

Abstract
Partial sciatic nerve ligation (pSNL) markedly increased glial fibrillary acidic protein immunoreactivity (GFAP-IR) 1 week after lesion in the L4–L5 spinal dorsal horn of wild-type, but not in dynorphin knock-out, mice lacking κ opioid receptors (KOR−/−) or in wild-type mice pretreated with the KOR antagonist nor-binaltorphimine (norBNI). A direct effect of KOR on glial cell proliferation was suggested by the findings that primary cultures of type II GFAP-immunoreactive astrocytes isolated from mouse spinal cord express KOR. Sustained treatment with the κ agonist U50,488 (trans-3,4-dichloro-N-methyl-N-[2-(1-pyrolytinil)-cyclohexyl]-benzeneacetamide methane sulfonate) significantly increased the proliferation rate of GFAP-immunoreactive astrocytes isolated from wild-type mice, and this effect was blocked by norBNI pretreatment. Proliferation of cultured type II astrocytes may have been stimulated by mitogen-activated protein kinase (MAPK) activation by KOR because (1) U50,488 treatment increased phospho-p38 MAPK-immunoreactivity 247 ± 44% over untreated cells, (2) the increase in phospho-p38 induced by U50,488 was blocked by norBNI and not evident in KOR−/− cultures, and (3) GFAP-immunoreactive astrocyte proliferation induced by U50,488 was blocked by the p38 MAPK inhibitor SB 203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole]. Similar mechanisms of astrocyte activation may also be responsible in vivo because intrathecal injection of SB 203580 blocked the increased GFAP-IR in lumbar spinal cord induced by pSNL. Although the relationship between κ-stimulated astrocyte proliferation and neuropathic pain mechanisms was not directly established in these studies, the results support the hypothesis that KOR activation induces spinal astrocyte proliferation, which may contribute to cellular reorganization after sciatic nerve damage.
Keywords: KOR, dynorphin, neuropathic pain, phospho-p38, MAPK kinase, G-protein-coupled receptor kinase
Introduction
Induction of painful peripheral neuropathy results in anatomical changes within the dorsal horn, including an increase in expression of glial fibrillary acidic protein (GFAP), an indicator of reactive gliosis after CNS injury (Kajander et al., 1990; Ma and Quirion, 2002). Although studies suggest that reactive gliosis is a major impediment to reestablishing neuronal pathways, little is known about the cellular and molecular signals controlling this process (Murray et al., 1990; Ma and Quirion, 2002). We reported previously that chronic pain induced by partial sciatic nerve ligation (pSNL) caused sustained dynorphin release that resulted in prolonged κ opioid receptor (KOR) activation in astroglial cells in the mouse spinal cord (Xu et al., 2004). Wang et al. (2001) have shown that dynorphin released in the spinal cord contributes strongly to the neuropathic pain response; however, the role of astroglial KOR activation after nerve damage remains unclear.
The type of glial cell responding to dynorphin release and subsequent KOR activation after pSNL was not established. Astrocytes and oligodendrocytes are the major classes of neuroglial cells in spinal cord and can be distinguished morphologically and antigenically (Hirano and Goldman, 1988; Misson et al., 1991; McMahon and McDermott, 2001). KOR is a member of the heptahelical, G-protein-coupled receptor opioid receptor family, and previous studies have shown that KOR is expressed by astroglial cells and can induce glial cell proliferation in vitro (Stiene-Martin and Hauser, 1991; Barg et al., 1993a; Eriksson et al., 1993; Ruzicka et al., 1995; Stiene-Martin et al., 1998). Accumulating evidence further suggests that KOR-selective agonists stimulate extracellular signal-regulated kinase (ERK) and p38 phosphorylation, activate phospholipase C in C6 glioma cells (Bohn et al., 2000; Belcheva et al., 2005; Bruchas et al., 2006), and increase DNA synthesis in cultures of mixed glial cells derived from fetal rat brain or rat spinal–dorsal root ganglion cocultures (Barg et al., 1993b). Nerve injury is known to activate mitogen-activated protein kinases (MAPKs), and this mechanism may mediate the response to opioids within the spinal cord during neuropathic pain (Jin et al., 2003).
MAPKs are a family of evolutionarily conserved proteins that play a critical role in cell signaling by transducing extracellular stimuli into intracellular responses (Chen et al., 2001). They are involved in cell proliferation and differentiation during development, neuronal plasticity, and injury responses (Ji and Woolf, 2001). Peripheral nerve lesions result in activation of MAPKs in microglia and astrocytes in the spinal cord, leading to the production of inflammatory mediators that sensitize dorsal horn neurons (Ma and Quirion, 2002; Jin et al., 2003; Zhuang et al., 2005). The primary objective of our study was to determine the cellular consequences of endogenous κ opioid system activation in mouse spinal cord after partial sciatic nerve ligation. κ receptor activation after chronic nerve injury produces sustained antinociceptive effects (Xu et al., 2004), and understanding the underlying mechanisms may have therapeutic implications.
Materials and Methods
Animals and housing.
Male C57BL/6 mice (Charles River Laboratories, Wilmington, MA) weighing 22–32 g were used in these experiments. Homozygous KOR, dynorphin, and G-protein-coupled receptor kinase 3 (GRK3) knock-out (−/−) mice were prepared by homologous recombination as described previously (Peppel et al., 1997; Hough et al., 2000; Sharifi et al., 2001) and provided for this study. Animals were backcrossed for >10 generations with C57BL/6 mice, and heterozygote breeding pairs were used to generate homozygotic “knock-out” mice of each type and paired wild-type (WT) littermate controls for this study. Individual mice were genotyped using DNA extracted from tail samples as a PCR template as described previously (Xu et al., 2004). The dynorphin, KOR, and GRK3 gene-disrupted animals show no discernible differences from WT littermates in growth, lifespan, or overt behavior. All mice were housed in groups of two to four in plastic cages (28 × 16 × 13 cm, length × width × height) using Bed-A-Cob for home bedding within the Animal Core Facility at the University of Washington and were maintained in pathogen-free housing units. Mice were transferred 1 week before training into a colony room adjacent to the testing room to acclimatize to the testing environment. The housing rooms were illuminated on a 12 h light/dark cycle with artificial lights on at 7:00 A.M. Lab chow and water were available ad libitum. All protocols with mice were approved by the institutional Animal Care and Use Committee in accordance with the 1996 National Institutes of Health Guide for the Care and Use of Laboratory Animals and guidelines for the International Association for the Study of Pain (Zimmermann, 1983). Mice were inspected regularly by veterinary staff to ensure compliance.
Surgical procedures.
The pSNL model of neuropathic pain used in this study has been described previously (Seltzer et al., 1990). The animals were anesthetized with pentobarbital sodium (80 mg/kg, i.p.). The right hindlegs were shaved, and the skin was sterilized with iodine. All surgical instruments were sterilized before surgery and then washed and heat treated (glass beads at 250°C) between animals. The right sciatic nerve was exposed, and approximately one-third to one-half the diameter of the nerve was tightly ligated with 7-0 silk suture (Surgical Specialties, Reading, PA). After checking hemostasis, the muscle and the adjacent fascia were closed with sutures, and the skin was closed with clips. The mice were killed by CO2 asphyxiation when experiments were completed.
To minimize the inflammatory response triggered by intrathecal implantation of a polyethylene catheter (DeLeo et al., 1997), a spinal cord puncture was made with a 30 gauge needle between L5 and L6 level to deliver the drug and vehicle to the spinal space around lumbosacral spinal cord (Hylden and Wilcox, 1980). A brisk flick of the tail was considered to be an indicator of the accuracy of each injection. The injection volume was 5 μl for intrathecal injection. One or 3 μg SB 203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole] (specific inhibitor of p38 MAPK) or saline was given 30 min before pSNL surgery and then once per 12 h after surgery for 7 consecutive days. SB 203580 HCl is water soluble, and the doses were based on previous studies (Zhang et al., 2005; Cui et al., 2006).
Behavioral tests for allodynia and hyperalgesia.
The mice were habituated to handling and testing equipment at least 20–30 min before experiments. Threshold for tactile allodynia was measured with a series of von Frey filaments (Semmes-Weinstein monofilaments; Stoelting, Wood Dale, IL). The mice stood on a metal mesh covered with a plastic dome. The plantar surface of the hindpaws was touched with different von Frey filaments having bending force from 0.166 to 3.63 g until the threshold that induced paw withdrawal was found. According to the specifications of the manufacturer, a 1 g filament induces a force of ∼9.8 mN. Unresponsive mice received a maximal score of 3.63 g. To assess thermal sensitivity (hyperalgesia), paw-withdrawal latencies to a radiant heat stimulus were measured using the paw-flick test apparatus (IITC Life Science, Woodland Hills, CA). Paw-withdrawal latency was determined as the average of three measurements per paw. The stimulus intensity was adjusted to give 8–10 s withdrawal latency in the normal mouse (baseline). The cutoff time in the absence of response was 15 s to prevent tissue damage.
Spinal astrocyte cultures.
Using sterile instruments and conditions, postnatal day 1–3 mice were decapitated, and spinal cords were rapidly dissected and placed in 2 ml culture medium (Neurobasal A supplemented with GlutaMAX, 2% B27, 50 U of penicillin, and 0.05 μg/ml streptomycin) (Invitrogen, Carlsbad, CA), at 4°C in a 35-mm-diameter dish. The spinal cord tissue was coarsely cut and transferred to sterile culture medium containing 40 U/ml active papain (Sigma, St. Louis, MO) at a volume of 1.5 ml/animal. The solution was then incubated at 30°C for 30 min on a platform rotating 120 rpm, keeping the segments suspended. The papain solution was then replaced with 2 ml of 30° culture medium and then triturated 15–20 times. After settling for 2 min, the supernatant was passed through a 70 μm nylon cell strainer (BD Biosciences, Bedford, MA). Twice more, the remaining sediment was suspended in 2 ml culture medium, triturated, and settled with the supernatant strained.
