

Abstract
Adenosine has been implicated as a modulator of retinohypothalamic neurotransmission in the suprachiasmatic nucleus (SCN), the seat of the light-entrainable circadian clock in mammals. Intracellular recordings were made from SCN neurons in slices of hamster hypothalamus using the in situ whole-cell patch clamp method. A monosynaptic, glutamatergic, excitatory postsynaptic current (EPSC) was evoked by stimulation of the optic nerve. The EPSC was blocked by bath application of the adenosine A1 receptor agonist cyclohexyladenosine (CHA) in a dose-dependent manner with a half-maximal concentration of 1.7 µM. The block of EPSC amplitude by CHA was antagonized by concurrent application of the adenosine A1 receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX). The adenosine A2A receptor agonist CGS21680 was ineffective in attenuating the EPSC at concentrations up to 50 µM. Trains of four consecutive stimuli at 25 ms intervals usually depressed the EPSC amplitude. However, after application of CHA, consecutive responses displayed facilitation of EPSC amplitude. The induction of facilitation by CHA suggested a presynaptic mechanism of action. After application of CHA, the frequency of spontaneous EPSCs declined substantially, while their amplitude distribution was unchanged or slightly reduced, again suggesting a mainly presynaptic site of action for CHA. Application of glutamate by brief pressure ejection evoked a long-lasting inward current that was unaffected by CHA at concentrations sufficient to reduce the evoked EPSC amplitude substantially (1 to 5 µM), suggesting that postsynaptic glutamate receptor-gated currents were unaffected by the drug. Taken together, these observations indicate that CHA inhibits optic nerve-evoked EPSCs in SCN neurons by a predominantly presynaptic mechanism.
Keywords: suprachiasmatic nucleus, circadian rhythm, entrainment, adenosine, presynaptic inhibition
INTRODUCTION
In mammals, daily (circadian) rhythms in physiology and behavior, including rhythms in sleep and wakefulness, are controlled by a biological clock located in the hypothalamic suprachiasmatic nucleus (SCN; for review, see Klein et al., 1991). Autonomous cellular oscillators within the SCN act in concert to drive rhythms in behavior (Welsh et al., 1995; Jagota et al., 2000). In addition, certain environmental stimuli “reset” the circadian clock on a daily basis to maintain synchrony with relevant environmental rhythms (Hastings et al., 1996; Rea, 1998). In humans, the significance of this phenomenon is particularly apparent after rapid transmeridian travel, which often results in a period of cognitive impairment and sleep disturbance (jet lag) that can last for several circadian cycles.
The most important environmental signal that synchronizes circadian and environmental rhythms is ambient light. The daily process by which this occurs is called photic entrainment (Rea, 1998). Exposure to light around dusk delays circadian phase, while similar exposure at dawn advances the circadian clock (DeCoursey, 1964; Nelson and Takahashi, 1991). Although photic information may reach SCN neurons by at least three distinct pathways, the retinohypothalamic tract (RHT) appears to be both necessary and sufficient to support photic entrainment (Moore and Lenn, 1972; Johnson et al., 1988). RHT axons arise from retinal ganglion cells and make monosynaptic connections with SCN neurons (Moore and Lenn, 1972). Physiological and pharmacological evidence points to glutamate as the excitatory neurotransmitter released at the RHT synapse in the SCN, and both glutamate- and light-induced phase shifts occur through the activation of AMPA and NMDA receptors in the SCN region (Colwell et al., 1990; Kim and Dudek, 1991; Rea et al., 1993; Ding et al., 1994).
The nucleoside messenger adenosine is a potent modulator of excitatory neurotransmission in the central nervous system (Dunwiddie, 1985). Adenosine is released from both neurons and glia and accumulates in the extracellular space during periods of high metabolic demand (Dunwiddie, 1985; White and Hoehn, 1991). Under these conditions, the nucleoside is thought to serve a neuroprotective role, primarily by inhibiting the release of glutamate (Dunwiddie, 1985). This effect of adenosine is mediated by presynaptic A1 receptors located on or near glutamatergic nerve terminals (Dunwiddie and Haas, 1985; Flagmeyer et al., 1997).
Although evidence for adenosine receptors in the SCN is limited (Chen and van den Pol, 1997), both behavioral and physiological evidence suggest that adenosine may directly inhibit photic input to the SCN (Watanabe et al., 1996; Elliott et al., 2001). Such an action may, in turn, underlie the effect of sleep deprivation on photic entrainment (Mistlberger et al., 1997), although a role for serotonin in this phenomenon has been implicated as well (Grossman et al., 2000; Challet et al., 2001). In this study, we have examined the effects of adenosine receptor agonists on RHT neurotransmission in the hamster SCN. The results suggest that adenosine has a direct effect on RHT synaptic transmission to SCN neurons that is mediated by A1, but not A2A, adenosine receptors.
METHODS
Male Syrian hamsters (Mesocricetus auratus; 20 days old) were obtained from Charles River Laboratories (Wilmington, MA) and used within 3 weeks of arrival. During this period, the hamsters were housed in groups of four to six per cage under a 14:10 light/dark (LD) cycle (lights on at 8:00 am) and were provided with rodent chow and water ad libitum. Between 8:30 and 9:00 am on the day of the experiment, hamsters were decapitated and the brains were rapidly removed and cut into blocks containing the basal hypothalamus, optic chiasm, and optic nerves. After chilling in ice-cold physiological saline, a single 400 µm thick horizontal brain slice containing the SCN and both optic nerves was prepared from each brain using a vibrating tissue slicer (Camden, Loughborough, UK), as described previously (Gannon et al., 1995). Prior to use, brain slices were incubated for at least 1.5 h in physiological saline under an atmosphere of 95% O2/5% CO2 at room temperature. The physiological saline used for both preincubation and subsequent perfusion of the slice contained (in mM): NaCl 122; KCl 3.8; MgSO4 1.2; KH2PO4 1.2; NaHCO3 25; CaCl2 2.5; dextrose 10; bubbled with 95% O2/5% CO2.
Individual brain slices were secured to the floor of an immersion chamber and perfused with physiological saline (0.8 mL/min) maintained at 33 ± 2°C. A diagram of the experimental preparation is shown in Figure 1(A). Slices were viewed either with a transilluminated Nikon dissecting microscope or with an Olympus BX-50WI upright microscope equipped under DIC optics and an infrared-sensitive Newvicon camera (Dage-MTI, Michigan City, IN). The entire preparation was mounted on an air table. Recordings were performed between 11:00 am and 6:00 pm (between 5 and 12 h after lights-on).
Figure 1.
