal latency (n = 4; data not shown). Pretreatment with this dose of nor-BNI blocked U50,488- but not morphine- or SNC-80-induced antinociception, suggesting that nor-BNI under these dosing conditions did not significantly occupy μ or δ opioid receptors. *Significantly different from baseline response; p < 0.05, as determined by Student's t test. Bars represent n = 4–10 mice.
Endogenous κ-opioid systems mediate the motor response to the second day of forced swim stress
Animals exposed to forced swimming display a characteristic immobility response as described in Materials and Methods. If endogenous κ opioids contribute to this response, the nor-BNI treatment effective in reducing SIA would also be expected to reduce the duration of immobility in the FST. Consistent with results using KOR knock-out mice presented by Filliol et al. (2000), C57Bl/6 WT mice pretreated 1 hr with vehicle or nor-BNI demonstrated no differences in time spent immobile on the first day of testing (Fig. 3A, left side, trial 1; F(1,44) = 0.136; p > 0.05). However, when mice were challenged with forced swim testing the next day, nor-BNI pretreatment significantly reduced (F(1,44) = 4.47; p < 0.05) the time spent immobile in the first trial of the day (the second swim trial overall) compared with the time of the vehicle-treated set (Fig. 3A, trial 2). The differences resulting from nor-BNI pretreatment were also significant on repeated trials (F(7,20,140) = 15.73; p < 0.05; Fig. 3A, trials 3–5). Although the reduction in immobility is small (15–20%), the effect is consistent with that produced by established antidepressant drugs (Lucki et al., 2001).
Figure 3.
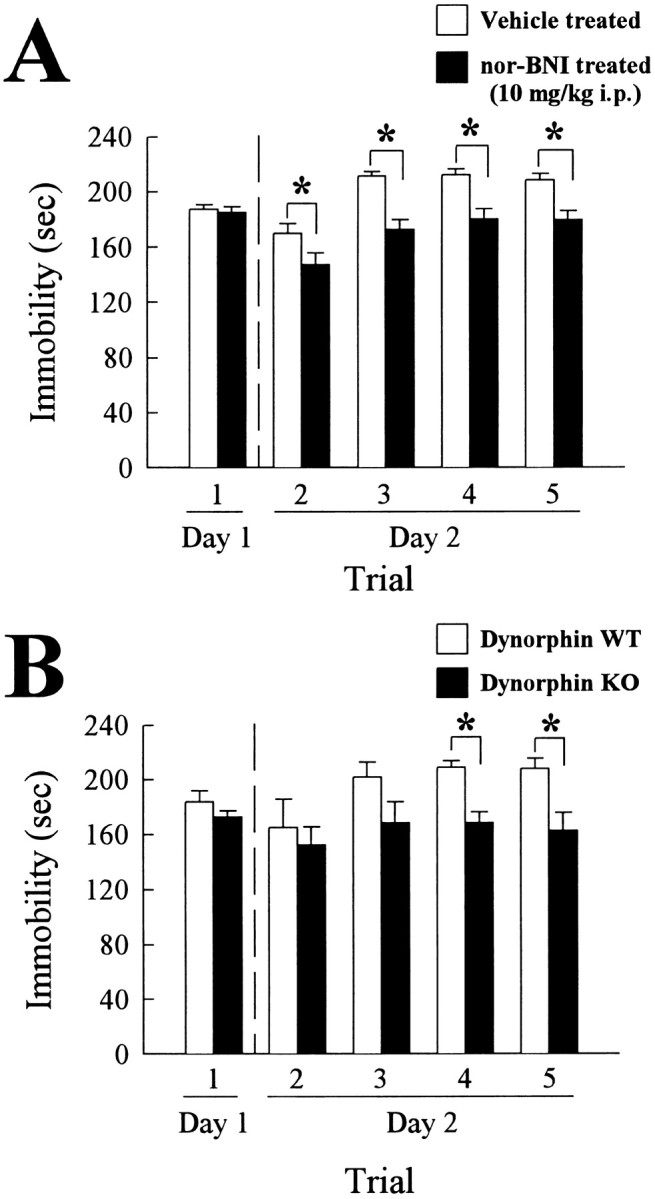
Immobility response to forced swim stress is reduced on the second day of testing by pretreatment with nor-BNI or by disruption of the prodynorphin gene. The time mice spent immobile during the last 4 min of the forced swim test was measured during multiple trials over 2 d. A, Mice received either vehicle (open bars) or nor-BNI (10 mg/kg, i.p., filled bars) in a bolus of 0.3 ml/30 gm of body weight 1 hr before daily swimming. Mice exposed to a single 15-min forced swim demonstrated no difference in immobility response on the first day, regardless of pretreatment (left-most bars). However, mice pretreated with nor-BNI spent significantly less time immobile in the FST on the second day during the four 6 min trials than vehicle-treated mice. Bars represent n = 20–24 animals. Note that mice did not demonstrate significant differences in body temperature 10 min after forced swimming on either day from unstressed mice, regardless of pretreatment. B, Wild-type, littermates (open bars) or prodynorphin gene knock-out mice (filled bars) were pretreated with vehicle in a bolus of 0.3 ml/30 gm of body weight 1 hr before daily swimming. Mice exposed to a single 15 min forced swim demonstrated no difference in immobility response on the first day, regardless of genotype. In contrast, on the second day, mice lacking dynorphin peptides through disruption of the prodynorphin gene spent significantly less time immobile in the last two 6 min trials of the FST than wild-type littermates. *Significant difference between immobility responses of stress-exposed vehicle-treated and nor-BNI treated mice (A) or between immobility responses of wild-type and prodynorphin gene-disrupted mice (B); p < 0.05, as determined by ANOVA followed by Student's t test. Bars represent n = 9 animals.
These results suggest that endogenous stimulation of KOR contributed to the immobility response during forced swimming. To test this, prodynorphin knock-out mice and their wild-type littermates were pretreated with vehicle and examined in the FST. There was no significant difference (F(1,16) = 1.49; p > 0.05) between the immobility responses of either the knock-out or wild-type mice after the first day of forced swimming (Fig. 3B, left side, trial 1). However, repeated trials on the second day of FST revealed a significant reduction (F(7,8,56) = 4.12; p < 0.01) in later trials in the duration of immobility displayed by the prodynorphin knock-out mice compared with their wild-type littermates (Fig. 3B, trials 4, 5). The significant reduction in immobility during the later forced swim trials evident for the prodynorphin knock-out mice (Fig. 3B) is consistent with the effects of nor-BNI on wild-type mice (Fig. 3A).