Cells were plated in 0.5 ml culture medium at a density of 2.5 × 105 cells/ml on 12 mm glass coverslips (PGC Scientifics, Gaithersburg, MD) placed in 24 well plates (BD Biosciences). Coverslips were acid washed, soaked in ethanol, and rinsed in sterile distilled water before a 24 h treatment with 50 μg/ml poly-d-lysine (135 kDa; Sigma). One hour after plating and incubation (5% CO2/95% air, 37°C; Forma Scientific, Marietta, OH), unattached cells and debris were aspirated and new culture medium was added. Cultures were initially shaken at 200 rpm overnight (VWR-OS-500 orbital shaker) to enrich for adherent cell types (e.g., astrocytes) and against neurons and microglia. Three days after plating and every 3 days thereafter, medium was replaced with complete medium containing 10% fetal bovine serum (Gemini Bio-Products, Woodland, CA). Coverslips were used for immunohistochemistry or immunoblotting 1–2 weeks after plating.
Immunohistochemistry in spinal sections.
Mice were injected intraperitoneally with bromodeoxyuridine (BrdU) (Sigma) solution in saline (100 mg of BrdU per kilogram of body weight) once a day for 7 d starting from the day of sciatic nerve ligation. BrdU is a thymidine analog that is specifically incorporated into DNA during DNA synthesis that can be used as a marker for proliferation. Mice were then anesthetized with isoflurane (Sigma) and intracardially perfused with 4% paraformaldehyde in phosphate buffer (PB) (0.1 m sodium phosphate, pH 7.4). The lumbar spinal cords were dissected, cryoprotected with solution of 30% (w/v) sucrose in PB at 4°C overnight, cut into 40 μm sections with a microtome, and processed for BrdU and GFAP immunostaining. Briefly, sections were washed three times in PBS, treated with 2N HCl for 60 min at 37°C, followed by two 10 min washes in 0.1 m borate buffer, pH 8.5, washed again three times in PBS, and blocked in PBS containing 0.1% Triton X-100 and 4% normal goat serum. Sections were then incubated overnight with primary BrdU monoclonal antibody (6 μg/ml; Millipore, Bedford, MA) and each of the following primary antibodies for double labeling: astroglia marker guinea pig GFAP (1:1000; Millipore), neuronal-specific nuclear protein NeuN (1:1000; Millipore), stem cell marker nestin (1:500; Abcam, Cambridge, MA), oligodendrocytes precursor marker NG2 (1:500; Millipore), and microglial marker Iba1 (ionized calcium-binding adaptor molecule 1) (1:300; Wako Pure Chemicals, Osaka, Japan). For double labeling of phospho-p38 and different cell type markers, the primary antibodies were as follows: phospho-p38 (1:200; Cell Signaling Technology, Beverly, MA), astroglial marker GFAP (1:1000; Millipore), microglial marker CD11b (1:500; Serotec Oxford, UK), and neuronal marker NeuN (1:1000; Millipore). Sections were then washed with PBS and then incubated (1 h, 37°C) with the fluorescent secondary antibodies FITC and rhodamine conjugate (1:250; Jackson ImmunoResearch, West Grove, PA) diluted in PBS containing 0.1% Triton X-100 and 4% normal goat serum. The sections were rinsed in PBS for 30 min and then mounted on gelatin-coated slides with Vectashield mounting medium (Vector Laboratories, Burlingame, CA) and sealed with nail polish for microscopy. The sections were viewed with a Nikon (Tokyo, Japan) Eclipse E600 fluorescence microscope or a Leica (Nussloch, Germany) confocal microscope located in the W. M. Keck Imaging Facility at the University of Washington. The BrdU-labeled astrocytes were counted in at least three cross sections. Data were presented as the average numbers of BrdU-labeled astrocytes per section.
Immunocytochemistry in spinal astrocyte cultures.
Spinal astrocyte proliferation was assessed using 10 μm BrdU incorporation into cells continuously exposed to the κ agonist U50,488 (trans-3,4-dichloro-N-methyl-N-[2-(1-pyrolytinil)-cyclohexyl]-benzeneacetamide methane sulfonate) (1 μm), U50,488 with the κ antagonist nor-binaltorphimine (norBNI), or U50,488 with the p38 inhibitor SB 203580 (100 or 500 nm) for 3 or 6 d. Freshly prepared drug solutions (dissolved in water) were added when the medium was changed every 3 d, and norBNI was added 30 min before U50,488. Cells were fixed with 4% paraformaldehyde in PB for 20 min, washed three times in PBS, treated with 2N HCl for 30 min at 37°C, followed by three 3 min washes in 0.1 m borate buffer, pH 8.5, washed again three times in PBS, and blocked in PBS containing 0.25% Triton X-100 and 0.3% gelatin. Cells were incubated with a mouse anti-BrdU monoclonal antibody (6 μg/ml; Millipore) and astroglial marker guinea pig anti-GFAP antiserum (diluted 1:1000; Millipore) overnight at 4°C. Sections were then washed with PBS, and detection was performed using the fluorescent secondary antibody FITC anti-mouse IgG (1:250; Jackson ImmunoResearch) and donkey anti-guinea pig IgG rhodamine conjugate (1:250; Jackson ImmunoResearch). Four to six coverslips were chosen in each group. The BrdU-labeled GFAP-positive cells were counted in at least four randomly chosen fields on each coverslip. The mean value of these fields was taken as the number of BrdU-labeled GFAP-positive cells on each coverslip.
Astrocytes express GFAP and can be divided into type I astrocytes, which are large, epithelioid cells, and type II, which have stellate processes and express a surface antigen (A2B5). Type II astrocytes and oligodendrocytes develop from O-2A progenitor cells (Raff et al., 1983); however, oligodendrocytes are not GFAP positive and can be identified by monoclonal antibody O1 (Sommer and Schachner, 1981). To identify type II astrocytes, we used primary antibodies directed against the cell surface marker A2B5 (5 μg/ml; Millipore) and the astrocyte marker GFAP. FITC anti-mouse IgG and donkey anti-guinea pig IgG rhodamine conjugate were used as secondary antibodies. To assess KOR activation in astrocytes, we used phosphoselective KOR antibody KOR-P (rabbit) (McLaughlin et al., 2003) and GFAP antibody (guinea pig) for double labeling, and FITC anti-rabbit IgG and donkey anti-guinea pig IgG rhodamine conjugate were used as secondary antibodies.
Phospho-p38 labeling of astrocytes was detected using a 1:200 dilution of mouse phospho-p38 monoclonal antibody (Cell Signaling Technology) and a 1:1000 dilution of guinea pig anti-GFAP antibody (Millipore). The secondary antibodies were FITC anti-mouse IgG (1:250; Jackson ImmunoResearch) and donkey anti-guinea pig IgG rhodamine conjugate (1:250; Jackson ImmunoResearch). The staining intensity was measured with NIH ImageJ on four to six coverslips in each group.
Western blotting.
Three days after culture in serum-free neurobasal media supplemented with B27 (Invitrogen), primary cultures of postnatal day 1–3 mouse astrocytes were plated on 35 mm Falcon dishes (Becton Dickinson, Franklin, NJ) in 10% fetal bovine serum (Sigma) for an additional 7–10 d until plates reached near-confluence. Astrocytes were treated for at least 24 h hour in serum-free DMEM/F-12 media and then treated for 10 min in vehicle (DMEM/F-12 media) or U50,488 (1 μm) in DMEM/F-12. Lysis and immunoblots were performed as described previously (Belcheva et al., 2001). Cells were immediately placed on ice, and media was aspirated and lysed in 650 μl of ice-cold lysis buffer (20 mm HEPES, 10 mm EGTA, 40 mm β-glycerophosphate, 2.5 mm MgCl2, and 1% Nonidet P-40) with protease inhibitors [50 μg/ml PMSF, 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 μg/ml pepstatin (Calbiochem, San Diego, CA)], a protein tyrosine phosphatase inhibitor (0.1 mg/ml, α-bromo-4-hydroxyacetophenone; Calbiochem), and a protein serine/threonine phosphatase inhibitor cocktail (Calbiochem) containing 2.5 mm (−)-p-bromotetramisole oxalate, 500 μm cantharidin, and 500 nm microcystin-LR. After 5 min on ice, lysates were scraped from each well by rubber policeman and then spun at 4°C for 20 min at 10,000 × g. Protein concentration was estimated using a bicinchoninic colorimetric assay kit (Pierce, Rockford, IL). Lysate concentration was estimated from a bovine serum albumin (BSA) standard curve generated by spectrophotometer at a visible absorbance of 562 nm. Lysates were diluted in lysis buffer to a final concentration of 15–20 μg/lane. Samples were boiled for 2 min in 20 μl of Laemmli sample buffer (100 mm Tris, pH 6.8, 2% SDS, 20% glycerol, 10% β-mercaptoethanol, and 0.1% bromophenol blue), resolved by SDS-PAGE using 10% bis-acrylamide nonreducing gels (Invitrogen) and transferred to nitrocellulose membranes. Blots were soaked in blocking buffer (5% BSA, 0.1% Tween 20, and TBS, pH 7.4) for 2 h at room temperature and incubated overnight at 4°C with a monoclonal primary antibody raised in rabbit recognizing phosphorylated p38 MAPK (Cell Signaling Technology) at a dilution of 1:500 in blocking buffer. Blots were washed, incubated for 1 h at room temperature in HRP-conjugated goat anti-rabbit secondary antibody (Bio-Rad, Hercules, CA) diluted 1:1000 in blocking buffer. Blots were washed, and then antibody labeling of proteins was visualized with an enhanced chemiluminescence detection kit (ECL; PerkinElmer, Boston, MA) on Eastman Kodak (Rochester, NY) BioMax XAR film. Blots were stripped by incubation for 60–90 min in erasure buffer (2% SDS, 62.5 mm Tris-HCl, pH 6.8 at 20°C, 100 mm β-mercaptoethanol) at 70°C, washed two times (10 min each) in TBS/0.1% Tween 20, blocked as described, and incubated overnight in total p38 MAPK (Cell Signaling Technology) diluted 1:1000 in dilution buffer. The secondary antibody used was HRP-conjugated goat anti-rabbit (Promega, Madison, WI) diluted 1:1000 in dilution buffer. Both primary p38 MAPK antibodies labeled a band of ∼50 kDa, consistent with a control cell lysate for phospho-p38 MAPK provided by Cell Signaling Technology.