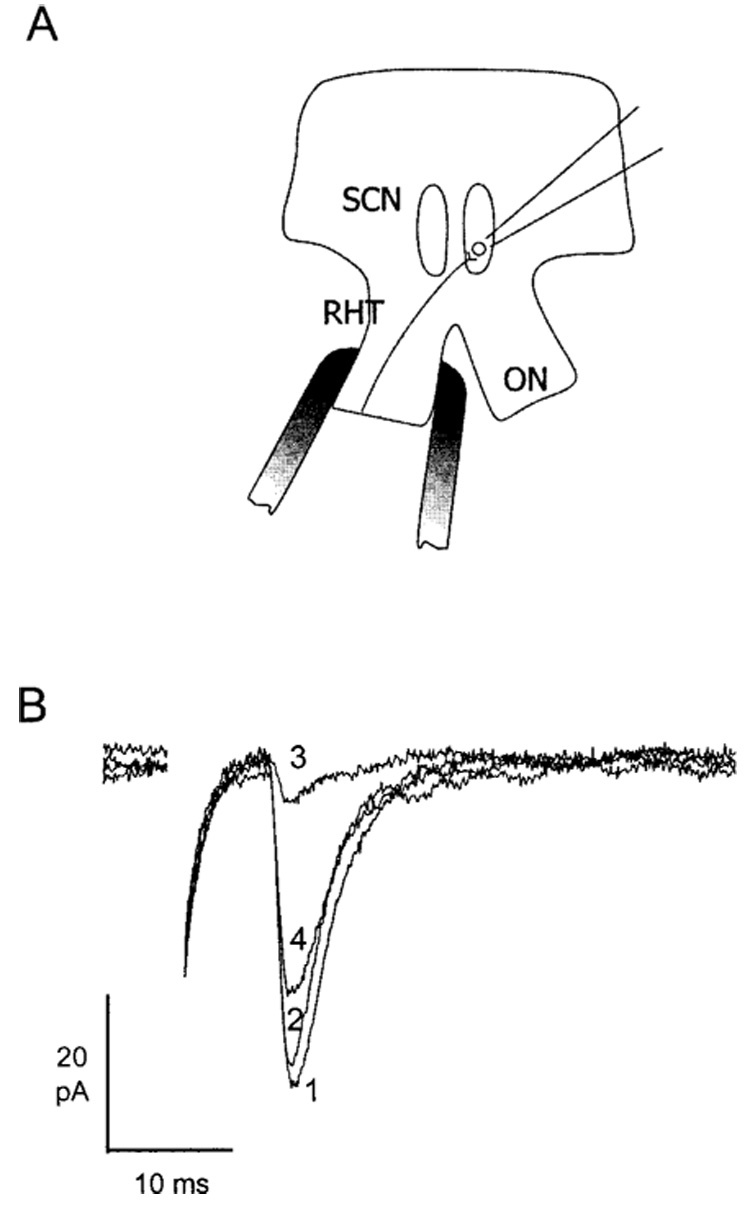
(A) Diagram of the hypothalamic slice preparation, showing the location of the recording pipette and stimulating electrode. SCN, suprachiasmatic nucleus; RHT, retinohypothalamic tract; ON, optic nerve. (B) Representative examples of EPSCs evoked by ON stimulation and recorded from a single hamster SCN neuron. Each trace is the average of 16 consecutive responses obtained (1) in control saline, (2) in 50 µM APV, (3) in 20 µM DNQX + 50 µM APV, (4) 30 min after washout of drugs. The holding potential was −70 mV.
The whole-cell patch clamp method was used to obtain voltage clamp recordings from SCN neurons (Blanton et al., 1989). Patch pipettes were pulled from borosilicate glass (A-M Systems, Carlsborg, WA) using a Flaming/Brown electrode puller and filled with a solution containing (in mM): EGTA 5; HEPES 10; MgCl2 1; K-gluconate 130; NaCl 1; CaCl2 1; K2ATP 2; adjusted to 275 mOsm and pH 7.2. Using an isolated constant current source (World Precision Instruments, Sarasota, FL) electrical stimulation of the optic nerve was achieved by means of either a silver/silver chloride suction electrode attached to the optic nerve or a twisted pair electrode made from fine Pt-Ir wire. Drugs were applied to the slice through the perfusion medium. In some experiments, glutamate, as a 20 mM solution in physiological saline buffered to pH 7.35 with 10 mM HEPES, was pressure-applied in the vicinity of a patched neuron via a second pipette using a PicoPump (World Precision Instruments).
Data acquisition was performed using an Axon Instruments Axopatch-1D amplifier controlled by an Intel Pentium-based computer running pClamp (v 6.0) software (Axon Instruments, Burlingame, CA). Cells were held at a potential of −60 to −80 mV to minimize involvement of NMDA receptor gated currents (Pennartz et al., 2001). Excitatory postsynaptic currents (EPSCs) were evoked by electrical stimulation of the contralateral or ipsilateral optic nerves at currents just sufficient to evoke a consistent EPSC (0.5–1.0 mA; 100 µs pulse). Signals were averaged over 16 presentations at 1 Hz. Paired pulse facilitation was tested by presentation of four consecutive stimuli at 25 ms intervals, and the evoked currents were averaged over 16 trials. Spontaneous synaptic currents were recorded for up to 300 s and analyzed using Axon Instruments software. Plotting and sigmoidal curve fitting were performed using Origin v.6 (Microcal, Northampton, MA). Cumulative probability functions were calculated using Microsoft Excel 97 and compared using the Kolmogorov-Smirnov two-sample nonparametric test (Steel and Torrie, 1980). Calculations for the Kolmogorov-Smirnov test were performed using the on-line statistics calculator at the College of St. Benedict, St. John’s University (http://bardeen.physics.csbsju.edu/stats).
Cell input resistance ranged from 200 MΩ to greater than 1 GΩ. Patch pipettes in the whole cell condition had apparent series resistances, based on the height of the transient response to a small depolarizing pulse, of no more than 20 MΩ. No series resistance compensation was performed, but the series resistance was carefully monitored throughout the experiment. Thus, it is likely that the effects of treatments on EPSC amplitude reported herein reflect changes in synaptic transmission rather than changes in electrode properties.
All salts (analytical grade or better) were obtained from Sigma Biochemicals (St. Louis, MO). N6-cyclohexyladenosine (CHA), 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), 6,7-dinitroquinoxaline-2,3-dione (DNQX), and CGS21680 were obtained from Research Biochemicals, Inc. (Natick, MA).
RESULTS
Effect of CHA on the RHT-Evoked EPSC
Electrical stimulation of the contralateral or ipsilateral optic nerve evoked a short-latency EPSC in approximately 25% of patched SCN neurons, which reversed at or near 0 mV. This EPSC was unchanged in the presence of the NMDA antagonist AP5 (50 µM), but was completely blocked by bath application of the AMPA-receptor antagonist DNQX [20 µM; Fig. 1(B)]. Bath application of the adenosine A1 receptor agonist CHA rapidly reduced EPSC amplitude [Fig. 2(A)]. The effect of CHA on EPSC amplitude was dose-dependent over a concentration range of 0.1–50 µM [Fig. 2(B)]. The average reduction of EPSC amplitude at each concentration was fitted to a single sigmoid function that yielded an apparent EC50 value for CHA of approximately 1.7 µM. The effect of CHA on EPSC amplitude was slowly reversible. However, subsequent application of the adenosine A1 receptor antagonist DPCPX (500 nM) together with CHA reduced the attenuation of EPSC amplitude by CHA [Fig. 2(A)]. DPCPX alone was without effect on the EPSC (500 nM; n = 4).
Figure 2.