Endogenous κ opioids mediating swim stress also potentiate the conditioned place preference response to cocaine
We next asked whether the forced swim stress would affect cocaine-conditioned place preference by a nor-BNI-sensitive or prodynorphin-dependent mechanism. A variety of stressors have been shown to potentiate cocaine-conditioned place preference (Haile et al., 2001; Kabbaj et al., 2001), and activation of the κ receptor has been demonstrated to regulate behavioral as well as mesolimbic dopaminergic responses to cocaine (Spanagel et al., 1992; Glick et al., 1995; Mello and Negus, 1998). Thus, we predicted that stress-induced activation of KOR by endogenous dynorphin would suppress cocaine-conditioned place preference.
In the conditioned place preference assay, C57Bl/6 mice spend greater amounts of time in environments associated with the rewarding effects of cocaine (Carr et al., 1989; Miner, 1997; Romieu et al., 2002). Initially, C57Bl/6 mice were tested in the conditioned place preference apparatus before drug treatment to obtain preconditioning responses for subsequent comparisons. Animals were exposed to forced swim stress over 2 d or left idle in home cages before conditioning on the afternoon of day 2 with cocaine (15 mg/kg, s.c.), followed by saline (see protocol; Fig. 4A). On day 4, unstressed control C57Bl/6 mice demonstrated a measurable increase in the difference in time spent in the drug-versus saline-paired chamber over the same difference shown before conditioning, an example of cocaine-conditioned place preference (307 ± 114 sec preference for the cocaine-paired chamber, significantly greater than the preconditioning response; F(1,21) = 15.43; p < 0.01; Fig. 4B). Mice pretreated with vehicle and exposed to forced swim stress before cocaine conditioning demonstrated a significant twofold greater cocaine CPP response over that shown by unstressed mice (F(2,36) = 4.17; p < 0.05; Fig. 4B). Notably, nor-BNI pretreatment of FST-exposed mice blocked the potentiation of the cocaine CPP response (Fig. 4B). Swim-stressed nor-BNI-pretreated mice demonstrated a significant cocaine CPP response over the preconditioned response (F(1,22) = 16.29; p < 0.01) that was not significantly different from the response produced by the unstressed mice (F(1,21) = 0.01; p > 0.05; Fig. 4B).
Figure 4.
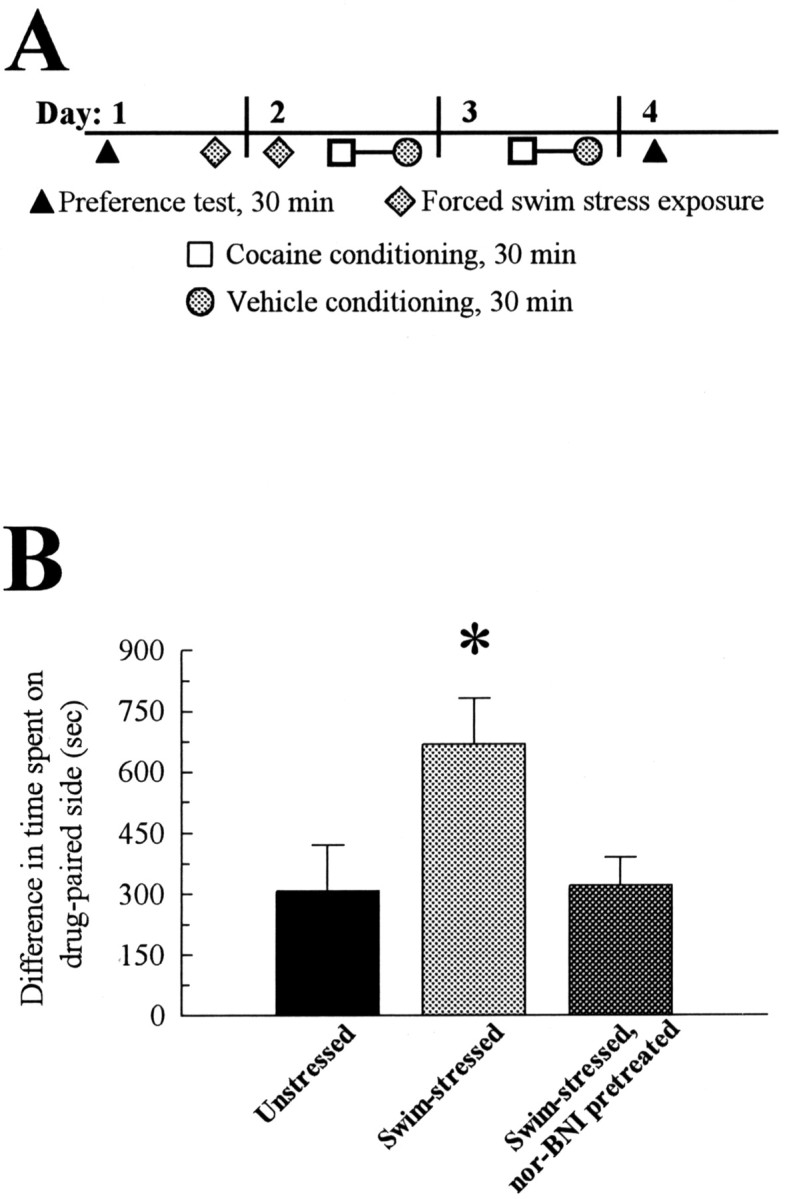
Exposure to forced swim stress produces a nor-BNI-sensitive potentiation of cocaine-conditioned place preference.A, Schematic of training paradigm. The CPP protocol was as described in Materials and Methods. Preference testing allowed mice to move freely for 30 min in the morning to measure preconditioning and subsequent responses for either of two conditioning chambers, as described in Materials and Methods (represented here by triangles). After assessment of preconditioning preference, mice were exposed to repeated forced swim stress over the next 24 hr, as detailed in Materials and Methods (diamonds), or allowed to remain in home cages without swimming. Within 10 min after forced swim testing on day 2, mice were administered cocaine (15 mg/kg, s.c.) and confined to the drug-paired box for a 30 min conditioning session (squares). Four hours later, mice were administered vehicle and confined to the vehicle-paired box for a 30 min conditioning session (circles). Cocaine and saline conditioning was repeated the next day, separated again by 4 hr (represented by joined square and circle, day 3). On day 4, the final preference test was performed blind to determine the effect of treatment and conditioning on place preference. B, Preference test data demonstrating a nor-BNI-sensitive, FST-induced potentiation of cocaine CPP. Preferences are given as the difference between time spent in the drug-paired chamber and time in the saline-paired chamber during the 30 min trial. A positive value represents time spent in the drug-paired chamber. Mice were divided into three groups. The first group was unstressed, remaining in home cages and not exposed to swim stress before 2 d of cocaine and saline conditioning, as described in Materials and Methods (black bar). The second group was administered vehicle and exposed to the forced swim stressor before 2 d of cocaine and saline conditioning (light gray bar). The third group was administered nor-BNI, exposed to the forced swim stressor as described above, and then conditioned over 2 d with cocaine and saline (dark gray bar). After conditioning, all three groups demonstrated an increase in time spent in the cocaine-paired chamber that was significantly greater than the time spent in that chamber before conditioning, an example of conditioned place preference. Control unstressed mice and nor-BNI-treated, FST-exposed mice demonstrated an equivalent degree of cocaine CPP. In contrast, vehicle-treated mice exposed to FST demonstrated a significant potentiation over the unstressed animals responses. Note that mice did not demonstrate significant differences in body temperature from unstressed mice immediately after forced swimming, immediately before place conditioning, or 30 min after cocaine administration. *Significant difference in cocaine CPP compared with CPP for both unstressed and nor-BNI-treated mice; p<0.05, as determined by ANOVA followed by Student's t test. Bars represent n = 11–16 mice.