Microglial staining in rat spinal cord sections.
As a positive control for p38 activation in microglia, peripheral nerve ligation was performed on two adult male rats (Sprague Dawley; Harlan, Livermore, CA), and spinal sections were double labeled with phospho-p38 and OX42 (microglia marker, purified mouse anti-rat antibody, 1:50 dilution; BD PharMingen, San Diego, CA).
Chemicals.
norBNI was obtained from the National Institute on Drug Abuse drug supply program (National Institutes of Health, Bethesda, MD). U50,488 and BrdU were from Sigma. SB 203580 HCl was from Calbiochem.
Statistical analysis.
In the immunohistochemical studies, cell numbers were counted and presented as the percentage of the control (e.g., contralateral side, sham, or vehicle treatment) for between-group differences. Similarly, the intensity of immunolabeling was compared for sections with the same optical parameters for the NIH Image or MetaMorph quantitation software (Molecular Devices, Palo Alto, CA). Significant results were demonstrated either by one-way ANOVA followed by Dunnett's test or Student's t test for significant pairwise comparisons. Response data are presented as means ± SEM of all groups, with significance set at p < 0.05.
Results
Spinal astrocytes were activated after pSNL in lumbar spinal sections
Consistent with previous results, GFAP immunoreactivity (IR) in lumbar spinal cord (L4–L5) was markedly increased in the lumbar spinal cord 7 d after pSNL (Fig. 1A,D) compared with sham-ligated mice (p < 0.05) (Fig. 1B,E) or untreated mice (data not shown). The GFAP-positive cells were located primarily in the lamina I–IV of the dorsal horn. Although an increase in GFAP-IR occurred bilaterally, staining appeared greater on the side of the spinal cord ipsilateral to the pSNL (right side). The GFAP-IR appeared to more fully fill the cells in the pSNL sections, suggesting that GFAP expression increased per cell (Fig. 1D). The increase in GFAP-IR in WT mice was blocked by pretreatment with the κ antagonist norBNI (10 mg/kg, i.p.) (Fig. 1C). In addition, mice lacking the dynorphin gene (DYN−/−) did not show increased GFAP-IR after pSNL (Fig. 1F) compared with sham-operated mice (Fig. 1B). Mice lacking κ opioid receptors (KOR−/−) also did not show increased GFAP-IR after pSNL (Fig. 1G). The consistent results from norBNI pretreatment and gene deletions of dynorphin or KOR suggest that the effect of pSNL on GFAP-IR induction was mediated by endogenous dynorphin peptide activation of the κ opioid receptor.
Figure 1.

GFAP-immunoreactive staining is increased in WT lumbar spinal cord 7 d after pSNL but not after sham ligation. The increase is blocked by norBNI and does not occur in Dyn−/− or KOR−/− mice. Transverse sections were taken of the lumbar spinal cord 7 d after pSNL. A–C,GFAP staining was significantly increased in WT pSNL mice compared with WT sham-operated mice (A, B), and this increase in GFAP-IR was blocked by the κ antagonist norBNI (10 mg/kg) (C). Mice lacking the dynorphin gene (Dyn−/−) did not show upregulated GFAP staining after pSNL (F). D and E are higher-magnification images of A and B, respectively. G, Graph showing mean ± SEM pixel intensity of GFAP staining in lamina I–IV of lumbar spinal sections. Image analysis of astrocytes expressing GFAP after pSNL, demonstrating approximately twofold higher signal intensity [114 ± 8.71 arbitrary units (AU); n = 4] than that of sham-ligated mice (57.9 ± 6.26 AU; n = 4) (*p < 0.001). In contrast, image analysis of sections from pSNL mice pretreated with norBNI (NBNI) did not show a significant increase over sham and were significantly lower than WT pSNL signal intensity (norBNI, 46.5 ± 0.85 AU; n = 4). Similarly, pSNL of Dyn−/− or KOR−/− mice did not result in significant increases in GFAP-immunoreactive pixel intensities over sham-ligated mice (Dyn−/−, 54.3 ± 0.84 AU, n = 4; KOR−/−, 48.0 ± 0.62 AU, n = 4). Scale bars: A–C, F, 400 μm; D, E, 100 μm.
To determine whether the apparent increase in the number of GFAP-positive cells was caused solely by increased GFAP expression in existing cells or by astrocyte proliferation, mice were injected with BrdU (100 mg/kg, i.p.) once daily for 7 d after pSNL. Seven days after pSNL, the number of BrdU-positive cells was increased 4.4-fold in the ipsilateral side of the spinal cord compared with contralateral side (p < 0.05) (Fig. 2A,D). The results suggest that pSNL induced both astrocyte proliferation and increased GFAP expression. Approximately 15–20% of BrdU-positive cells were GFAP positive (Fig. 2C,F), suggesting that only a portion of the proliferative response after pSNL was attributable to GFAP-immunoreactive astrocyte proliferation. Although κ antagonism completely blocked the increase in GFAP-IR (Fig. 1G), the pSNL-induced increase in BrdU staining was not fully blocked by norBNI or KOR−/− (data not shown). The results suggest that KOR-independent mechanisms were simultaneously occurring in other cell types.
Figure 2.
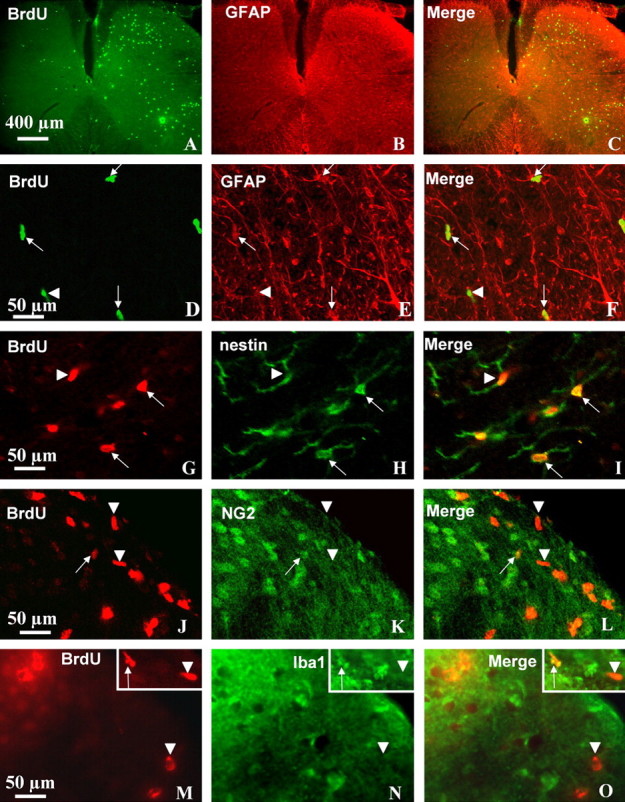
The increase in GFAP-IR after pSNL is caused by astrocyte proliferation and hypertrophy. Transverse sections through the lumbar spinal cord segment were taken 7 d after pSNL. BrdU was administered 100 mg/kg intraperitoneally once daily for 7 d. Double-immunohistochemical labeling was viewed by laser-scanning confocal microscopy. A, BrdU-positive cells (green) were markedly increased on the ipsilateral side (right) of the spinal cord compared with the contralateral side (left). The immunoreactive cells were preferentially located in the lamina II–IV and VII. B, GFAP staining occurred on both sides, with a significant increase on the ipsilateral side. F, GFAP staining was more pronounced in astrocyte processes, whereas staining for BrdU was more pronounced in cell bodies (nucleus). D–F, Some of the dividing BrdU-labeled cells were GFAP-positive astrocytes (arrows), and others were not GFAP positive (arrowheads). The increased GFAP staining in astrocytes that were not BrdU positive suggested that these cells only increased their sizes (hypertrophy). G–I, Many BrdU-positive cells were obviously double labeled with nestin, a stem cell marker (arrows), but a small fraction of BrdU-positive cells were not nestin-positive stem cells (arrowheads). J–O, Similarly, a small fraction of BrdU-labeled cells were colabeled by antibodies against NG2, an oligodendrocyte precursor marker (J–L, arrows) or colabeled by antibodies against Iba 1, a microglial marker (M–O, arrows). Scale bars: A–C, 400 μm; D–O, 50 μm.
To further characterize the BrdU-positive cells that did not express GFAP-IR, we performed dual-labeling of BrdU and nestin (stem cell marker), BrdU and NG2 (oligodendrocytes precur-sor marker), BrdU and Iba1 (microglia marker), or BrdU and NeuN (neuronal marker). The majority (∼70%) of the BrdU-positive cells were nestin-positive stem cells (Fig. 2G–I). Only a very small portion of BrdU-positive cells (<10%) were double-labeled microglia (Fig. 2M–O) or oligodendrocytes (Fig. 2J–L). We did not detect BrdU-positive cells that double labeled with NeuN (data not shown), suggesting that proliferation of neurons was not evident 7 d after pSNL.
Primary astrocytes cultured from spinal cord were used to determine whether κ-opioid activation directly induced astrocyte proliferation. Previous studies showed that κ opioid receptors are expressed by brain and spinal cord astrocytes (Barg et al., 1993a; Ruzicka et al., 1995; Xu et al., 2004), and we found that primary astrocytes isolated from mouse spinal cord expressed functional κ opioid receptors. Treatment of primary astrocyte cultures using the κ agonist U50,488 (1 μm for 1 h at 37°C) significantly increased κ opioid receptor labeling detected by a phosphoselective antibody KOR-P compared with vehicle control (p < 0.05) (Fig. 3A,B). KOR-P is an affinity-purified antibody shown previously to detect KOR phosphorylated on serine 369 by GRK3 after agonist activation (McLaughlin et al., 2003; Xu et al., 2004). Coincubation with norBNI (10 μm) blocked the U50,488-induced increase in KOR-P labeling of astrocytes (Fig. 3C). Preabsorption of the KOR-P antibody with the corresponding cognate phospho-peptide prevented antibody labeling after 1 μm U50,488 treatment (data not shown). Treatment with norBNI alone had no effect on KOR-P labeling (data not shown). The results suggested that cultured spinal cord astrocytes express functional κ opioid receptors.