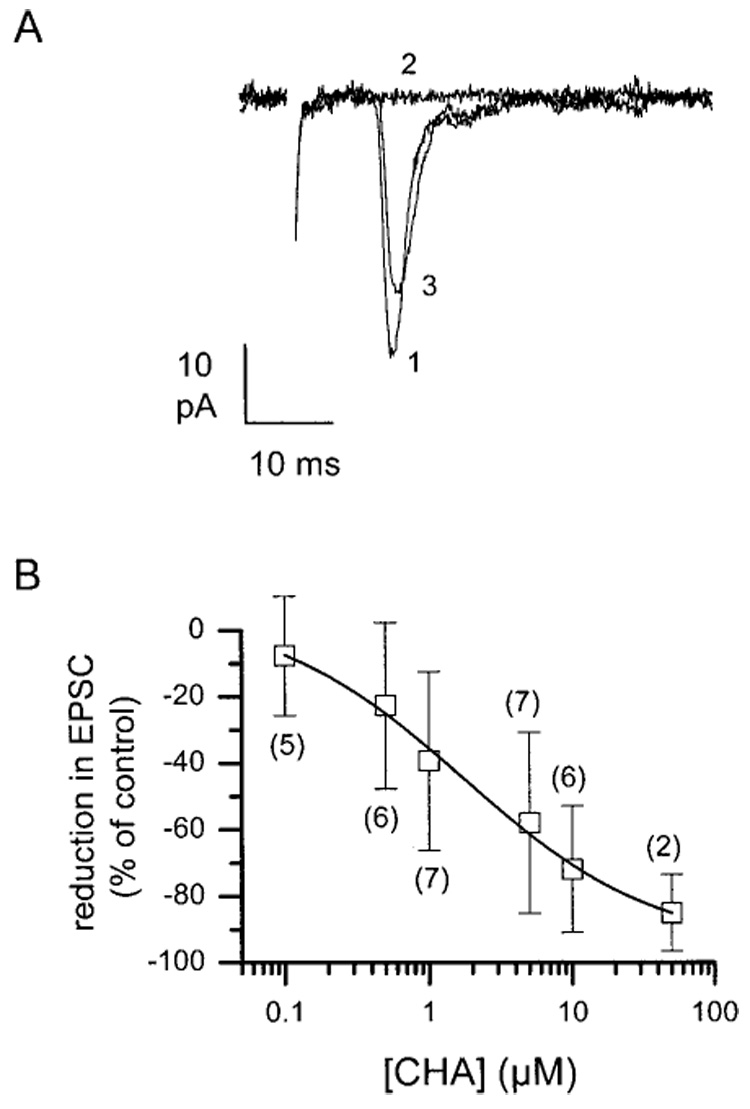
Application of the selective adenosine A1 receptor agonist CHA dose-dependently attenuates EPSC amplitude. (A) Representative examples of EPSCs evoked by optic nerve stimulation (1) prior to drug exposure, (2) 20 min after application of 5 µM CHA, and (3) 20 min after application of 0.5 µM DPCPX in the presence of CHA, showing nearly complete restoration by the antagonist. Data were acquired from the same cell during sequential treatments. (B) Dose-response function for the reduction in the EPSC amplitude by CHA. Data (open squares) represent the mean ±S.E.M. of two to six determinations. The number of cells studied at each concentration is indicated beside the data point. The data were fitted to a sigmoidal dose response function yielding a half-maximally effective concentration of 1.70 µM (slope 0.59).
Application of the adenosine A2A agonist CGS21680 failed to affect the amplitude of the EPSC [Fig. 3(A)]. Exposures as long as 30 min at concentrations of up to 50 µM were ineffective in four cells tested [Fig. 3(B)].
Figure 3.
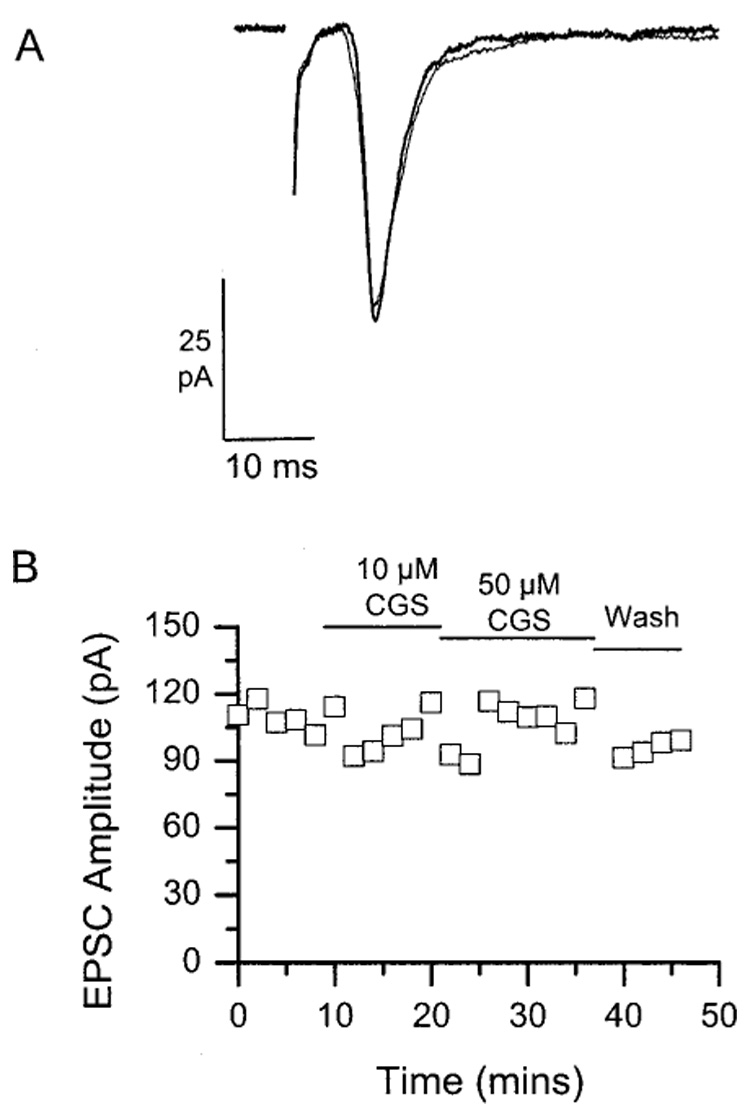
Lack of effect of the selective adenosine A2 receptor agonist on RHT-evoked EPSCs. (A) Representative examples of EPSCs evoked by optic nerve stimulation and recorded in the same cell in the absence (thick line) and presence (thin line) of 50 µM CGS 21680. (B) Time course showing lack of effect of CGS 21680 (10 and 50 µM) on the amplitude of RHT-evoked EPSCs recorded from a single SCN neuron.
Does CHA Act through a Presynaptic Mechanism to Attenuate RHT-Evoked EPSCs?
We investigated whether CHA acts through a presynaptic mechanism to attenuate RHT-evoked EPSCs in the SCN using three different approaches. First, we observed the effect of CHA on the currents evoked by short trains of depolarizing pulses (interpulse interval 25 ms) presented to the optic nerves to elicit either facilitation or short-term depression, which are thought to be presynaptic phenomena (Zucker, 1993). In the absence of CHA, the EPSC amplitude either decreased during the pulse train or remained unchanged. However, after perfusion of the slice with CHA (1 to 5 µM), the EPSC amplitude was clearly and reproducibly facilitated during the train, even though the amplitude of each EPSC was substantially reduced relative to the pretreatment values [Fig. 4(A); 2.5 µM CHA]. This effect was observed in 16 cells at CHA concentrations ranging from 1 to 20 µM. Figure 4(B) shows the results obtained from six cells in 2.5 µM CHA, a concentration near the EC50 value for the drug. The change in EPSC amplitude during the pulse train was significantly greater (p < .001) in the presence of CHA compared to that observed for the corresponding pulse in the control condition. Finally, perfusion of the slice with DPCPX together with CHA resulted in either a reduction of the degree of facilitation or a return to depression (n = 9; Fig. 5). These data suggest that CHA acts presynaptically to attenuate RHT-evoked EPSC amplitude.
Figure 4.