To determine whether the potentiating effect of stress worked to increase the rate of acquisition for cocaine CPP, we modified the paradigm to measure CPP at intermediate times. In this modified protocol, cocaine and saline conditioning was performed separately on sequential days, with preference testing performed each morning before the conditioning sessions. Mice received cocaine conditioning after the second FST exposure on day 2 and then were tested the morning of the next day (day 3) for place preference. In the afternoon of day 3, mice received vehicle conditioning in the opposite chamber and were tested the morning of the next day (day 4) for place preference. This alternation was repeated on days 4 (cocaine conditioning) and 5 (vehicle conditioning) with testing for place preference each morning. Place preference was assessed on day 6 to end the experiment (see protocol; Fig. 5A). Control mice were treated by the same cocaine-conditioning protocol but not exposed to forced swimming. Although the modified (repeated testing) paradigm might contribute to extinction, it has the advantage of assessing how stress affects the rate of acquisition of cocaine place preference.
Figure 5.

Exposure to forced swim stress results in rapid, long-lasting nor-BNI-sensitive potentiation of cocaine conditioned place preference. A, Schematic of modified CPP paradigm. The CPP protocol was modified to allow one preference test and one conditioning session per day, alternating between cocaine (15 mg/kg, s.c.; squares) and saline (circles), such that the study extended over 6 d. Preference testing allowed mice to move freely for 30 min in the morning to measure preconditioning and subsequent responses for either of two conditioning chambers, as described in Materials and Methods (represented here by triangles). After assessing preconditioning preferences, some of the mice were exposed to repeated forced swim stress over the next 24 hr, as detailed in Materials and Methods (diamonds). On day 2, unstressed mice or FST-treated mice within 10 min after forced swim were administered cocaine (15 mg/kg, s.c.; squares), confined to the drug-paired box for a 30 min conditioning session, and then returned to the home cage overnight. Preference testing followed the next day (triangle, day 3), with animals then administered vehicle and confined to the vehicle-paired box for a 30 min conditioning session (circle). The monitoring of cocaine CPP acquisition continued on days 4 and 5 to ascertain a steady-state response, with one more cocaine-conditioning session (day 4) and vehicle-conditioning session (day 5) preceding the final preference test on day 6. B, Daily preference test data demonstrating acquisition of cocaine CPP and nor-BNI-sensitive potentiation by exposure to FST. Summarized results of daily preference tests (represented by triangles in A) are plotted in seconds to highlight time spent on the drug-paired side of the apparatus. Grouped mice on day 1 demonstrate a 94-sec preconditioning preference for the chamber that would subsequently be vehicle-paired. Mice were then divided into three groups. The first group was administered vehicle and exposed to the forced swim stressor (open circles); the second group was administered nor-BNI 60-min before FST (open squares); and the third group was returned to home cages and not exposed to swim stress (filled circles). All mice were subsequently used in cocaine CPP testing, with daily preference testing done blind to monitor the acquisition of cocaine CPP as detailed in A. Both control, unstressed mice and swim-stressed mice pretreated with nor-BNI demonstrated rapid acquisition of cocaine CPP on day 3, indicated by a time spent in the drug-paired chamber that was significantly (p < 0.05) greater after cocaine conditioning than before. Moreover, the preferences shown in subsequent tests with these animals were not significantly different from the day 3 response (filled circles). Vehicle-treated mice exposed to FST demonstrated the same rapid acquisition of cocaine CPP, also reaching a stable, peak response by day 5, but the response on each day was significantly potentiated twofold to fourfold over the unstressed animals responses. *Significant difference between cocaine CPP mice and preconditioning preference, as determined by ANOVA followed by Student's t test. Points represent means for six to eight animals.