Figure 3.
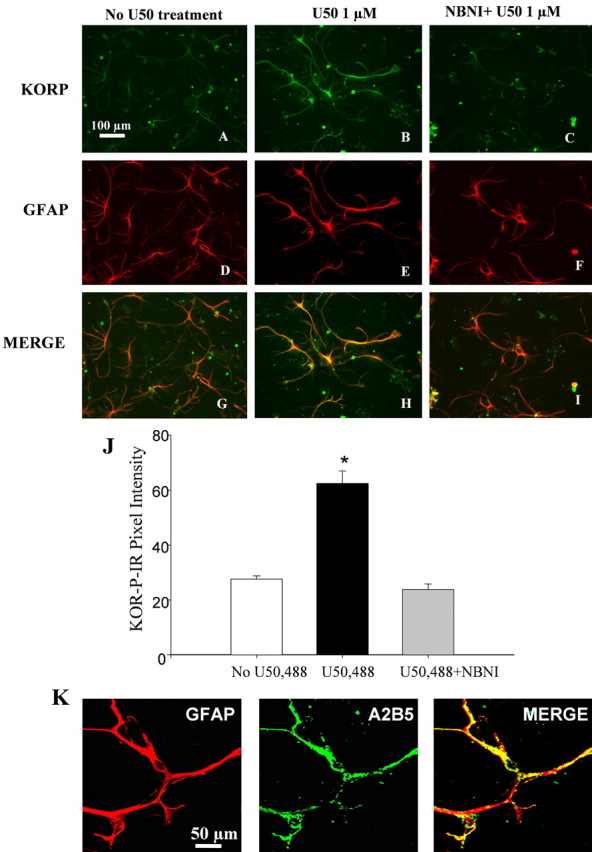
KOR activation increases KOR-P staining in astrocyte culture. A–I, Astrocyte cultures were pretreated for 1 h at 37°C with vehicle (A, D, G), 1 μm U50,488 (B, E, H), or 10 μm norBNI with 1 μm U50,488 (C, F, I). Cells were fixed, double labeled with affinity-purified KOR-P antibody (10 μg/ml) and anti-GFAP antibody (1:1000), and then visualized with a confocal microscope. The KOR-P antibody did not label untreated cells (A), whereas incubation with U50,488 (1 μm) for 1 h produced an increase in KOR-P antibody labeling that colocalized with the GFAP labeling (B, H). Coincubation with the KOR-selective antagonist norBNI (10 μm) blocked the U50,488-induced increase of KOR-P labeling (C, I). GFAP staining did not change significantly after 1 h U50,588 treatment (D–F). J, Mean ± SEM pixel intensity of KOR-P staining in astrocyte cultures. Image analysis of the astrocytes expressing KOR-P treated with U50,488 demonstrated a 2.3-fold higher signal intensity (62.48 ± 4.52 AU; n = 4) than that of untreated cells (27.56 ± 1.2 AU; n = 4) (*p < 0.001). Image analysis of the astrocytes expressing KOR-P coincubated with both U50,488 and norBNI showed no significant difference in signal intensity (23.8 ± 2.04; n = 4) compared with untreated cells. K, Dual labeling of anti-A2B5 (green) and anti-GFAP (red) showed that these astrocytes were type II astrocytes. Scale bars: A–I, 100 μm; K, 50 μm.
KOR activation increases the rate of type II astrocyte proliferation in culture
The cultured cells expressing κ opioid receptors were also immunolabeled by an antibody (anti-A2B5) specific for type II astrocytes. These type II astrocytes showed a stellate process-bearing morphology in culture (Fig. 3K) and closely resembled radial glia in the spinal cord (McMahon and McDermott, 2001). Incubation with 1 μm U50,488 for 3 or 6 d caused a robust increase in BrdU labeling of type II astrocytes compared with untreated cultures (p < 0.01) (Fig. 4). This increase was blocked by the inclusion of norBNI (10 μm) in the culture medium (Fig. 4C,F), suggesting that exogenous κ agonists can activate KOR on type II astrocytes and induce proliferation. Treatment with norBNI alone had no effect on BrdU labeling (data not shown).
Figure 4.
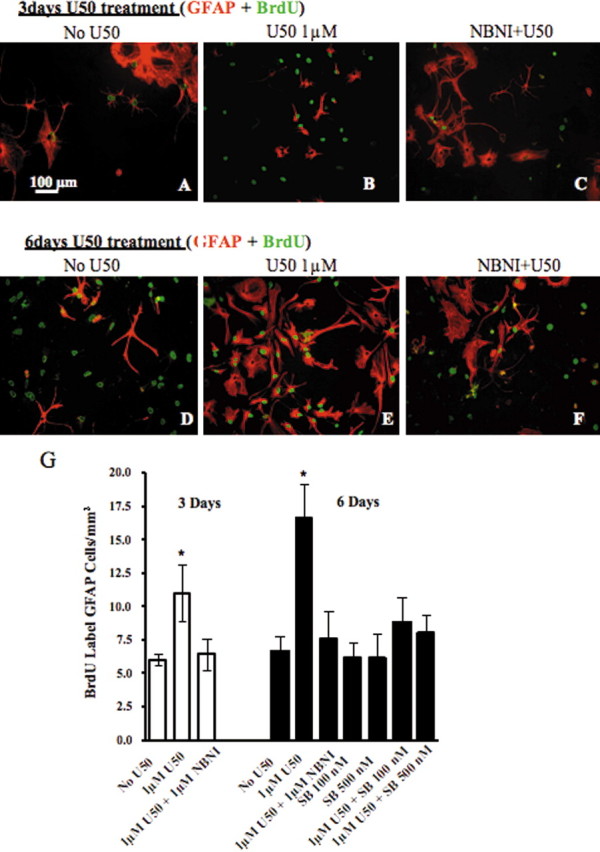
KOR activation increases the rate of astrocyte proliferation in culture. Cells were pretreated with the drugs listed above each image. A, D, In cell culture with vehicle treatment, there was a greater increase in the number of BrdU-positive cells after 6 d culture than after 3 d culture (A, D), and some BrdU-positive cells were not GFAP-positive (D). B, E, When the cultures were incubated with U50,488 (U50) (1 μm) in media for 3 or 6 d, there was a significant increase in BrdU labeling in type II astrocytes at 6 d compared with that of 3 d. C, F, This increase was blocked by the addition of 10 μm norBNI (NBNI). G, Graph showing mean ± SEM BrdU labeling of type II astrocytes per square millimeter in the astrocyte culture. BrdU labeling of type II astrocytes after 6 d of U50,488 treatment (16.64 ± 2.6; n = 4) was 1.6-fold at >3 d of treatment (11 ± 2.45; n = 4) (*p < 0.05). The U50,488-induced type II astrocyte proliferation was also blocked with cotreatment of the p38 inhibitor SB 203580. Scale bars: A–F, 100 μm; K, 50 μm.
KOR activation stimulated p38 MAPK in cultured astrocytes
The U50,488-induced type II astrocyte proliferation was blocked by cotreatment with the p38 inhibitor SB 203580 (100 or 500 nm) (Fig. 4G), suggesting that KOR activation induced astrocyte proliferation through the p38 MAPK pathway. Consistent with previous reports, Western blotting experiments demonstrated that both 100 and 500 nm of the selective p38 inhibitor SB 203580 (Gallagher et al., 1997; Young et al., 1997; Bhat et al., 1998) were sufficient to block anisomycin (50 μm, 15 min) induced p38 activation in primary spinal cord astrocytes (data not shown). To test whether KOR mediates p38 activation in spinal cord astrocytes, we assessed the effects of U50,488 treatment on phospho-p38 MAPK levels in primary astrocyte cultures. A low basal level of phospho-p38 MAPK-IR was evident in the absence of agonist (Fig. 5A), and phospho-p38-IR was markedly increased after 1 μm U50,488 for 10 min (Fig. 5B,D). The U50,488-induced increase in phospho-p38 staining was blocked by 10 μm norBNI (Fig. 5C). Treatment with norBNI alone had no effect on phospho-p38-immunoreactive labeling (data not shown). The cultured cells responding to U50,488 with an increase in phospho-p38-IR were also GFAP-IR as shown by the double labeling (Fig. 5D–F). Quantitation of phospho-p38 staining intensities showed that the significant increase in intensity caused by U50,488 was blocked by norBNI (Fig. 5G). The in vitro results suggest that the increase in GFAP-IR caused by pSNL may have also been mediated by a p38 MAPK mechanism.
Figure 5.