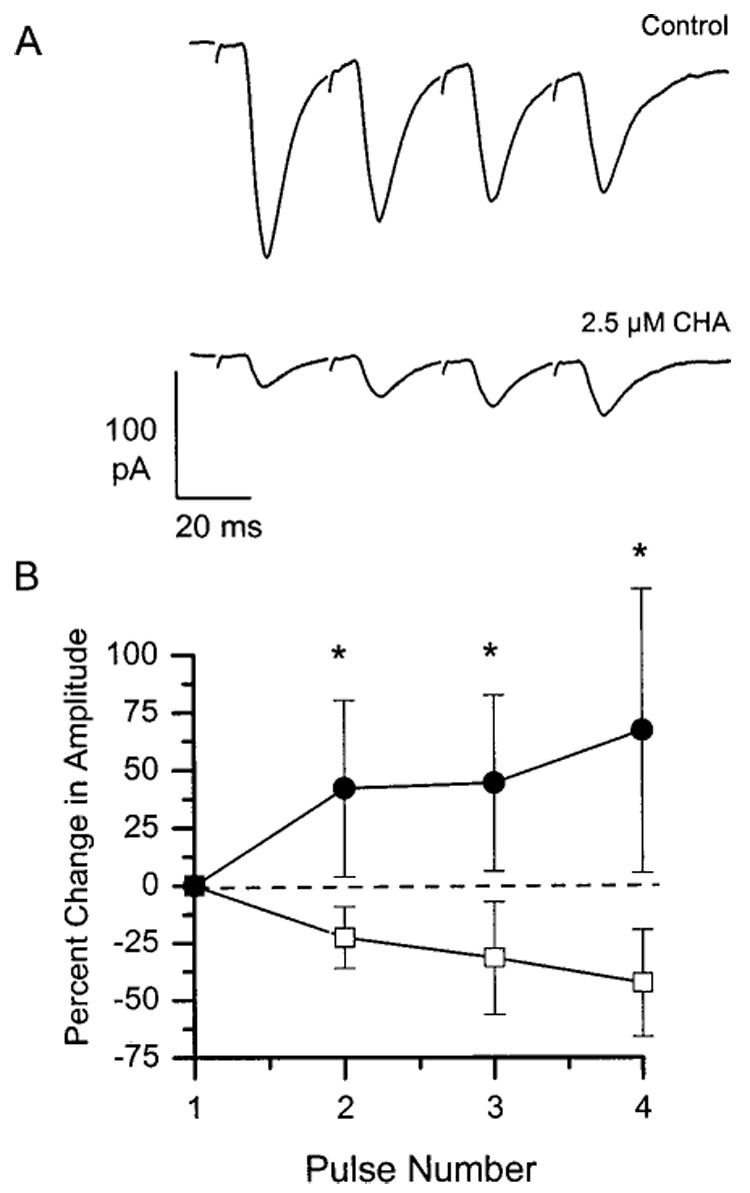
Application of 2.5 µM CHA reveals paired-pulse facilitation of RHT-evoked EPSCs. (A) Representative examples of sequential EPSCs evoked by a train of four stimuli presented at 25 ms intervals to the contralateral optic nerve in a single SCN neuron. Traces are the average of 16 trials performed in the absence (upper trace) or in the presence (lower trace) of 2.5 µM CHA. In control medium (upper trace), the amplitudes of consecutive EPSCs were usually slightly reduced, while, in the presence of 2.5 µM CHA (lower trace), the amplitudes were consistently facilitated. (B) Average change in EPSC amplitude relative to the first EPSC in the train, expressed as a percentage, performed in the absence (open squares) and in the presence (closed circles) of 2.5 µM CHA. Data represent the mean ±S.E.M. of six trials. Asterisks (*) indicate statistically significant (p < .005) differences from the corresponding pulse in the control condition as determined by the Student’s t test.
Figure 5.
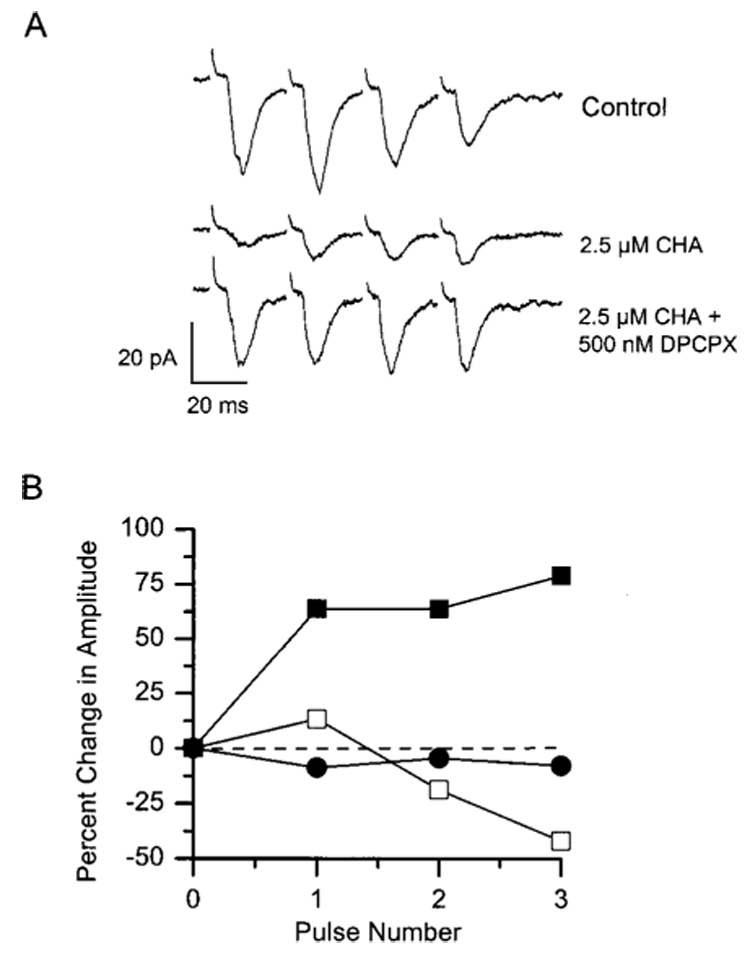
The appearance of paired pulse facilitation in the presence of CHA is blocked by DPCPX. Representative data obtained from a single cell are shown. (A) The effect of CHA (2.5 µM) alone (middle trace) and in combination (bottom trace) with DPCPX (500 nM) on paired pulse facilitation is shown. In control medium (upper waveform), the amplitudes of consecutive EPSCs were typically depressed. In the presence of 2.5 µM CHA (middle trace), the EPSC amplitude was reduced, but successive EPSCs were facilitated. In the presence of 2.5 µM CHA together with 500 nM DPCPX (lower waveform), the initial EPSC amplitude was partially restored and facilitation of subsequent EPSCs was lost. (B) Plot of the percentage change in EPSC amplitude from the same experiment as in (A). Open squares = control; filled squares = 2.5 µM CHA; filled circles = 2.5 µM CHA plus 500 nM DPCPX.