Unstressed control C57Bl/6 mice demonstrated a measurable place preference on day 3 (147 ± 135 sec preference for drug-paired side; F(1,15) = 10.0; p < 0.01; Fig. 5B). After the second conditioning sessions with cocaine and saline, place preference reached a stable level on day 6 (358 ± 85 sec) that was not significantly different from that on days 3–5 (F(3,28) = 0.62; p > 0.05; Fig. 5B). The modified conditioning paradigm produced the same level of cocaine-conditioned place preference as the original paradigm, suggesting that extinction was not a significant effect. In contrast, mice exposed to the forced swim stressor over 2 d demonstrated significantly greater cocaine-conditioned place preference on each day of testing than demonstrated by the untreated control mice (p < 0.05 each day; Fig. 5B). This twofold to fourfold potentiation of place preference occurred rapidly and remained significantly elevated over the preference responses of unstressed mice, reaching 693 ± 105 sec on day 6 (F(1,12) = 7.42; p < 0.05; Fig. 5B). Importantly, swim stress induced a fourfold potentiation of cocaine CPP on day 3, after the first cocaine conditioning session, and the potentiation was not significantly increased by subsequent cocaine or saline training sessions (F(3,20) = 1.00; p > 0.05).
Pretreatment of mice with nor-BNI (10 mg/kg, i.p.) on days 1 and 2 before forced swim testing blocked the potentiation of subsequent conditioned place preference in this modified paradigm (Fig. 5B). There was no significant difference between stress-exposed, nor-BNI-treated mice and unstressed mice in the cocaine-conditioned place preference trials on any day. In the absence of stress, mice pretreated with nor-BNI did not show a significant difference in cocaine CPP compared with untreated mice (F(1,10) = 0.11; p > 0.05; Fig. 6A). Thus, in the absence of stress, nor-BNI does not affect cocaine place preference in this conditioning paradigm.
Figure 6.
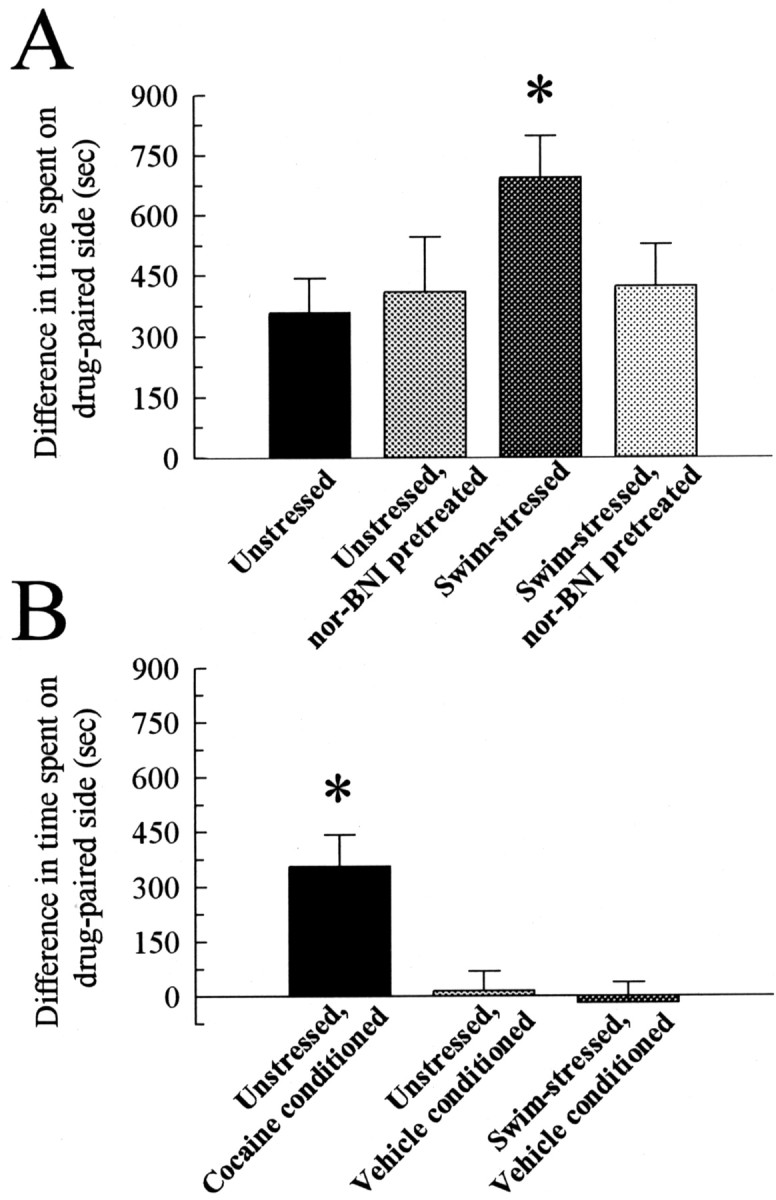
Control experiments. A, nor-BNI has no effect on cocaine CPP in the absence of stress Mice were pretreated twice over 2 d with vehicle or nor-BNI (10 mg/kg, i.p.) and either exposed to the forced swim stressor as detailed in Material and Methods or allowed to sit in their home cages before the development of cocaine-conditioned place preference as described in Figure 5. By the final day of preference testing, nor-BNI pretreatment of unstressed mice showed cocaine CPP that was not significantly different from the cocaine CPP of unstressed, untreated control mice. *Significant difference in matching cocaine CPP response of unstressed mice; p < 0.05 for all, as determined by ANOVA followed by Student's t test. B, Swim stress alone does not produce place preference in the absence of cocaine. Three sets of mice were pretreated with vehicle and either exposed to the forced swim stressor or left in their home cages. Conditioned place preference testing was performed as described in Figure 5A, but vehicle was substituted for cocaine in all conditioning sessions for the two sets of mice. Mice conditioned with saline on both sides of the apparatus did not show place preference significantly different from the animal's preconditioning responses. *Significant difference in time spent on the drug paired side compared with time spent on the drug paired side by saline-conditioned animals; p < 0.05 for all as determined by ANOVA followed by Student's t test. Bars represent n = 3–8 animals.
It was possible that pairing the termination of stress with a chamber could, by itself, lead to a place preference. To assess the possibility that removal from the swim test and immediate placement in a conditioning chamber could create a preference for that chamber, animals were conditioned with saline in both chambers (i.e., no cocaine-training sessions but placement in one of the chambers 10 min after the termination of FST). As shown, vehicle-conditioned mice did not develop significant place preference when either unstressed (F(1,5) = 0.01; p > 0.05) or exposed to FST (F(1,6) = 0.08; p > 0.05; Figure 6B). Thus, nor-BNI in the absence of stress did not affect cocaine CPP, and exposure to the forced swim stress in the absence of subsequent cocaine did not produce CPP. Surprisingly, the nor-BNI data suggest that a stress-induced release of endogenous opioids may have potentiated rather than suppressed cocaine CPP.