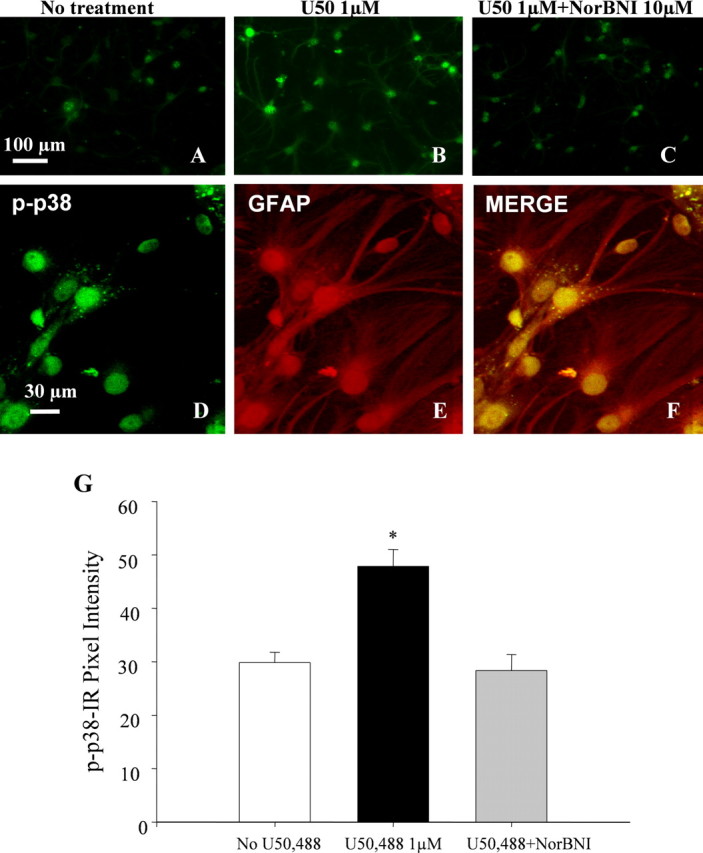
U50,488 activates phospho-p38 MAPK pathway in spinal astrocyte cultures. Cells were pretreated for 10 min at 37°C with the drugs listed above each image. A, B, In vehicle-treated cultures, some basal phospho-p38 staining was evident (A), and cells treated with 1 μm U50,488 (U50) showed a marked increased in the level of phospho-p38-IR (B). C, The increase in phospho-p38 staining was not evident in cultures incubated with both norBNI and U50,488. D–F, Double labeling with phospho-p38 and GFAP using laser-scanning confocal microscopy revealed that phospho-p38 MAPK staining was localized to the cytoplasm and nucleus of GFAP-immunoreactive astrocytes. F, Mean ± SEM pixel intensity of phospho-p38 staining in astrocyte cultures. Image analysis of the astrocytes treated with U50,488 demonstrated a 1.6-fold higher signal intensity of phospho-p38 (47.85 ± 3.16 AU; n = 4) than in untreated cell cultures (29.85 ± 1.94 AU; n = 4) (*p < 0.01). Image analysis of the astrocytes coincubated with U50,488 and norBNI showed no significant increase in phospho-p38 signal intensity (28.38 ± 2.98; n = 4). Scale bars: A–C, 100 μm; D–F, 30 μm.
p38 MAPK was activated in spinal astrocytes after pSNL in lumbar spinal sections
Although several studies have shown that p38 was activated in different pain models in rats and p38 is activated exclusively in microglia (Jin et al., 2003; Svensson et al., 2003), p38 MAPK activation in mice during neuropathic pain has not been demonstrated previously. In this study, we used phospho-p38 antibody staining in mice lumbar spinal sections 7 d after pSNL. Phospho-p38 antibody staining was increased in the ipsilateral dorsal horn compared with the contralateral dorsal horn (Fig. 6A, inset). Most of the p38 MAPK staining was in the nucleus of astrocytes (Fig. 6A–C) or neurons (Fig. 6K–L). There was no evidence of phospho-p38 staining in CD11b-positive microglia (Figs. 6D–F); a series of 80 sections obtained from five mice at 7 d after pSNL were scanned. As a positive control for microglial double labeling, we showed the phospho-p38 staining in microglia in rat spinal cord tissue after chronic nerve injury (Figs. 6G–I), suggesting that there may be important differences in the cellular responses of rats and mice. Nevertheless, the present study was focused on the role of κ receptors in GFAP-immunoreactive type II astrocyte activation and did not attempt to further resolve this apparent microglial discrepancy.
Figure 6.
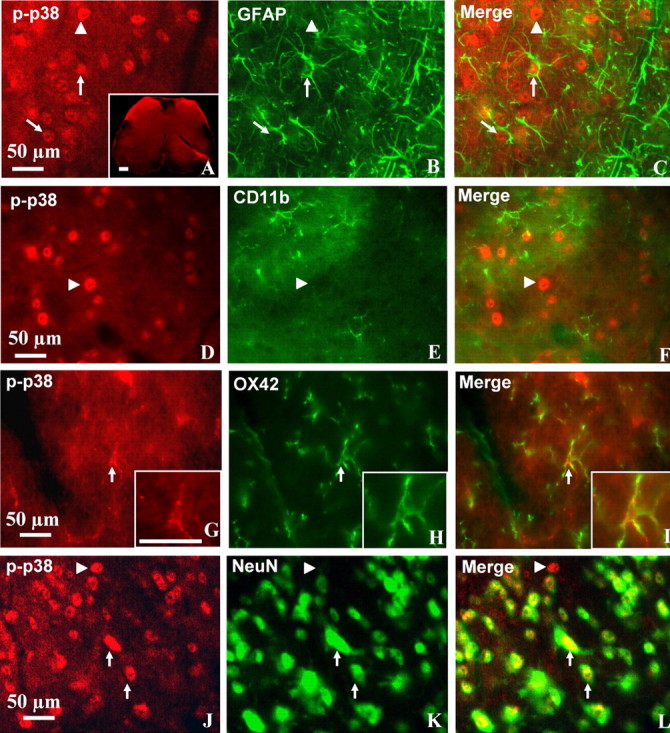
p38 MAPK was activated in astrocytes after pSNL in lumbar spinal sections. Transverse sections through the lumbar spinal cord segment 7 d after pSNL were labeled with antibody against phospho-p38 (p-p38, red) and double labeled with GFAP, CD11b, OX42, or NeuN antibodies (green). Double-immunohistochemical labeling was viewed by laser-scanning confocal microscopy (Leica SL). A, Phospho-p38 staining was increased on the ipsilateral side dorsal horn (left) of the spinal cord compared with the contralateral side (right) (inset). A–L, The phospho-p38-immunoreactive staining was preferentially located in the nucleus of GFAP-positive (A–C) and NeuN-positive (J–L) cells. A–C, Some of the phospho-p38-labeled cells were GFAP-positive astrocytes (arrows), and others were not GFAP positive (arrowheads). J–L, Many phospho-p38-labeled cells were clearly double labeled with NeuN antisera, a neuronal marker (arrows), although some phospho-p38-labeled cells were not NeuN-positive neurons (arrowheads). D–I, No colocalization was detected for phospho-p38 and CD11b, a microglia marker (D–F, arrowheads) in mouse tissue, but, as a positive control, colocalization was detected for phospho-p38 and microglia marker OX42 in rat spinal cord sections (G–I, arrows). The insets for G–I show a two times enlargement of the double-labeled microglial cell indicated by the arrow. Scale bars: A–L, 50 μm.
Multiple intrathecal injections of SB 203580 reduced the spinal astrocyte proliferation after pSNL as well as the hyperalgesia and allodynia induced by pSNL
We intrathecally injected either saline or SB 203580 (1 or 3 μg) in a volume of 5 μl once per 12 h for 7 d after pSNL. Paw-flick latency and von Frey hair force sensitivity was measured 30 min before the intrathecal injections on days 1, 3, 5, and 7. On day 7, the mice were fixative perfused, and GFAP-IR was assessed in the spinal cord sections. Astrocyte proliferation within the lumbar spinal cord caused by pSNL was inhibited by 1 μg (Fig. 7A–F) and 3 μg (data not shown) of SB 203580. As evident from A and B, SB 230580 treatment substantially reduced the GFAP-IR in the lamina I–VI. There was no effect of SB 230580 on the GFAP-IR in the ventral white matter. Higher-magnification images show that the SB 230580-sensitive phospho-p38 staining was predominantly in the deeper lamina of the spinal cord (Fig. 7C–F).
Figure 7.
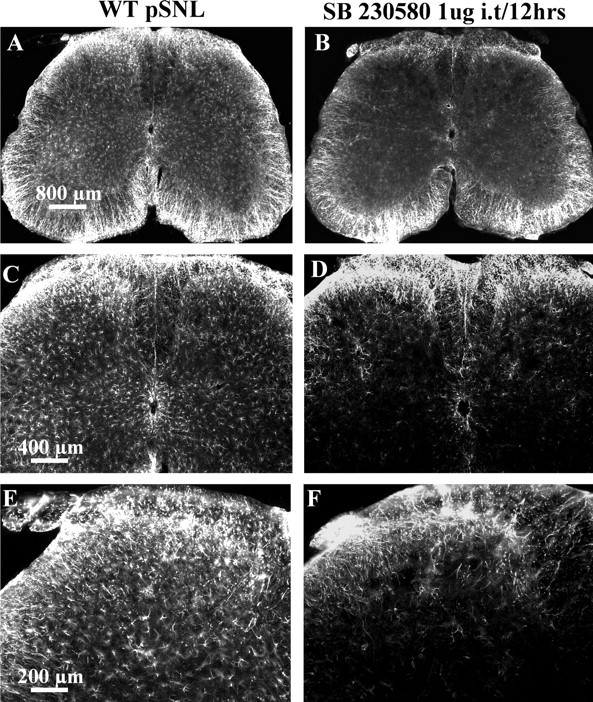
pSNL-induced GFAP proliferation was reduced by a specific p38 MAPK inhibitor, SB 203580. Mice were intrathecally injected with either saline or 1 μg of SB 203580 in 5 μl, 30 min before pSNL and twice per day after pSNL for 7 d. After perfusion with 4% paraformaldehyde, lumbar spinal sections were stained with GFAP antisera. A, B, The GFAP staining was significantly lower in the SB 203580-treated mice (B) compared with the saline-treated mice (A). A, C, There was a significant increase of GFAP staining in the deeper layers on the ipsilateral side (left) compared with contralateral side (right) in saline-injected mice. C, E and D, F are higher-magnification images of A and B, respectively. SB 203580 significantly reduced the increase in GFAP staining after pSNL in both ipsilateral and contralateral regions of the deeper layers of the spinal cord (B, D, F). Scale bars: A, B, 800 μm; C, D, 400 μm; E, F, 800 μm.
On day 7 after pSNL, the saline-injected mice showed significantly increased sensitivity to thermal and mechanical stimulation in the ipsilateral hindpaw compared with the baseline values before lesion (Fig. 8A,C) (p < 0.05). The intrathecal administration of SB 203580 at either 1 or 3 μg/dose modestly reduced the allodynic effects of pSNL in both the ipsilateral (Fig. 8C) and contralateral (Fig. 8D) paws. The significantly increased threshold response to mechanical stimulation suggested that there were anti-allodynic effects of this compound (p < 0.05). The higher dose of SB 203580 (3 μg) increased paw-withdrawal latency in the thermal analgesia assay (an anti-hyperalgesic effect), whereas the 1 μg dose of SB 203580 did not significantly affect paw-withdrawal latencies (Fig. 8A). Similar to our previous findings (Xu et al., 2004), pSNL did not induce hyperalgesia in the contralateral paw (Fig. 8B), but robust allodynia developed that was significantly reduced by 1 and 3 μg of SB 203580 on day 7 (Fig. 8D).