As a second approach, we investigated the effect of CHA on the frequency and amplitude distribution of spontaneous EPSCs in retinorecipient SCN neurons. In many cells, a substantial number of spontaneous EPSCs were observed. These EPSCs showed reversal potentials at or near 0 mV and had waveforms similar to the evoked EPSCs. Because the RHT is the major source of excitatory input to the SCN, the EPSCs were presumed to originate from RHT axons making synaptic contacts with the observed SCN neuron, including, but probably not limited to, those axons excited by RHT stimulation. Presynaptic inhibition of neurotransmitter release should affect the number of spontaneous EPSCs, but not their amplitudes. On the other hand, postsynaptic inhibition of EPSC amplitude would be revealed as a reduction in spontaneous EPSC amplitude without a change in frequency. In 11 experiments, continuous samples of spontaneous EPSCs were recorded for up to 200 s in control conditions and subsequently in the presence of CHA (1–20 µM). All of these cells also demonstrated a robust RHT-evoked EPSC. Examples of spontaneous EPSCs in an individual cell are shown in Figure 6(A). Although the frequency of EPSCs was clearly reduced in the presence of CHA, large amplitude EPSCs were still apparent in the record [Fig. 6(A)]. The amplitudes of spontaneous EPSCs collected over a 50 s interval were measured and plotted as a frequency histogram, and a cumulative probability plot of the amplitude histograms was constructed [Fig. 6(B)]. In 8 of 11 cells observed, the frequency of spontaneous EPSCs was reduced substantially by CHA while the amplitude distribution was not significantly altered (Kolmogorov-Smirnov test; p > .05). The observation of a substantial reduction in the frequency of spontaneous EPSCs by CHA without a change in amplitude distribution is consistent with a presynaptic mechanism of action of the drug. In two other cells, significant (p < .05) leftward shifts in the presence of CHA were observed, and in one cell a rightward shift in amplitude distribution occurred.
Figure 6.
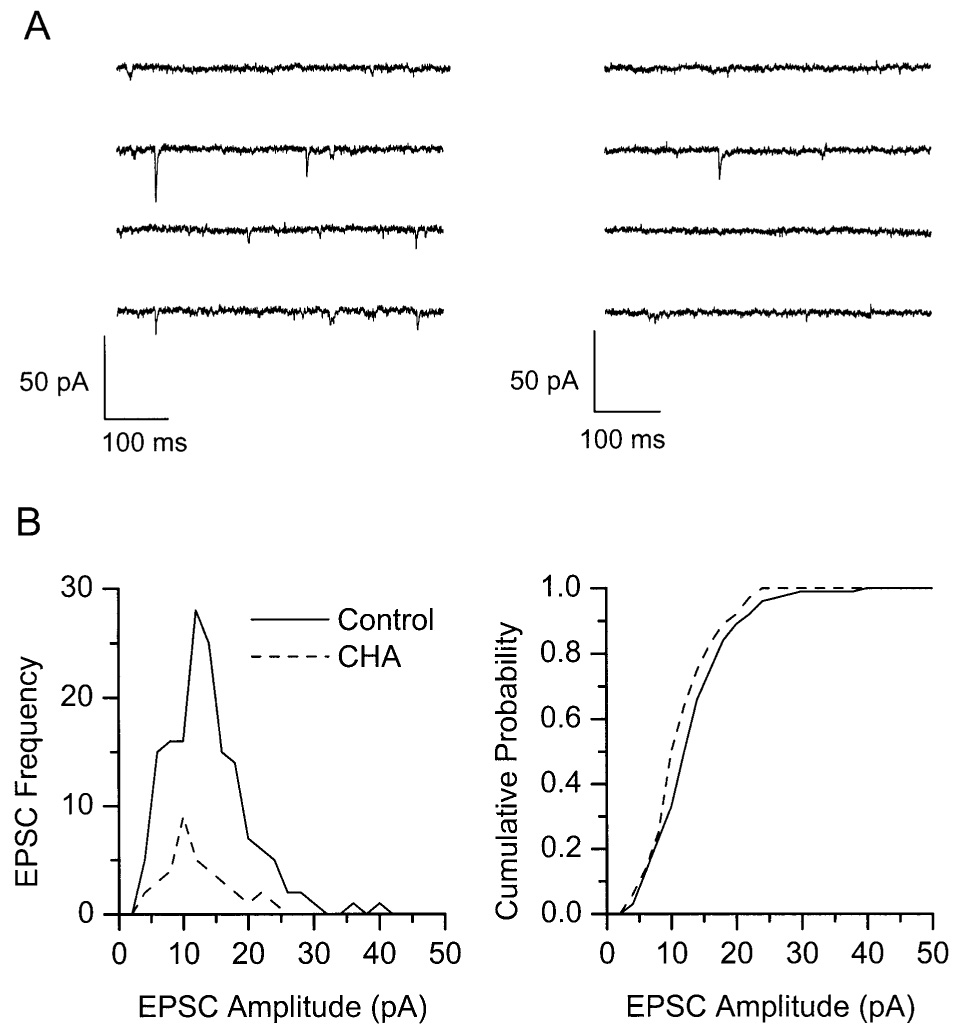
CHA does not alter spontaneous EPSC amplitude. (A) Continuous samples of spontaneous EPSCs (0.5 s per trace) recorded in a single SCN neuron in the absence (left) and presence (right) of 5 µM CHA. In this cell, CHA reduced the frequency of spontaneous EPSCs by 78%. (B) Amplitude histogram (left) and cumulative probability plot (right) for 50 s of continuous recording in each condition for the same cell as in (A). Solid lines represent the control condition; dashed lines represent the presence of CHA. The amplitude distributions were not significantly different, as determined by the Kolmogorov-Smirnov test (D = 0.19; p = .225).
The third approach to address the site of action of CHA was to examine the effect of the drug on currents evoked by application of the excitatory RHT neurotransmitter, glutamate. Postsynaptic effects of CHA on glutamatergic RHT neurotransmission would be expected to reduce glutamate-evoked currents in parallel with the reduction of the RHT-evoked EPSCs. The EPSC evoked by RHT stimulation in a typical cell is shown in Figure 7(B). We found that pressure application of a glutamate solution (20 mM) evoked a long-lasting inward current [Fig. 7(B)]. This current was increased by hyperpolarization, decreased by depolarization, and completely blocked by the AMPA receptor antagonist DNQX (20 µM), consistent with an inward current mediated by AMPA-type glutamate receptors. Low concentrations of CHA substantially reduced the RHT-evoked EPSC [Fig. 7(A)], while the glutamate-evoked current remained unchanged in amplitude [Fig. 7(B)]. Finally, both the RHT-evoked EPSC and the glutamate-evoked EPSC were completely blocked by addition of the AMPA antagonist DNQX (20 µM) to the bath (Fig. 7). In a group of four cells, 20 µM CHA reduced EPSC amplitude by 77 ± 13% (mean ± S.E.M.), but the reduction in the glutamate-evoked current was only 5 ± 18% [Fig. 7(C)]. Similar results were obtained for six other cells at lower concentrations of CHA (1–5 µM; not shown). Thus, CHA had little or no effect on the postsynaptic glutamate receptor-gated currents, even at concentrations that markedly attenuated the RHT-evoked EPSCs.
Figure 7.