The possible involvement of the endogenous κ opioid system in the stress response was assessed using prodynorphin knockout mice and their wild-type littermates. Neither group showed significant chamber preference before cocaine conditioning (F(1,19) = 0.006; p > 0.05; Fig. 7, left pair of bars). Unstressed prodynorphin knock-out mice and their wild-type littermates used in cocaine CPP assays demonstrated similar place preference responses (Fig. 7) that were not statistically different from each other (F(1,7) = 0.01; p > 0.05; center pair of bars) or from the response of unstressed C57Bl/6 mice (prodynorphin KO mice, F(1,11) = 0.003; p > 0.05; wild-type littermates, F(1,11) = 0.005; p > 0.05). Consistent with the previous data, wild-type littermates exposed to 2 d forced swim stress developed significantly greater cocaine CPP than unstressed wild-type littermate mice, as measured by comparing the differences in time spent in the drug- and saline-paired chambers between the two sets of animals (F(1,8) = 7.57; p < 0.05). The cocaine CPP of swim-stressed prodynorphin wild-type littermate mice was not significantly different from that of swim-stressed control C57Bl/6 mice on day 6 (F(1,10) = 0.10; p > 0.05). In contrast, prodynorphin knock-out mice exposed to 2 d swim stress developed cocaine CPP that was not significantly different from the preference displayed by the unstressed prodynorphin knock-out littermates (F(1,9) = 0.07; p > 0.05) or C57Bl/6 mice (F(1,11) = 0.003; p > 0.05) but that was significantly smaller than their swim-stressed wild-type littermates (F(1,10) = 10.8; p < 0.01; Fig. 7, right pair of bars). Thus, a normal prodynorphin gene was required for stress-induced potentiation of cocaine CPP.
Figure 7.

Disruption of the prodynorphin gene prevents the forced swim stress-induced potentiation of cocaine-conditioned place preference. Prodynorphin gene knock-out or wild-type littermate mice were exposed to 2 d forced swim stress or left in their home cages and then used in cocaine-conditioned place preference assays as detailed in Figure 5A and Materials and Methods. All animals demonstrated cocaine CPP by day 6 that was significantly different from matching preconditioning responses displayed on day 1. However, FST-exposed wild-type littermates demonstrated cocaine CPP on day 6 that was double the preference responses of the unstressed wild-type littermates or swim-stressed prodynorphin knock-out mice. The horizontal dashed line designates for comparison the cocaine CPP response of unstressed C57Bl/6 mice obtained on the same testing day (see Fig. 6). *Significant difference in time spent on the drug paired side after cocaine conditioning compared with baseline response; ζsignificant difference in matching cocaine CPP response of FST-exposed prodynorphin wild-type versus unstressed wild-type or FST-exposed knock-out mice; p<0.05 for all as determined by ANOVA followed by Student's t test. Bars represent n = 4–10 animals.
Discussion
The principal findings of this study were that repeated swim stress resulted in immobility, analgesia, and potentiation of cocaine CPP that were blocked by nor-BNI and prodynorphin gene disruption. One explanation of these results is that stress induced the release of the endogenous dynorphins to activate the κ opioid receptor to mediate these behavioral effects, although additional work is required to establish this hypothesis. First, the conclusion that endogenous opioid peptides were released requires a direct measure of peptide release evoked by swim stress. Second, establishing the role of κ opioid receptors in mediating the response requires a more direct demonstration of activation by swim stress. Third, although disruption of the prodynorphin gene has been shown to prevent expression of the dynorphin peptides (Sharifi et al., 2001) thought to be endogenous ligands for the κ opioid receptor (Chavkin et al., 1982), expression of other prodynorphin products is also blocked. Moreover, interpretation of knock-out studies must be done cautiously because gene deletion can have other nonspecific effects (Tronche et al., 2002). Nevertheless, if verified by additional work, the hypothesis would have important implications, as discussed below.
First, the suggestion that dynorphin–KOR interactions can contribute to stress-induced analgesia would extend our understanding of the diverse mechanisms controlling nociception. Although exposure to a stressor has been demonstrated to increase dynorphin levels (Przewlocki et al., 1987; Nabeshima et al., 1992), an antinociceptive role for the dynorphin–KOR systems has been underappreciated. For example, using a single, brief exposure to a forced swim, κ opioid receptor knock-out mice were found to have normal SIA (Filliol et al., 2000). However, work with rats using the established 2 d procedure of repeated forced swim exposure (Porsolt et al., 1977b) suggested that nor-BNI could reduce immobility in the forced swim test (Pliakas et al., 2001). Drawing on this report, the present study extended the forced swim test to the 2 d procedure using mice to demonstrate the sensitivity of the stress-induced immobility and analgesia to nor-BNI or prodynorphin gene disruption. The repeated, prolonged nature of the stressor used in this study could account for the differences with previous findings to implicate chronic stress-induced KOR activation by dynorphin peptides.
Other endogenous opioid systems have also been shown to mediate the response to stressors. When exposed to inescapable tail shock or forced swimming, rats and mice show SIA blocked by the opioid antagonists naloxone and naltrexone (Maier et al., 1980; Mogil et al., 1996). Therefore, an alternative explanation is that the dynorphin–κ system acts indirectly to modulate the actions of another endogenous opioid peptide, such as β-endorphin. Previous reports have noted that SIA induced after a forced-swim test stressor was absent in mice lacking β-endorphin (Rubinstein et al., 1996), and concentrations of this endogenous opioid were elevated in the periaqueductal gray region of mice demonstrating SIA after exposure to forced walking stress (Nakagawasai et al., 1999). Another alternative possibility is that a prodynorphin-derived peptide was released to activate either the δ or μ opioid receptor. Prodynorphin has been shown to be a precursor for Leu5enkephalin in brain (Zamir and Quirion, 1985). However, the pharmacological data we obtained suggest that nor-BNI acted selectively at the κ receptor. In addition, the KOR selectivity of nor-BNI is supported by studies using mouse analgesic assays and recent functional studies using guanosine-5′-O-(3-thio)triphosphate binding (Horan et al., 1992; Thomas et al., 2002). Overall, the most parsimonious explanation of these results is that a prolonged forced swim stress induced the release of dynorphin peptides to stimulate the κ opioid receptor and to mediate the analgesia observed; however, this requires further studies.