Figure 8.

Intrathecal injection of SB 203580 partially blocked pSNL-induced hyperalgesia and mechanical allodynia. Paw-flick latency or von Frey hair force mechanical threshold were measured on days 1, 3, 5, and 7 after pSNL for mice injected with saline or 1 or 3 μg of SB 203580. A, On day 7 after pSNL, the saline-injected mice showed hyperalgesia in the ipsilateral hindpaw as evident from the significantly decreased thermal latencies compared with before pSNL; this hyperalgesia was not evident in mice injected with 3 μg of SB 203580. B, The contralateral hindpaw did not develop hyperalgesia, and SB 203580 did not affect the thermal sensitivity of the contralateral hindpaw. C, D, However, both ipsilateral and contralateral hindpaws developed mechanical allodynia after saline intrathecal injection, and this allodynia was significantly reduced by both 1 and 3 μg of SB 203580 (*p < 0.05; one-way ANOVA, followed by Dunnett's multiple comparisons test). Unlike the hyperalgesic response to pSNL, the allodynic response was not completely blocked by SB 203580 treatment.
Western blotting experiments in primary mouse spinal cord astrocyte cultures were used to extend the immunohistochemistry analysis. Cell lysates prepared 10 min after 1 μm U50,488 addition in vitro showed increased phospho-p38 MAPK-IR (195 ± 32% of vehicle-treated cells). The increase in phospho-p38 induced by U50,488 was not evident in KOR−/− cultures (Fig. 9A,B). Because we showed previously that KOR activation induced phosphorylation of p38 MAPK in transfected cells and primary cultured mouse astrocytes by GRK3 and arrestin-dependent mechanism (Bruchas et al., 2006), we next asked whether pSNL of GRK3−/− mice would show increased GFAP-IR. Basal phospho-p38-IR in astrocytes from GRK3−/− mice was significantly lower than in astrocytes from wild-type mice (54 ± 20% of vehicle-treated controls) (Fig. 9B). Furthermore, U50,488 treatment of GRK3−/− astrocyte cultures did not increase phospho-p38-IR compared with vehicle-treated controls. Equal protein loading was confirmed by stripping each blot and reprobing with a monoclonal antibody raised in rabbit to total p38 MAPK protein (Fig. 9A). Consistent with the lack of U50,488-induced increase in phospho-p38 in cultured astrocytes from GRK3−/− mice, pSNL did not cause an increase in GFAP-IR in GRK3−/− mice (Fig. 9C).
Figure 9.

agonist activation enhances phospho-p38 MAPK-IR in primary astrocyte cultures by a mechanism requiring GRK3. A, Representative gels showing phospho-p38-IR after U50,488 (U50) treatment in cell lysates prepared from spinal cord astrocytes cultured from WT (+/+), KOR−/−, or GRK3−/− mice. Top, Phospho-p38-IR; bottom, loading control; Anis, anisomycin. B, U50,488 treatment of WT astrocytes (left bars) increased phospho-p38 MAPK-IR to 195 ± 32% of vehicle-treated controls. In contrast, phospho-p38-IR in KOR−/− astrocyte cultures did not change after 1 μm U50,488 treatment (middle bars). Basal phospho-p38-IR in GRK3−/− astrocyte cultures was significantly lower than in GRK3+/+ astrocytes (*p < 0.05), and phospho-p38-IR did not significantly increase in GRK3−/− cultures after U50,488 treatment [right bars, 54 ± 20% (black) and 43 ± 37% (gray) of WT (+/+) vehicle-treated controls, respectively] (*p < 0.05 compared with vehicle-treated WT (+/+) controls; n = 6–10 replicates). C, Consistent with the lack of U50,488-induced increase in phospho-p38 in astrocyte cultures, GRK3−/− mice did not increase GFAP-IR in lumbar spinal cord 7 d after pSNL.
Discussion
The principal finding of this study is that κ opioid receptor activation mediates a component of pSNL-induced reactive gliosis in mouse spinal cord. Proliferation of type II astrocytes after pSNL was blocked by pretreatment with the KOR antagonist norBNI and did not occur in mice genetically lacking either prodynorphin-derived peptides or κ opioid receptors. Numerous studies have established that the endogenous dynorphin opioid peptides are released by neuropathic pain and contribute to the pain response (Wang et al., 2001; Xu et al., 2004). In addition, considerable evidence supports the idea that enhanced expression of endogenous dynorphin within the spinal cord promotes morphine tolerance (Vanderah et al., 2000) and is pronociceptive (Wang et al., 2001). The dynorphins act as endogenous agonists at the opioid receptors (KORs) (Chavkin et al., 1982), and the increased dynorphin expression in neuropathic pain also leads to a sustained activation of KOR (Xu et al., 2004). Several studies have documented antinociceptive effects of κ agonists (Nakazawa et al., 1991). Thus, the endogenous dynorphin-derived opioids seem to have both antinociceptive and pronociceptive actions. In our previous report, we showed that neuropathic pain induced a sustained release of endogenous prodynorphin-derived opioid peptides and increased KOR phosphorylation in GABAergic neurons and astrocytes (Xu et al., 2004). These results suggested that pSNL activated the KOR system in mouse spinal cord and induced opioid receptor tolerance. Similar results in the μ opioid system have shown that μ opioid receptor (MOR) phosphorylation increased after induction of neuropathic pain (Narita et al., 2004b), and MOR tolerance caused an increase in GFAP-IR in the spinal cord of both rats and mice (Song and Zhao, 2001; Narita et al., 2004a). However, the consequences of κ receptor activation on astrocyte function and how astrocytes may mediate the effects of dynorphin in the neuropathic pain condition remained unclear.
Among the hallmarks of reactive gliosis is an increase in GFAP-IR, hypertrophy of astrocytic cell bodies, and proliferation of astrocytes (Faulkner et al., 2004). Reactive gliosis is likely to play an important role in neuronal survival and functional recovery after central or peripheral injury (Murray et al., 1990; Liu et al., 1998). pSNL induced a proliferation of GFAP-immunoreactive cells in the dorsal and ventral spinal cord ipsilateral to the lesion. The ligation-induced proliferation was not restricted to type II astrocytes, and ∼80% of the BrdU-positive cells were not GFAP positive; these other dividing cells might be progenitors or microglia (Ishii et al., 2001; Tanga et al., 2004). In this study, we found that ∼70% of the BrdU-positive cells were stem cells (nestin staining positive), suggesting that these dividing cells were still in their early development stage. The ultimate differentiation states of these progenitors were not established in this study. The astrocyte activation observed is consistent with previous findings showing that astrocytes in the spinal cord become activated after spinal nerve injury. The novel observation was that endogenous κ opioid receptor activation played a key role. Other mediators have also been shown to be important, and previous reports demonstrated that interleukin release from both astrocytes and microglial cells results in reactive astrogliosis after spinal cord injury that can be blocked by interleukin-6 receptor antagonism (Okada et al., 2004). Although studies have suggested that the reactive gliosis in response to injury is important in reestablishing neuronal pathways, little is understood about the cellular and molecular signals that regulate this process.
Primary spinal astrocyte cultures are potentially less complex, and in vitro studies allow clearer resolution of the underlying mechanisms; however, these cultures are still heterogeneous, and cells in culture derived from neonates may represent a less differentiated state. Nevertheless, in our in vitro preparation, we demonstrated that κ opioid receptor activation could directly stimulate proliferation of type II astrocytes expressing both KOR-IR and GFAP-IR. These results are consistent with previous studies showing κ receptors are expressed by astrocytes (Barg et al., 1993a; Eriksson et al., 1993) and are consistent with a previous study demonstrating that κ agonists increased thymidine incorporation in spinal cord–dorsal root ganglion cocultures (Barg et al., 1993b). κ opioid effects differ from μ opioid effects, which have been shown to inhibit type I astrocyte proliferation in cultures derived from rat brain (Hauser et al., 1996).
The astrocyte proliferation caused by U50,488 treatment was blocked by cotreatment with SB 203580, suggesting that p38 MAPK activation was required. The three MAPK pathways identified to date include ERK (1 and 2), p38 (α, β, γ, and δ), and c-Jun N-terminal protein kinase (JNK) (encoded by three genes, JNK1, JNK2, and JNK3) (McDonald et al., 2000). All three pathways have been implicated in pain transmission and mechanisms of opioid analgesic tolerance in the spinal cord (Ji, 2004; Narita et al., 2004a, b; Obata and Noguchi, 2004). ERK is involved in both cell proliferation and differentiation during development and in neuronal plasticity. p38 and JNK are stress-activated protein kinases and participate in the injury responses (Ji and Woolf, 2001). ERK and p38 both have distinct roles in response to pain (Watkins et al., 2001). Ozaki et al. (2004) recently found that basally active p38 in the ventral tegmental area of the brain can be suppressed by sciatic nerve ligation. Other evidence has shown that p38 and ERK were activated within microglia and astrocytes after peripheral nerve injury (Jin et al., 2003; Zhuang et al., 2005). Results from the present study suggest that KOR activation of p38 contributes to the proliferative response of type II astrocytes after pSNL. Numerous publications have shown that intrathecal administration of SB 203580 significantly reduced neuropathic pain behavior, including hyperalgesia and allodynia (Obata et al., 2004; Zhang et al., 2005), which is similar to our finding. No previous anatomical studies have shown the effects of p38 inhibitors on astrocyte proliferation. In our study, we demonstrated that 1 μg of SB 203580 intrathecal administration twice a day for 7 d significantly inhibited the astrocyte proliferation induced by pSNL. These results suggest that p38 MAPK pathway may play an important role in pSNL-induced astrocyte proliferation. However, the effect of p38 MAPK activation on cell proliferation in vivo may also function indirectly through the activation of microglia. Studies have shown that p38 was activated on microglia after inflammatory pain (Svensson et al., 2003) and neuropathic pain (Jin et al., 2003), and it is possible that SB 203580 may have inhibited astrocyte proliferation indirectly.