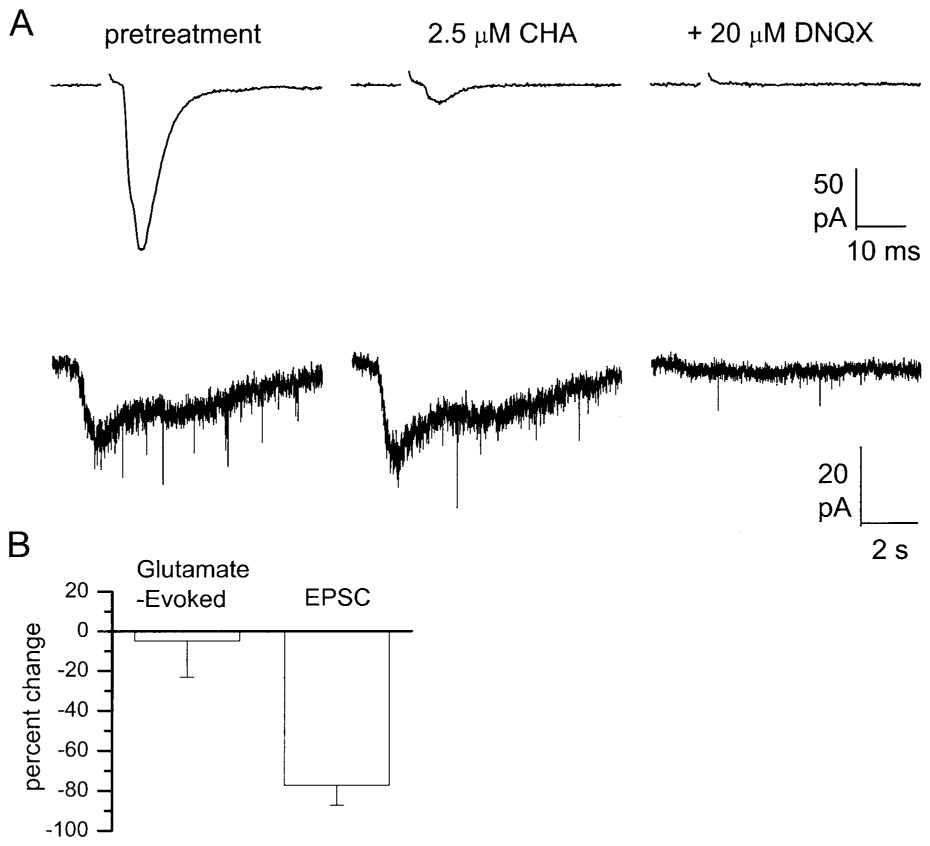
CHA fails to attenuate glutamate-evoked currents in retinorecipient SCN neurons. (A) Shown are representative examples of RHT-evoked (top traces) and glutamate-evoked (bottom traces) EPSCs recorded from the same cell in control saline (left panels), 2.5 µM CHA (middle panels), and after addition of 20 µM DNQX (right panels). Traces showing RHT-evoked EPSCs are the average of 16 trials, while the glutamate-evoked traces are single trials. (B) Effect of 2.5 µM CHA on glutamate- and RHT-evoked EPSC amplitude. Data are expressed as percent change from the pretreatment value and represent the mean ±S.E.M. of six experiments. CHA significantly (p < .05) reduced RHT-evoked EPSC amplitude by approximately 80%, but did not alter the glutamate-evoked EPSC.
DISCUSSION
Adenosine Blocks Photic Regulation of Circadian Phase
Adenosine A1 receptors have been implicated in the regulation of circadian phase by ambient light. Systemic administration of adenosine A1 receptor agonists dose-dependently attenuates light-induced phase shifts of the circadian activity rhythm in hamsters and rats, and this effect is blocked by the selective adenosine A1 receptor antagonist DPCPX (Watanabe et al., 1996; Elliott et al., 2001). Furthermore, local infusion of the selective adenosine A1 receptor agonist CHA also attenuates light-induced phase shifts, indicating that the site of action of these drugs is localized to the SCN region.
The present study provides new data concerning a possible mechanism through which endogenous adenosine might regulate light-induced adjustments in circadian phase. We show that bath application of CHA dose-dependently attenuates AMPA-receptor dependent EPSCs evoked in SCN neurons by optic nerve stimulation. As observed with light-induced phase shifts (Elliott et al., 2001), attenuation of EPSC amplitudeby CHA was blocked by the selective antagonist DPCPX. Furthermore, three lines of evidence are presented that support a presynaptic site of action of adenosine A1 agonists. Specifically, application of CHA at concentrations that substantially reduced the EPSC, induced facilitation of EPSC amplitude to a stimulus train, reduced the rate of spontaneous EPSCs without substantial effect on their amplitude distribution, and did not attenuate responses of retinorecipient SCN neurons to application of exogenous glutamate, the excitatory neurotransmitter released at RHT terminals.Taken together, these data suggest that presynaptic adenosine A1-like receptors located on RHT nerve terminals attenuate the release of glutamate in the hamster SCN, and that this mechanism may be responsible for the reduction in light-induced phase shifts by systemically administered adenosine receptor agonists.
Adenosine Effects in Other Systems
The reduction in synaptic transmission in the SCN by CHA is consistent with observations of adenosine and adenosine receptor agonists in other preparations. Adenosine, acting primarily through adenosine A1-like receptors, has been shown to reduce excitatory synaptic transmission in the hippocampus (Dunwiddie and Hoffer, 1980), in periaqueductal gray neurons (Bagley et al., 1999), and in striatal neurons (Flagmeyer et al., 1997). A reduction of inhibitory synaptic transmission has also been reported in cultured SCN neurons (Chen and van den Pol, 1997), in the substantia nigra (Wu et al., 1995), and in the thalamus (Ulrich and Huguenard, 1995). In the supraoptic nucleus of the hypothalamus, both inhibitory and excitatory synaptic transmission was attenuated by adenosine (Oliet and Poulain, 1999). However, it should be pointed out that Hirai and Okada (1994, 1995) reported enhanced field potentials and increased glutamate release in brain slices containing the superior colliculus, and Nishimura et al. (1990) have reported both excitatory and inhibitory effects of adenosine in hippocampal slices, depending on the concentration.
In most systems for which data are available, adenosine acts presynaptically to inhibit neurotransmission. In the hippocampus, adenosine reduces EPSCs in dentate gyrus neurons without reducing the amplitude of spontaneous miniature EPSCs (Prince and Stevens, 1992). Similarly, Lupica et al. (1992) found that the quantal content of hippocampal EPSPs was reduced by adenosine without altering quantal size. Further, adenosine induces or enhances facilitation of closely spaced synaptic events in the hippocampus (Dunwiddie and Haas, 1985). Flagmeyer et al. (1997), in the striatum, reported paired-pulse facilitation of a low-level excitatory postsynaptic potential but depression for a high level of stimulation. Similarly, in the current report, we show that the synaptic depression usually observed in retinorecipient SCN cells during repeated stimulation is either reduced or reversed (i.e., facilitated) in the presence of adenosine A1 receptor agonists (Fig. 4 and Fig. 5).
The conversion of paired pulse depression to facilitation by adenosine, even as the amplitude of the first pulse was reduced, is intriguing. Presynaptic facilitation and depression at synapses are phenomena thought to reflect differences in intraterminal calcium dynamics during a pulse train (Zucker, 1993). One possible presynaptic action of adenosine is to reduce the inward calcium current (Chen and van den Pol, 1997). It is therefore possible that adenosine reduces depolarization-induced calcium influx into RHT terminals such that, during a brief pulse train, the intraterminal calcium concentration never reaches a level at which calcium buffering is significant. As a result, the concentration of free calcium in the terminal would gradually rise during the pulse train, facilitating the release of glutamate in response to subsequent stimuli.
Possible Postsynaptic Action of CHA
The preponderance of data reported in the current article supports a presynaptic site of action for the effects of adenosine on the RHT neurotransmission. However, in experiments where effects of drugs on the amplitude of spontaneous EPSCs were determined, 2 of 11 cells showed a clear reduction in average EPSC amplitude in the presence of CHA. This observation suggests that SCN neurons may also express postsynaptic adenosine receptors capable of responding to CHA. Obreitan et al. (1995) reported that most hypothalamic neurons in dispersed cell culture showed both pre- and postsynaptic effects of adenosine. However, all of the cells studied in the current report were retinorecipient, while those investigated by Obrietan et al. were largely of undetermined phenotype. It is possible that fewer retinorecipient SCN neurons express postsynaptic A1 receptors than do hypothalamic neurons that differentiate under cell culture conditions.