The forced swim test is an established and predictive animal model for the study of depression, with antidepressants typically reducing the duration of the immobility exhibited (Porsolt et al., 1977a,b; Dalvi and Lucki, 1999; Lucki et al., 2001). Pliakas et al. (2001, 2002) have previously shown that nor-BNI blocked the swim stress-induced immobility and suggested that κ receptor antagonists may also be effective antidepressants by blocking endogenous dynorphin function. The reduction of immobility caused by the disruption of the prodynorphin gene further suggests a mediating role for dynorphin in behavioral depression and supports the suggestion that KOR-selective antagonists may provide a new therapeutic approach for the treatment of depression.
Forced swim stress potentiated cocaine-conditioned place preference, possibly mediated by the induced release of dynorphin and the activation of κ opioid receptors. Results from this study showed that blockade of endogenous κ opioid systems by nor-BNI and prodynorphin gene disruption prevented the potentiation. Stress-induced potentiation of the conditioned place preference of rewarding drugs has been noted previously, such as the potentiation of morphine CPP after foot shock stress (Will et al., 1998). However, a positive relationship between dynorphin activation of KOR and the potentiation of cocaine CPP would be surprising, given reports that KOR agonists actually reduce self-administration of reinforcing drugs. For instance, peripheral administration of κ agonists U50,488 and spiradoline produced dose-dependent decreases in morphine and cocaine self-administration in rats, and a series of κ agonists dose-dependently decreased cocaine self-administration in monkeys (Glick et al., 1995; Mello and Negus, 1998). This reduction in drug self-administration is likely attributable to a κ-opioid-mediated suppression of dopaminergic signaling in the putative dopamine reward pathway because in vivo microdialysis detected reductions in dopamine released in the nucleus accumbens (NAc) of anesthetized rats in response to administration of κ agonists (Spanagel et al., 1992). This reduction in NAc dopamine release was associated with the activation of κ opioid receptors located on synaptic terminals in the NAc (Acri et al., 2001). These reports predict that endogenous κ opioid agonists should suppress conditioned place preference for cocaine. One way to reconcile the observation that stress-induced release of endogenous dynorphin peptides resulted in a potentiation of cocaine response might be to presume that the repeated stress actually depleted dynorphin to reduce a tonic κ system tone. However, stress-induced increases in the tail withdrawal latency immediately before cocaine conditioning were blocked by nor-BNI and prodynorphin gene disruption, suggesting the presence, not absence, of dynorphin. Moreover, neither nor-BNI pretreatment nor the prodynorphin gene-disrupted mice showed an elevated cocaine CPP response, as would be expected if decreased dynorphin levels were in fact responsible for the potentiation. Additionally, there are many important spatial and temporal differences between coadministration of a κ drug with cocaine and preactivation of the endogenous dynorphin–κ system by a prolonged stress exposure preceding cocaine administration. For example, the specific activation by the stressor of the dynorphin–κ system in a subset of brain circuits may differ from a global activation of the κ system. Furthermore, if the activation of a dynorphin–κ circuit contributes to the stress response, cocaine might have an enhanced rewarding value because it may counteract the dynorphin-mediated stress response. This has been recently demonstrated behaviorally, in which rats exposed to chronic, unpredictable stress demonstrated a leftward shift in the dose–response relationship of cocaine in CPP and locomotor activity testing (Haile et al., 2001).
Not only is the conditioned place preference test an assay of the rewarding properties of a stimulus, it is also a learning task requiring that the animal form an association between the rewarding drug and the compartment-specific cues (Carr et al., 1989). Thus, the potentiation of cocaine CPP observed in this study could have resulted from an enhancement of the acute effects of cocaine or by affecting a step in the learning process. Further analysis of the cellular and molecular mechanism of the nor-BNI-sensitive and prodynorphin-dependent potentiation is required to determine whether dynorphin activation of κ opioid receptors affects the acute response to cocaine or has a more general effect on learning mechanisms. For example, an acute enhancement of the cocaine response might result from a synergistic action of the dynorphin–κ system or an effect on the cocaine sensitization processes. Examination of the effects of stress-induced release of dynorphin peptides in the brain would help address this question.
In conclusion, the mediation of swim stress-induced behaviors was shown to be sensitive to nor-BNI pretreatment or prodynorphin gene disruption, suggesting that stress induced a release of dynorphin to activate the κ opioid receptor. The findings further our understanding of the neurobiological mechanisms underlying the response to stress and illuminate a possible κ opioid connection among chronic stress, depression, and drug abuse. Moreover, because stress has been demonstrated to increase both the rewarding potential and self-administration of drugs of abuse, the demonstration of endogenous κ opioid involvement might lead to new therapeutic approaches to the problems of depression and drug abuse.
Footnotes
The work was supported by United States Public Health Service grants RO1-DA11672, PO1-DA15916, and T32-DA07278 from the National Institute on Drug Abuse. We thank Dr. Ilene Bernstein for critical reading and suggestions on this manuscript. Dr. Uwe Hochgeschwender generously provided the prodynorphin knock-out mice. Sumit Sud performed the mouse genotyping. Theodore A. Chavkin helped perform some of the behavioral assays.