The results showing that GRK3 contributes to the phosphorylation of p38 in primary astrocyte cultures are intriguing. GRK and β-arrestin mediate receptor desensitization and resensitization (Liu and Anand, 2001). When KOR is activated by agonist, the receptor becomes a substrate for GRK that specifically phosphorylates the serine 369 residue in the C-terminal domain of the receptor and initiates arrestin-dependent receptor internalization and desensitization (Appleyard et al., 1999). There are currently seven known GRKs and four arrestins (Pitcher et al., 1998; Miller et al., 2003). Previous results from our laboratory suggest that GRK3 and β-arrestin mediate κ opioid receptor phosphorylation and subsequent homologous desensitization in response to sustained endogenous opioid release (McLaughlin et al., 2003; Xu et al., 2004). GRK is activated by opioid receptor stimulation and phosphorylates the agonist-occupied receptor, a process that further induces desensitization and tolerance (Pitcher et al., 1998).
Evidence suggests that GRKs are targets for MAPK signaling. For example, GRK2 can be inactivated by ERK-dependent phosphorylation of serine 670 of GRK2 (Pitcher et al., 1999). GRK2 was also shown to be a target for the ERK MAPK cascade by studies demonstrating that ERK inhibition impairs phosphorylation and desensitization of A3 adenosine receptors, possibly by impairing translocation of GRK2 to the plasma membrane (Trincavelli et al., 2002). Conversely, GRKs can activate ERK1/2 MAPK, contributing to the development of opioid tolerance and withdrawal (Liu and Anand, 2001). β-Arrestin association with β2 adrenergic receptors after their phosphorylation by GRK resulted in c-src recruitment, thereby initiating ERK1/2 MAPK pathway activation (Luttrell et al., 1999), a result applicable to other G-protein-coupled receptors (Ahn et al., 1999). Thus, in addition to their traditionally acknowledged role in desensitization, GRKs and arrestins may also serve as adapters for the initiation of alternative signaling routes (Luttrell et al., 1999; Miller et al., 2003). For instance, angiotensin receptors use arrestin scaffolds to couple to MAPK family members, including ERK and JNK (McDonald et al., 2000).
An association between p38 MAPK and GRK is also suggested by previous studies as well (Bruchas et al., 2006). The human cytomegalovirus receptor US28 is a Gq-coupled receptor that is hyperphosphorylated by GRK5, resulting in constitutive β-arrestin association at the plasma membrane and resulting in maximal p38 MAPK phosphorylation and activation (Miller et al., 2003). If a similar mechanism may be involved in the KOR activation of astrocytes observed in the present study, the GRK3 gene deletion might result in basally lower p38 MAPK phosphorylation and may prevent the KOR-mediated activation of p38 observed.
A better understanding of the mechanisms underlying chronic neuropathic pain is likely to suggest therapeutic strategies that could benefit the many people with this condition. Results from the present study suggest that endogenous κ opioids induce the activation of astrocytes within the spinal cord. How that contributes to the neuropathic pain response is not yet clear, but astrocytosis is likely to result in persistent changes in neuronal function and may contribute to the expression of the spontaneous pain, mechanical and thermal allodynia, and hyperalgesia responses characteristic of chronic pain syndromes.
Footnotes
This work was supported by United States Public Health Service Grants R37DA11672, PO1DA15916, and F32DA20430 from the National Institute on Drug Abuse. We thank Dr. John Pintar for KOR knock-out mice, Dr. Uwe Hochgeschwender for prodynorphin knock-out mice, and Drs. Marc Caron and Robert Lefkowitz for the GRK3 knock-out mice. We thank Drs. Megumi Aita and Margaret R. Byers for help with chronic nerve injury in rats.
References
- Ahn S, Maudsley S, Luttrell LM, Lefkowitz RJ, Daaka Y. Src-mediated tyrosine phosphorylation of dynamin is required for beta2-adrenergic receptor internalization and mitogen-activated protein kinase signaling. J Biol Chem. 1999;274:1185–1188. doi: 10.1074/jbc.274.3.1185. [DOI] [PubMed] [Google Scholar]
- Appleyard SM, Celver J, Pineda V, Kovoor A, Wayman GA, Chavkin C. Agonist-dependent desensitization of the kappa opioid receptor by G protein receptor kinase and beta-arrestin. J Biol Chem. 1999;274:23802–23807. doi: 10.1074/jbc.274.34.23802. [DOI] [PubMed] [Google Scholar]
- Barg J, Belcheva MM, Rowinski J, Coscia CJ. kappa-Opioid agonist modulation of [3H]thymidine incorporation into DNA: evidence for the involvement of pertussis toxin-sensitive G protein-coupled phosphoinositide turnover. J Neurochem. 1993a;60:1505–1511. doi: 10.1111/j.1471-4159.1993.tb03314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barg J, Nah SY, Levy R, Saya D, Vogel Z. Modulation of thymidine incorporation by kappa-opioid ligands in rat spinal cord-dorsal root ganglion co-cultures. Brain Res. 1993b;629:109–114. doi: 10.1016/0006-8993(93)90488-9. [DOI] [PubMed] [Google Scholar]
- Belcheva MM, Szùcs M, Wang D, Sadee W, Coscia CJ. μ-Opioid receptor-mediated ERK activation involves calmodulin-dependent epidermal growth factor receptor transactivation. J Biol Chem. 2001;276:33847–33853. doi: 10.1074/jbc.M101535200. [DOI] [PubMed] [Google Scholar]
- Belcheva MM, Clark AL, Haas PD, Serna JS, Hahn JW, Kiss A, Coscia CJ. Mu and kappa opioid receptors activate ERK/MAPK via different protein kinase C isoforms and secondary messengers in astrocytes. J Biol Chem. 2005;280:27662–27669. doi: 10.1074/jbc.M502593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-α gene expression in endotoxin-stimulated primaryglialcultures. J Neurosci. 1998;18:1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Belcheva MM, Coscia CJ. Mu-opioid agonist inhibition of kappa-opioid receptor-stimulated extracellular signal-regulated kinase phosphorylation is dynamin-dependent in C6 glioma cells. J Neurochem. 2000;74:574–581. doi: 10.1046/j.1471-4159.2000.740574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Macey TA, Lowe JD, Chavkin C. Kappa opioid receptor activation of p38 MAPK is GRK3 and arrestin dependent in neurons and astrocytes. J Biol Chem. 2006;281:18081–18089. doi: 10.1074/jbc.M513640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. 1982;215:413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH. MAP kinases. Chem Rev. 2001;101:2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- Cui Y, Chen Y, Zhi JL, Guo RX, Feng JQ, Chen PX. Activation of p38 mitogen-activated protein kinase in spinal microglia mediates morphine antinociceptive tolerance. Brain Res. 2006;1069:235–243. doi: 10.1016/j.brainres.2005.11.066. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Colburn RW, Rickman AJ, Yeager MP. Intrathecal catheterization alone induces neuroimmune activation in the rat. Eur J Pain. 1997;1:115–122. doi: 10.1016/s1090-3801(97)90069-0. [DOI] [PubMed] [Google Scholar]
- Eriksson PS, Nilsson M, Wagberg M, Hansson E, Ronnback L. Kappa-opioid receptors on astrocytes stimulate L-type Ca2+ channels. Neuroscience. 1993;54:401–407. doi: 10.1016/0306-4522(93)90261-d. [DOI] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher TF, Seibel GL, Kassis S, Laydon JT, Blumenthal MJ, Lee JC, Lee D, Boehm JC, Fier-Thompson SM, Abt JW, Soreson ME, Smietana JM, Hall RF, Garigipati RS, Bender PE, Erhard KF, Krog AJ, Hofmann GA, Sheldrake PL, McDonnell PC. Regulation of stress-induced cytokine production by pyridinylimidazoles; inhibition of CSBP kinase. Bioorg Med Chem. 1997;5:49–64. doi: 10.1016/s0968-0896(96)00212-x. [DOI] [PubMed] [Google Scholar]
- Hauser KF, Stiene-Martin A, Mattson MP, Elde RP, Ryan SE, Godleske CC. mu-Opioid receptor-induced Ca2+ mobilization and astroglial development: morphine inhibits DNA synthesis and stimulates cellular hypertrophy through a Ca2+-dependent mechanism. Brain Res. 1996;720:191–203. doi: 10.1016/0006-8993(96)00103-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano M, Goldman JE. Gliogenesis in rat spinal cord: evidence for origin of astrocytes and oligodendrocytes from radial precursors. J Neurosci Res. 1988;21:155–167. doi: 10.1002/jnr.490210208. [DOI] [PubMed] [Google Scholar]
- Hough LB, Nalwalk JW, Chen Y, Schuller A, Zhu Y, Zhang J, Menge WM, Leurs R, Timmerman H, Pintar JE. Improgan, a cimetidine analog, induces morphine-like antinociception in opioid receptor-knockout mice. Brain Res. 2000;880:102–108. doi: 10.1016/s0006-8993(00)02776-1. [DOI] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- Ishii K, Toda M, Nakai Y, Asou H, Watanabe M, Nakamura M, Yato Y, Fujimura Y, Kawakami Y, Toyama Y, Uyemura K. Increase of oligodendrocyte progenitor cells after spinal cord injury. J Neurosci Res. 2001;65:500–507. doi: 10.1002/jnr.1180. [DOI] [PubMed] [Google Scholar]
- Ji RR. Mitogen-activated protein kinases as potential targets for pain killers. Curr Opin Investig Drugs. 2004;5:71–75. [PubMed] [Google Scholar]
- Ji RR, Woolf CJ. Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol Dis. 2001;8:1–10. doi: 10.1006/nbdi.2000.0360. [DOI] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. 2003;23:4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajander KC, Sahara Y, Iadarola MJ, Bennett GJ. Dynorphin increases in the dorsal spinal cord in rats with a painful peripheral neuropathy. Peptides. 1990;11:719–728. doi: 10.1016/0196-9781(90)90187-a. [DOI] [PubMed] [Google Scholar]