The results of this study do not exclude the possibility of a postsynaptic action by adenosine on membrane currents. For example, adenosine may modify the activity of postsynaptic NMDA receptors, which are known to play a role in light-induced phase shifts (Colwell et al., 1990; Rea et al., 1993), and whose sensitivity was recently shown to be under circadian regulation (Colwell, 2001; Pennartz et al., 2001). Because the conditions employed in the current study probably excluded the activation of most NMDA receptors [Fig. 1(B)], a postsynaptic effect of CHA on NMDA receptor-gated currents would not have been observed. Similarly, enhancement of a depolarization-activated potassium current by adenosine would not have been detected under our voltage-clamp conditions employed in the current study, but would have nonetheless shunted an excitatory postsynaptic potential evoked by RHT activation in vivo. To address this question, we examined, post hoc, a series of 12 consecutive recordings from SCN neurons and found no net change in input resistance elicited by CHA at the normal holding potential for concentrations from 1 to 50 µM. However, the possibility of postsynaptic effects of CHA at more depolarized membrane potentials remains to be systematically explored.
Modulation of Sleep and Circadian Rhythms by Adenosine
Considerable evidence supports a role for adenosine as a natural sleep promoting substance in the mammalian brain. Infusion of adenosine or selective adenosine A1 receptor agonists into the basal forebrain cholinergic region decreases wakefulness and increases slow wave sleep (Portas et al., 1997). Similarly, introduction of adenosine into the lateral tegmental nucleus decreases wakefulness (Portas et al., 1997). Both systemic and intraventricular administration of an adenosine A1 receptor agonist promote changes in the EEG resembling those observed during sleep deprivation (Bennington et al., 1995). Furthermore, adenosine A1 receptor antagonists, including caffeine and theophylline, promote alertness even after prolonged sleep deprivation (Snyder et al., 1981; Radulovacki et al., 1984). In addition, adenosine A2 agonists were found to increase sleep when introduced near the rostral forebrain (Satoh et al., 1998).
Adenosine accumulates in the basal forebrain during sustained wakefulness, where it appears to promote sleep through the activation of adenosine A1 receptors (Porkka-Heiskanen et al., 1997). Sleep deprivation blocks or attenuates light-induced phase delays in circadian rhythm induced by light exposure during the early subjective night (Mistlberger et al., 1997). We propose that accumulation of adenosine in the SCN region during periods of sustained wakefulness attenuates light-induced phase shifts by inhibiting glutamate release from RHT terminals. Through this mechanism, adenosine could provide information to the circadian clock regarding the state of fatigue of the organism, resulting in an altered response to the daily, light-induced phase adjustment of the clock. In this way, the temporal program of wakefulness and sleep, which is under control of the circadian clock, could be adjusted to accommodate day-to-day variation in the quantity and/or quality of sleep.
Acknowledgments
R.H. was a Senior Fellow of the National Research Council for part of this study. We thank Keith Kelleher for expert technical assistance.
Contract grant sponsor: Air Force Office of Scientific Research; contract grant number: F49620-00-0058 (M.A.R.).
Contract grant sponsor: N.I.M.H.; contract grant number: MH62490 (M.A.R.).
Contract grant sponsor: NINDS; contract grant number: NS36982 (C.C.).
REFERENCES
- Bagley EE, Vaughan CW, Christie MJ. Inhibition by adenosine receptor agonists of synaptic transmission in rat periaqueductal grey neurons. J Physiol. 1999;516:219–225. doi: 10.1111/j.1469-7793.1999.219aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennington JH, Kodali SH, Heller CH. Stimulation of A1 adenosine receptors mimics the electroencephalographic effects of sleep deprivation. Brain Res. 1995;692:79–85. doi: 10.1016/0006-8993(95)00590-m. [DOI] [PubMed] [Google Scholar]
- Blanton MG, Lo Turco JJ, Kriegstein AR. Whole cell recording from neurons in slices of reptilian and mammalian cerebral cortex. J Neurosci Meth. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- Challet E, Turek FW, Laute M, Van Reeth O. Sleep deprivation decreases phase-shift responses of circadian rhythms to light in the mouse: role of serotonergic and metabolic signals. Brain Res. 2001;909:81–91. doi: 10.1016/s0006-8993(01)02625-7. [DOI] [PubMed] [Google Scholar]
- Chen G, van den Pol AN. Adenosine modulation of calcium currents and presynaptic inhibition of GABA release in suprachiasmatic and arcuate nucleus neurons. J Neurophysiol. 1997;77:3035–3047. doi: 10.1152/jn.1997.77.6.3035. [DOI] [PubMed] [Google Scholar]
- Colwell CS. NMDA-evoked calcium transients and currents in the suprachiasmatic nucleus: Gating by the circadian system. Eur J Neurosci. 2001;13:1420–1428. doi: 10.1046/j.0953-816x.2001.01517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell CS, Ralph MR, Menaker M. Do NMDA receptors mediate the effects of light on circadian behavior? Brain Res. 1990;523:117–120. doi: 10.1016/0006-8993(90)91643-u. [DOI] [PubMed] [Google Scholar]
- DeCoursey PJ. Function of a light response rhythm in hamsters. J Cell Comp Physiol. 1964;63:189–196. doi: 10.1002/jcp.1030630208. [DOI] [PubMed] [Google Scholar]
- Ding JM, Chen D, Weber ET, Faiman LE, Rea MA, Gillette MU. Resetting the biological clock: mediation of nocturnal circadian shifts by glutamate and NO. Science. 1994;266:1713–1717. doi: 10.1126/science.7527589. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV. The physiological role of adenosine in the central nervous system. Int Rev Neurobiol. 1985;27:63–139. doi: 10.1016/s0074-7742(08)60556-5. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Haas HL. Adenosine increases synaptic transmission in the in vitro rat hippocampus: Evidence for a presynaptic site of action. J Physiol. 1985;9:365–377. doi: 10.1113/jphysiol.1985.sp015907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Hoffer BJ. Adenine nucleotides and synaptic transmission in the in vitro rat hippocampus. Brit J Pharmacol. 1980;69:59–68. doi: 10.1111/j.1476-5381.1980.tb10883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott KJ, Weber ET, Rea MA. Adenosine A1 receptors regulate the response of the hamster circadian clock to light. Euro J Pharmacol. 2001;414:45–53. doi: 10.1016/s0014-2999(01)00786-5. [DOI] [PubMed] [Google Scholar]
- Flagmeyer I, Haas HL, Stevens DR. Adenosine A1 receptor-mediated depression of corticostriatal and thalamostriatal glutamatergic synaptic potentials in vitro. Brain Res. 1997;778:178–185. doi: 10.1016/s0006-8993(97)01060-3. [DOI] [PubMed] [Google Scholar]