Correspondence should be addressed to Dr. Charles Chavkin, Department of Pharmacology, Box 357280, University of Washington, Seattle, WA 98195-7280. E-mail: cchavkin@u.washington.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/235674-10$15.00/0
References
- Acri JB, Thompson AC, Shippenberg T ( 2001) Modulation of pre- and postsynaptic dopamine D2 receptor function by the selective kappa-opioid receptor agonist U69593. Synapse 39: 343–350. [DOI] [PubMed] [Google Scholar]
- Banbury Conference ( 1997) Mutant mice and neuroscience: recommendations concerning genetic background. Neuron 19: 755–759. [DOI] [PubMed] [Google Scholar]
- Bardo MT, Rowlett JK, Harris MJ ( 1995) Conditioned place preference using opiate and stimulant drugs: a meta-analysis. Neurosci Biobehav Rev 19: 39–51. [DOI] [PubMed] [Google Scholar]
- Broom DC, Jutkiewicz EM, Folk JE, Traynor JR, Rice KC, Woods JH ( 2002) Nonpeptidic δ-opioid receptor agonists reduce immobility in the forced swim assay in rats. Neuropsychopharmacology 26: 744–755. [DOI] [PubMed] [Google Scholar]
- Carlezon Jr WA, Thome J, Olsen VG, Lane-Ladd SB, Brodkin ES, Hirol N, Duman RS, Neve RL, Nestler EJ ( 1998) Regulation of cocaine reward by CREB. Science 282: 2272–2275. [DOI] [PubMed] [Google Scholar]
- Carr GD, Fibiger HC, Phillips AG ( 1989) Conditioned place preference as a measure of drug reward. In: The neuropharmacological basis of reward (Liebman JM, Cooper SJ, eds), pp 264–319. New York: Oxford UP.
- Chavkin C, James IF, Goldstein A ( 1982) Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science 215: 413–415. [DOI] [PubMed] [Google Scholar]
- Dalvi A, Lucki I ( 1999) Murine models of depression. Psychopharmacology (Berl) 147: 14–16. [DOI] [PubMed] [Google Scholar]
- Filliol D, Ghozland S, Chluba J, Martin M, Matthes HWD, Simonin F, Befort K, Gaveriaux-Ruff C, Dierich A, LeMeur M, Valverde O, Maldonado R, Kieffer BL ( 2000) Mice deficient for δ- and μ-opioid receptors exhibit opposing alterations of emotional responses. Nat Genet 25: 195–200. [DOI] [PubMed] [Google Scholar]
- Glick SD, Maisonneuve IM, Raucci J, Archer S ( 1995) Kappa opioid inhibition of morphine and cocaine self-administration in rats. Brain Res 681: 147–152. [DOI] [PubMed] [Google Scholar]
- Haile CN, GrandPre T, Kosten TA ( 2001) Chronic unpredictable stress, but not chronic predictable stress, enhances the sensitivity to the behavioral effects of cocaine in rats. Psychopharmacology (Berl) 154: 213–220. [DOI] [PubMed] [Google Scholar]
- Horan P, Taylor J, Yamamura HI, Porreca F ( 1992) Extremely long-lasting antagonistic actions of nor-binaltorphimine (nor-BNI) in the mouse tail-flick test. J Pharmacol Exp Ther 260: 1237–1243. [PubMed] [Google Scholar]
- Kabbaj M, Norton CS, Kollack-Walker S, Watson SJ, Robinson TE, Akil H ( 2001) Social defeat alters the acquisition of cocaine self-administration in rats: role of individual differences in cocaine-taking behavior. Psychopharmacology (Berl) 158: 382–387. [DOI] [PubMed] [Google Scholar]
- Katoh A, Nabeshima T, Kameyama T ( 1990) Behavioral changes induced by stressful situations: effects of enkephalins, dynorphin, and their interactions. J Pharmacol Exp Ther 253: 600–607. [PubMed] [Google Scholar]
- Kosten TA, Miserendino MJD, Kehoe P ( 2000) Enhanced acquisition of cocaine self-administration in adult rats with neonatal isolation stress experience. Brain Res 875: 44–50. [DOI] [PubMed] [Google Scholar]
- Kosten TR, Rounsaville BJ, Kleber HD ( 1986) A 2.5-year follow-up of depression, life crises, and treatment effects on abstinence among opioid addicts. Arch Gen Psychiatry 43: 733–738. [DOI] [PubMed] [Google Scholar]
- Kreek MJ, Koob GF ( 1998) Drug dependence: stress and dysregulation of brain reward pathways. Drug Alcohol Depend 51: 23–47. [DOI] [PubMed] [Google Scholar]
- Lucki I, Dalvi A, Mayorga AJ ( 2001) Sensitivity to the effects of pharmacologically selective antidepressants in different strains of mice. Psychopharmacology (Berl) 155: 315–322. [DOI] [PubMed] [Google Scholar]
- Maier SF, Davies S, Grau JW, Jackson RL, Morrison DH, Moye T, Madden IV J, Barchas JD ( 1980) Opiate antagonists and long-term analgesic reaction induced by inescapable shock in rats. J Comp Physiol Psychol 94: 1172–1183. [DOI] [PubMed] [Google Scholar]
- Mello NK, Mendelson JH ( 1997) Cocaine's effects on neuroendocrine systems: clinical and preclinical studies. Pharmacol Biochem Behav 57: 571–599. [DOI] [PubMed] [Google Scholar]
- Mello NK, Negus SS ( 1998) Effects of kappa opioid agonists on cocaine- and food-maintained responding by rhesus monkeys. J Pharmacol Exp Ther 286: 812–824. [PubMed] [Google Scholar]
- Menendez L, Andres-Trelles F, Hidalgo A, Baamonde A ( 1993) Involvement of spinal kappa opioid receptors in a type of footshock induced analgesia in mice. Brain Res 611: 264–271. [DOI] [PubMed] [Google Scholar]
- Miner LL ( 1997) Cocaine reward and locomotor activity in C57BL/6J and 129/SvJ mice and their F1 cross. Pharmacol Biochem Behav 58: 25–30. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Sternberg WF, Balian H, Liebeskind JC, Sadowski B ( 1996) Opioid and nonopioid swim stress-induced analgesia: a parametric analysis in mice. Physiol Behav 59: 123–132. [DOI] [PubMed] [Google Scholar]
- Nabeshima T, Katoh A, Wada M, Kameyama T ( 1992) Stress-induced changes in brain Met-enkephalin, Leu-enkephalin and dynorphin concentrations. Life Sci 51: 211–217. [DOI] [PubMed] [Google Scholar]
- Najavits LM, Gastfriend DR, Barber JP, Reif S, Muenz LR, Blaine J, Frank A, Crits-Christoph P, Thase M, Weiss RD ( 1998) Cocaine dependence with and without PTSD among subjects in the National Institute on Drug Abuse Collaborative Cocaine Treatment Study. Am J Psychiatry 155: 214–219. [DOI] [PubMed] [Google Scholar]