- Liu JG, Anand KJ. Protein kinases modulate the cellular adaptations associated with opioid tolerance and dependence. Brain Res Brain Res Rev. 2001;38:1–19. doi: 10.1016/s0165-0173(01)00057-1. [DOI] [PubMed] [Google Scholar]
- Liu L, Persson JK, Svensson M, Aldskogius H. Glial cell responses, complement, and clusterin in the central nervous system following dorsal root transection. Glia. 1998;23:221–238. [PubMed] [Google Scholar]
- Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- Ma W, Quirion R. Partial sciatic nerve ligation induces increase in the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in astrocytes in the lumbar spinal dorsal horn and the gracile nucleus. Pain. 2002;99:175–184. doi: 10.1016/s0304-3959(02)00097-0. [DOI] [PubMed] [Google Scholar]
- McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, Lefkowitz RJ. Beta-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science. 2000;290:1574–1577. doi: 10.1126/science.290.5496.1574. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Xu M, Mackie K, Chavkin C. Phosphorylation of a carboxy-terminal serine within the kappa opioid receptor produces desensitization and internalization. J Biol Chem. 2003;278:34631–34640. doi: 10.1074/jbc.M304022200. [DOI] [PubMed] [Google Scholar]
- McMahon SS, McDermott KW. Proliferation and migration of glial precursor cells in the developing rat spinal cord. J Neurocytol. 2001;30:821–828. doi: 10.1023/a:1019693421778. [DOI] [PubMed] [Google Scholar]
- Miller WE, Houtz DA, Nelson CD, Kolattukudy PE, Lefkowitz RJ. G-protein-coupled receptor (GPCR) kinase phosphorylation and beta-arrestin recruitment regulate the constitutive signaling activity of the human cytomegalovirus US28 GPCR. J Biol Chem. 2003;278:21663–21671. doi: 10.1074/jbc.M303219200. [DOI] [PubMed] [Google Scholar]
- Misson JP, Austin CP, Takahashi T, Cepko CL, Caviness VS., Jr The alignment of migrating neural cells in relation to the murine neopallial radial glial fiber system. Cereb Cortex. 1991;1:221–229. doi: 10.1093/cercor/1.3.221. [DOI] [PubMed] [Google Scholar]
- Murray M, Wang SD, Goldberger ME, Levitt P. Modification of astrocytes in the spinal cord following dorsal root or peripheral nerve lesions. Exp Neurol. 1990;110:248–257. doi: 10.1016/0014-4886(90)90036-r. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Furuya Y, Kaneko T, Yamatsu K. Spinal kappa receptor-mediated analgesia of E-2078, a systemically active dynorphin analog, in mice. J Pharmacol Exp Ther. 1991;256:76–81. [PubMed] [Google Scholar]
- Narita M, Suzuki M, Yajima Y, Suzuki R, Shioda S, Suzuki T. Neuronal protein kinase C gamma-dependent proliferation and hypertrophy of spinal cord astrocytes following repeated in vivo administration of morphine. Eur J Neurosci. 2004a;19:479–484. doi: 10.1111/j.0953-816x.2003.03119.x. [DOI] [PubMed] [Google Scholar]
- Narita M, Kuzumaki N, Suzuki M, Oe K, Yamazaki M, Yajima Y, Suzuki T. Increased phosphorylated-mu-opioid receptor immunoreactivity in the mouse spinal cord following sciatic nerve ligation. Neurosci Lett. 2004b;354:148–152. doi: 10.1016/j.neulet.2003.09.077. [DOI] [PubMed] [Google Scholar]
- Obata K, Noguchi K. MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci. 2004;74:2643–2653. doi: 10.1016/j.lfs.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Kobayashi K, Dai Y, Mizushima T, Katsura H, Fukuoka T, Tokunaga A, Noguchi K. Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci. 2004;24:10211–10222. doi: 10.1523/JNEUROSCI.3388-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Mikami Y, Shimazaki T, Mihara M, Ohsugi Y, Iwamoto Y, Yoshizaki K, Kishimoto T, Toyama Y, Okano H. Blockade of interleukin-6 receptor suppresses reactive astrogliosis and ameliorates functional recovery in experimental spinal cord injury. J Neurosci Res. 2004;76:265–276. doi: 10.1002/jnr.20044. [DOI] [PubMed] [Google Scholar]
- Ozaki S, Narita M, Narita M, Ozaki M, Khotib J, Suzuki T. Role of extracellular signal-regulated kinase in the ventral tegmental area in the suppression of the morphine-induced rewarding effect in mice with sciatic nerve ligation. J Neurochem. 2004;88:1389–1397. doi: 10.1046/j.1471-4159.2003.02272.x. [DOI] [PubMed] [Google Scholar]
- Peppel K, Boekhoff I, McDonald P, Breer H, Caron MG, Lefkowitz RJ. G protein-coupled receptor kinase 3 (GRK3) gene disruption leads to loss of odorant receptor desensitization. J Biol Chem. 1997;272:25425–25428. doi: 10.1074/jbc.272.41.25425. [DOI] [PubMed] [Google Scholar]
- Pitcher JA, Freedman NJ, Lefkowitz RJ. G protein-coupled receptor kinases. Annu Rev Biochem. 1998;67:653–692. doi: 10.1146/annurev.biochem.67.1.653. [DOI] [PubMed] [Google Scholar]
- Pitcher JA, Tesmer JJ, Freeman JL, Capel WD, Stone WC, Lefkowitz RJ. Feedback inhibition of G protein-coupled receptor kinase 2 (GRK2) activity by extracellular signal-regulated kinases. J Biol Chem. 1999;274:34531–34534. doi: 10.1074/jbc.274.49.34531. [DOI] [PubMed] [Google Scholar]
- Raff MC, Miller RH, Noble M. A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature. 1983;303:390–396. doi: 10.1038/303390a0. [DOI] [PubMed] [Google Scholar]
- Ruzicka BB, Fox CA, Thompson RC, Meng F, Watson SJ, Akil H. Primary astroglial cultures derived from several rat brain regions differentially express mu, delta and kappa opioid receptor mRNA. Brain Res Mol Brain Res. 1995;34:209–220. doi: 10.1016/0169-328x(95)00165-o. [DOI] [PubMed] [Google Scholar]
- Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990;43:205–218. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- Sharifi N, Diehl N, Yaswen L, Brennan MB, Hochgeschwender U. Generation of dynorphin knockout mice. Brain Res Mol Brain Res. 2001;86:70–75. doi: 10.1016/s0169-328x(00)00264-3. [DOI] [PubMed] [Google Scholar]
- Sommer I, Schachner M. Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces: an immunocytological study in the central nervous system. Dev Biol. 1981;83:311–327. doi: 10.1016/0012-1606(81)90477-2. [DOI] [PubMed] [Google Scholar]
- Song P, Zhao ZQ. The involvement of glial cells in the development of morphine tolerance. Neurosci Res. 2001;39:281–286. doi: 10.1016/s0168-0102(00)00226-1. [DOI] [PubMed] [Google Scholar]
- Stiene-Martin A, Hauser KF. Glial growth is regulated by agonists selective for multiple opioid receptor types in vitro. J Neurosci Res. 1991;29:538–548. doi: 10.1002/jnr.490290415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiene-Martin A, Zhou R, Hauser KF. Regional, developmental, and cell cycle-dependent differences in mu, delta, and kappa-opioid receptor expression among cultured mouse astrocytes. Glia. 1998;22:249–259. [PMC free article] [PubMed] [Google Scholar]
- Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Freshwater JD, Catalano R, Feng Y, Protter AA, Scott B, Yaksh TL. Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem. 2003;86:1534–1544. doi: 10.1046/j.1471-4159.2003.01969.x. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Raghavendra V, DeLeo JA. Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochem Int. 2004;45:397–407. doi: 10.1016/j.neuint.2003.06.002. [DOI] [PubMed] [Google Scholar]
- Trincavelli ML, Tuscano D, Marroni M, Klotz KN, Lucacchini A, Martini C. Involvement of mitogen protein kinase cascade in agonist-mediated human A(3) adenosine receptor regulation. Biochim Biophys Acta. 2002;1591:55–62. doi: 10.1016/s0167-4889(02)00248-3. [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Gardell LR, Burgess SE, Ibrahim M, Dogrul A, Zhong CM, Zhang ET, Malan TP, Jr, Ossipov MH, Lai J, Porreca F. Dynorphin promotes abnormal pain and spinal opioid antinociceptive tolerance. J Neurosci. 2000;20:7074–7079. doi: 10.1523/JNEUROSCI.20-18-07074.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gardell LR, Ossipov MH, Vanderah TW, Brennan MB, Hochgeschwender U, Hruby VJ, Malan TP, Jr, Lai J, Porreca F. Pronociceptive actions of dynorphin maintain chronic neuropathic pain. J Neurosci. 2001;21:1779–1786. doi: 10.1523/JNEUROSCI.21-05-01779.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Spinal cord glia: new players in pain. Pain. 2001;93:201–205. doi: 10.1016/S0304-3959(01)00359-1. [DOI] [PubMed] [Google Scholar]
- Xu M, Petraschka M, McLaughlin JP, Westenbroek RE, Caron MG, Lefkowitz RJ, Czyzyk TA, Pintar JE, Terman GW, Chavkin C. Neuropathic pain activates the endogenous κ opioid system in mouse spinal cord and induces opioid receptor tolerance. J Neurosci. 2004;24:4576–4584. doi: 10.1523/JNEUROSCI.5552-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young PR, McLaughlin MM, Kumar S, Kassis S, Doyle ML, McNulty D, Gallagher TF, Fisher S, McDonnell PC, Carr SA, Huddleston MJ, Seibel G, Porter TG, Livi GP, Adams JL, Lee JC. Pyridinyl imidazole inhibitors of p38 mitogen-activated protein kinase bind in the ATP site. J Biol Chem. 1997;272:12116–12121. doi: 10.1074/jbc.272.18.12116. [DOI] [PubMed] [Google Scholar]
- Zhang FE, Cao JL, Zhang LC, Zeng YM. Activation of p38 mitogen-activated protein kinase in spinal cord contributes to chronic constriction injury-induced neuropathic pain. Sheng Li Xue Bao. 2005;57:545–551. [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]