- Gannon RL, Cato MJ, Kelley KH, Armstrong DL, Rea MA. GABAergic modulation of optic nerve-evoked field potentials in the rat suprachiasmatic nucleus. Brain Res. 1995;694:264–270. doi: 10.1016/0006-8993(95)00854-j. [DOI] [PubMed] [Google Scholar]
- Grossman GH, Mistlberger RE, Antle MC, Ehlen JC, Glass JD. Sleep deprivation stimulates serotonin release in the suprachiasmatic nucleus. NeuroReport. 2000;11:1929–1932. doi: 10.1097/00001756-200006260-00024. [DOI] [PubMed] [Google Scholar]
- Hastings MH, Best JD, Ebling FJ, Maywood ES, McNulty S, Schurov I, Selvage D, Sloper P, Smith KL. Entrainment of the circadian clock. Prog Brain Res. 1996;111:147–174. doi: 10.1016/s0079-6123(08)60406-9. [DOI] [PubMed] [Google Scholar]
- Hirai H, Okada Y. Adenosine facilitates glutamate release in a protein kinase-dependent manner in superior colliculus slices. Euro J Pharmacol. 1994;256:65–71. doi: 10.1016/0014-2999(94)90617-3. [DOI] [PubMed] [Google Scholar]
- Hirai H, Okada Y. Adenosine facilitates in vivo neurotransmission in the superior colliculus of the rat. J Neurophysiol. 1995;74:950–960. doi: 10.1152/jn.1995.74.3.950. [DOI] [PubMed] [Google Scholar]
- Jagota A, De La Iglesia HO, Schwartz WJ. Morning and evening circadian oscillations in the suprachiasmatic nucleus in vitro. Nature Neurosci. 2000;3:372–376. doi: 10.1038/73943. [DOI] [PubMed] [Google Scholar]
- Johnson RF, Moore RY, Morin LP. Retinohypothalamic projections in the rat and hamster demonstrated using cholera toxin. Brain Res. 1988;462:301–312. doi: 10.1016/0006-8993(88)90558-6. [DOI] [PubMed] [Google Scholar]
- Kim YI, Dudek FE. Intracellular electrophysiological study of suprachiasmatic nucleus in rodents: Excitatory synaptic mechanisms. J Physiol. 1991;444:269–287. doi: 10.1113/jphysiol.1991.sp018877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein DC, Moore RY, Reppert SM. New York: Oxford University Press; 1991. Suprachiasmatic nucleus: the mind’s clock; 230 pp. [Google Scholar]
- Lupica CR, Proctor WR, Dunwiddie TV. Presynaptic inhibition of excitatory synaptic transmission by adenosine in rat hippocampus: Analysis of unitary EPSP variance measured by whole-cell recording. J Neurosci. 1992;12:3753–3764. doi: 10.1523/JNEUROSCI.12-10-03753.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistlberger RE, Landry GJ, Marchant EG. Sleep deprivation can attenuate light-induced phase shifts of circadian rhythms in hamsters. Neurosci Lett. 1997;238:5–8. doi: 10.1016/s0304-3940(97)00815-x. [DOI] [PubMed] [Google Scholar]
- Moore RY, Lenn NJ. A retinohypothalamic projection in the rat. J Comp Neurol. 1972;146:1–14. doi: 10.1002/cne.901460102. [DOI] [PubMed] [Google Scholar]
- Nelson DE, Takahashi JS. Sensitivity and integration in a visual pathway for circadian entrainment in the hamster (Mesocricetus auratus) J Physiol. 1991;439:115–145. doi: 10.1113/jphysiol.1991.sp018660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura S, Mohri M, Okada Y, Mori M. Excitatory and inhibitory effects of adenosine on the neurotransmitters in the hippocampal slices of guinea pig. Brain Res. 1990;525:165–169. doi: 10.1016/0006-8993(90)91335-e. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Belousov AB, Heller HC, van den Pol AN. Adenosine pre- and postsynaptic modulation of glutamate-dependent calcium activity in hypothalamic neurons. J Neurophysiol. 1995;74:2150–2162. doi: 10.1152/jn.1995.74.5.2150. [DOI] [PubMed] [Google Scholar]
- Oliet SHR, Poulain DA. Adenosine-induced presynaptic inhibition of IPSCs and EPSCs in rat hypothalamic supraoptic nucleus neurones. J Physiol. 1999;520:815–825. doi: 10.1111/j.1469-7793.1999.00815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennartz CM, Hamstra R, Geurtsen AM. Enhanced NMDA receptor activity in retinal inputs to the rat suprachiasmatic nucleus during the subjective night. J Physiol. 2001;532:181–194. doi: 10.1111/j.1469-7793.2001.0181g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science. 1997;276:1265–1268. doi: 10.1126/science.276.5316.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portas CM, Thakkar M, Rainnie DG, Greene RW, McCarley RW. Role of adenosine in behavioral state modulation: a microdialysis study in the freely moving cat. Neurosci. 1997;79:225–235. doi: 10.1016/s0306-4522(96)00640-9. [DOI] [PubMed] [Google Scholar]
- Prince DA, Stevens CF. Adenosine decreases neurotransmitter release at central synapses. Proc Natl Acad Sci USA. 1992;89:8586–8590. doi: 10.1073/pnas.89.18.8586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovacki M, Virus RM, Djuricic-Nedelson M, Green RD. Adenosine analogs and sleep in rats. J Pharm Exp Ther. 1984;228:268–274. [PubMed] [Google Scholar]
- Rea MA. Photic entrainment of circadian rhythms in rodents. Chronobiol Int. 1998;15:395–423. doi: 10.3109/07420529808998699. [DOI] [PubMed] [Google Scholar]
- Rea MA, Buckley B, Lutton LM. Local administration of EAA antagonists blocks light-induced phase shifts and c-fos expression in the hamster SCN. Am J Physiol. 1993;265:R1191–R1198. doi: 10.1152/ajpregu.1993.265.5.R1191. [DOI] [PubMed] [Google Scholar]
- Satoh S, Matsumura H, Hayaishi O. Involvement of adenosine A2A receptor in sleep promotion. Euro J Pharmacol. 1998;351:155–162. doi: 10.1016/s0014-2999(98)00302-1. [DOI] [PubMed] [Google Scholar]
- Snyder SH, Katims JJ, Annau Z, Bruns RF, Daly JW. Adenosine receptors and behavioral actions of methylxanthines. Proc Natl Acad Sci USA. 1981;78:3260–3264. doi: 10.1073/pnas.78.5.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel RGD, Torrie JH. New York: McGraw-Hill; 1980. Principles and Procedures of Statistics; 672 pp. [Google Scholar]
- Ulrich D, Huguenard JR. Purinergic inhibition of GABA and glutamate release in the thalamus: implications for thalamic network activity. Neuron. 1995;15:909–918. doi: 10.1016/0896-6273(95)90181-7. [DOI] [PubMed] [Google Scholar]
- Watanabe A, Moriya T, Nisikawa Y, Araki T, Hamada T, Shibata S, Watanabe S. Adenosine A1-receptor agonist attenuates the light-induced phase shifts and fos expression in vivo and optic nerve stimulation-evoked field potentials in the suprachiasmatic nucleus in vitro. Brain Res. 1996;740:329–336. doi: 10.1016/s0006-8993(96)00881-5. [DOI] [PubMed] [Google Scholar]
- Welsh DK, Logothetis DE, Meister M, Reppert SM. Individual neurons dissociated from rat suprachiasmatic nucleus express independently phased circadian firing rhythms. Neuron. 1995;14:697–706. doi: 10.1016/0896-6273(95)90214-7. [DOI] [PubMed] [Google Scholar]
- White TD, Hoehn K. Release of adenosine and ATP from nervous tissue. In: Stone TW, editor. Adenosine in the nervous system. London: Academic Press; 1991. pp. 173–195. [Google Scholar]
- Wu YN, Mercuri NB, Johnson SW. Presynaptic inhibition of gamma-aminobutyric acid B-mediated synaptic current by adenosine recorded in vitro in midbrain dopamine neurons. J Pharm Exp Therapeut. 1995;273:576–581. [PubMed] [Google Scholar]
- Zucker RS. Calcium and transmitter release at nerve terminals. Biochem Soc Trans. 1993;21:395–401. doi: 10.1042/bst0210395. [DOI] [PubMed] [Google Scholar]