- Nakagawasai O, Tadano T, Tan No K, Niijima F, Sakurada S, Endo Y, Kisara K ( 1999) Changes in beta-endorphin and stress-induced analgesia in mice after exposure to forced walking stress. Methods Find Exp Clin Pharmacol 21: 471–476. [PubMed] [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, Emrich HM ( 1986) Psychotomimesis mediated by kappa opiate receptors. Science 233: 774–776. [DOI] [PubMed] [Google Scholar]
- Piazza PV, Deminiere JM, Le Moal M, Simon H ( 1990) Stress- and pharmacologically induced behavioral sensitization increases vulnerability to acquisition of amphetamine self-administration. Brain Res 514: 22–26. [DOI] [PubMed] [Google Scholar]
- Pliakas AM, Carlson RR, Neve RL, Konradi C, Nestler EJ, Carlezon Jr WA ( 2001) Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element-binding protein expression in nucleus accumbens. J Neurosci 21: 7397–7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliakas AM, Mague SD, Tomasiewicz HT, Portoghese PS, Carlezon Jr WA ( 2002) Immobility behavior in the forced swim test mediated by kappa opioid receptors in rats. Soc Neurosci Abstr 28: 102.1. [Google Scholar]
- Porsolt RD, Bertin A, Jalfre M ( 1977a) Behavioural despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn 229: 327–336. [PubMed] [Google Scholar]
- Porsolt RD, Le Pichon M, Jalfre M ( 1977b) Depression: a new animal model sensitive to antidepressant treatments. Nature 266: 730–732. [DOI] [PubMed] [Google Scholar]
- Przewlocki R, Lason W, Hollt V, Silberring J, Herz A ( 1987) The influence of chronic stress on multiple opioid peptide systems in the rat: pronounced effects upon dynorphin in spinal cord. Brain Res 413: 213–219. [DOI] [PubMed] [Google Scholar]
- Rhoads DL ( 1983) A longitudinal study of life stress and social support among drug abusers. Int J Addict 18: 195–222. [DOI] [PubMed] [Google Scholar]
- Romieu P, Phan V-L, Martin-Fardon R, Maurice T ( 2002) Involvement of the sigma1 receptor in cocaine-conditioned place preference: possible dependence on dopamine uptake blockade. Neuropsychopharmacology 26: 444–455. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Mogil JS, Japon M, Chan EC, Allen RG, Low MJ ( 1996) Absence of opioid stress-induced analgesia in mice lacking beta-endorphin by site-directed mutagenesis. Proc Natl Acad Sci USA 93: 3995–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham Y, Stewart J ( 1994) Exposure to mild stress enhances the reinforcing efficacy of intravenous heroin self-administration in rats. Psychopharmacology (Berl) 114: 523–527. [DOI] [PubMed] [Google Scholar]
- Sharifi N, Diehl N, Yaswen L, Brennan MB, Hochgeschwender U ( 2001) Generation of dynorphin knockout mice. Mol Brain Res 86: 70–75. [DOI] [PubMed] [Google Scholar]
- Schenk S, Partridge B, Shippenberg TS ( 1999) U69,593, a kappa-opioid agonist, decreases cocaine self-administration and decreases cocaine-produced drug-seeking. Psychopharmacology (Berl) 144: 339–346. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Herz A, Shippenberg TS ( 1992) Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci USA 89: 2046–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starec M, Rosina J, Malek J, Krsiak M ( 1996) Influence of dynorphin A (1–13) and dynorphin A (1–10) amide on stress-induced analgesia. Physiol Res 45: 433–438. [PubMed] [Google Scholar]
- Takahashi M, Senda T, Tokuyama S, Kaneto H ( 1990) Further evidence for the implication of a kappa-opioid receptor mechanism in the production of psychological stress-induced analgesia. Jpn J Pharmacol 53: 487–494. [DOI] [PubMed] [Google Scholar]
- Takemori AE, Ho BY, Naeseth JS, Portoghese PS ( 1988) Norbinaltorphimine, a highly selective kappa-opioid antagonist in analgesic and receptor binding assays. J Pharmacol Exp Ther 246: 255–258. [PubMed] [Google Scholar]
- Thomas JB, Atkinson RN, Namdev N, Rothman RB, Gigstad KM, Fix SE, Mascarella SW, Burgess JP, Vinson NA, Xu H, Dersch CM, Cantrell BE, Zimmerman DM, Carroll FI ( 2002) Discovery of an opioid kappa receptor selective pure antagonist from a library of N-substituted 4beta-methyl-5-(3-hydroxyphenyl)morphans. J Med Chem 45: 3524–3530. [DOI] [PubMed] [Google Scholar]
- Tronche F, Casanova E, Turiault M, Sahly I, Kellendonk C ( 2002) When reverse genetics meets physiology: the use of site-specific recombinases in mice. FEBS Lett 529: 116–121. [DOI] [PubMed] [Google Scholar]
- Vaught JL, Takemori AE ( 1979) Differential effects of leucine and methionine enkephalin on morphine-induced analgesia, acute tolerance and dependence. J Pharmacol Exp Ther 208: 86–90. [PubMed] [Google Scholar]
- Watanabe Y, Weiland NG, McEwen BS ( 1995) Effects of adrenal steroid manipulations and repeated restraint stress on dynorphin mRNA levels and excitatory amino acid receptor binding in hippocampus. Brain Res 680: 217–225. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Wiertelak EP, Maier SF ( 1992) Kappa opioid receptors mediate tail-shock induced antinociception at spinal levels. Brain Res 582: 1–9. [DOI] [PubMed] [Google Scholar]
- Will MJ, Watkins LR, Maier SF ( 1998) Uncontrollable stress potentiates morphine's rewarding properties. Pharmacol Biochem Behav 60: 655–664. [DOI] [PubMed] [Google Scholar]
- Zamir N, Quirion R ( 1985) Dynorphinergic pathways of Leu-enkephalin production in the rat brain. Neuropeptides 5: 441–444. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Mantsch JR, Schlussman SD, Ho A, Kreek MJ ( 2002) Conditioned place preference after single doses or “binge” cocaine in C57BL/6J and 129/J mice. Pharmacol Biochem Behav 73: 655–662. [DOI] [PubMed] [Google Scholar]