

Abstract
CIITA is a positive regulator of class II major histocompatibility complex gene transcription that has been found to be defective in one of the five complementation groups of class II major histocompatibility complex-negative cell lines. Its N-terminal region is capable of activating transcription from a reporter gene when fused to a DNA binding domain. We have investigated the mechanism of transactivation mediated by the CIITA activation domain by studying its role in the process of transcription initiation and elongation. Specifically the altered specificity TBP (TATA box binding protein) assay has been used to analyze the response of the CIITA activation domain to mutations in TBP known to disrupt its interaction with its associated general factors. Transactivation by CIITA was extremely sensitive to a mutation in TBP that in yeast is known to abolish VP16-mediated transcription but leaves basal transcription unaffected. A TBP mutant defective in interaction with TBP-associated factor TAFII250 also failed to mediate transactivation through the CIITA activation domain. Certain interactions between TBP and general factors that are specifically required for acidic activation domains were also required for CIITA-mediated transactivation to reach its full potential. Finally, like VP16, CIITA was able to stimulate elongation of transcription. Overall the mechanism of transactivation by the human B-cell-specific CIITA is very similar to that mediated by the herpes virus transactivator VP16 in the ways that have been tested.
The human class II major histocompatibility complex (MHC) genes encode cell surface proteins that present peptide antigens to helper T cells for the generation of antibody response (1). The regulation of expression of these genes is extremely important as their inappropriate expression may lead to autoimmune disease, and absence of expression results in type II immunodeficiency or bare lymphocyte syndrome (2, 3). The expression of class II MHC genes is subject to tight developmental and tissue-specific regulation (4). In B cells, expression is initiated after Ig rearrangement and surface expression (5) and is lost with the differentiation of B cells into antibody-secreting plasma cells (6). Certain cell types such as macrophages become competent for antigen presentation after induction of class II MHC genes under the influence of cytokines such as interferon γ (7). Class II MHC gene expression is controlled at the level of transcription. Proximal promoters of all class II genes contain sequence motifs (X, Y, J, and S boxes) with conserved nucleotide sequences, and a number of proteins that bind to some of these promoter elements and regulate transcription either positively or negatively have been described (8, 9). The availability of MHC class II-negative cell lines, patient derived as well as generated in vitro, that fall into different genetic complementation groups (10–13) has led to the identification of two genes, CIITA (14) and RFX5 (15), by complementation cloning that were also defective in patient-derived cell lines. Although RFX5 is a DNA binding protein, CIITA is not and, hence, is presumed to function via protein–protein interactions, i.e., as a coactivator. In an accompanying paper, the direct interaction of CIITA with RFX5 is shown (16). The N-terminal region of CIITA has been shown to possess an activation domain and can communicate with the general transcription machinery to drive transcription (17, 18). Some mechanistically important aspects of this latter interaction have been studied herein.
Transcription factors are proposed to function by physically interacting with one or more proteins in the basal transcription apparatus that contains several basal transcription factors including TFIIA, TFIIB, TFIID [TATA box binding protein (TBP) with TBP-associated factors or TAFs], TFIIE, TFIIF, and TFIIH (for review, see refs. 19 and 20). Among the proteins in the basal apparatus that have been shown to interact with different activators are TBP (21, 22), TFIIB (23), TAFs (24–26), RAP74, the large subunit of TFIIF (27), TFIIH (28), etc. Transcription activation is a multistep process that includes initiation, elongation, and termination (29). Also, the expression of some of the general factors could be developmentally regulated. For example, the small subunit of TFIIA is transiently up-regulated in Drosophila photoreceptor precursors and this up-regulation is independent of and is required for the Ras-activated pathway of photoreceptor determination (30). Therefore, it is essential to characterize the nature of interaction of an activator with the basal transcription machinery.
This study addresses the role of CIITA in the step of transcription initiation and elongation. CIITA belongs to a class of activators called acidic activators because the transactivation domain of CIITA contains a high percentage of acidic residues (17). Although several activators have been grouped on the basis of the nature of the amino acids found predominantly in their activation domains, the functional basis of this classification is not understood. Furthermore, there may be differences in the ways different activators from a particular class, for example, the acidic activators, interact with the general transcription complex (31). It has been shown that class II MHC promoters could be stimulated by the acidic activation domains of the viral transactivators VP16 (herpes simplex) and E1a (adenovirus) but not by those of the mammalian and yeast transactivators ATF-2 and GAL4, respectively (32). This could be due to specific structural differences or to differences in the strength of the individual activation domains. Therefore, it is important to study the interaction of the human B-cell-specific CIITA activation domain with the general transcription complex. To our knowledge, this study represents the first step in that direction and the approach taken is to make use of several TBP mutations to perturb transactivation by CIITA and thereby attempt to understand its mechanism of action. TBP in view of its association with other general factors, such as TAFs, TFIIB, and TFIIA, occupies a central place in this complex. TBP and associated factors have both been shown to function as targets of activators (21, 33). Therefore, mutations in different regions of TBP can be expected to differentially effect its interaction with one or more proteins with which it is associated and accordingly influence the outcome of the response of an activator. While some of these interactions are disrupted significantly by single (34) or double (35) point mutation(s), others require specific combinations of mutations on the surface of the molecule (36). Mutations in TBP were used that specifically affect its interaction with TFIIB, the TAF complex, and TFIIA to individually assess the importance of each TBP-associated proteins for transactivation by CIITA activation domain. Also, the ability of CIITA to stimulate transcription elongation was studied by testing whether it could act synergistically with HIV Tat in transactivation.
MATERIALS AND METHODS
Cells and Cell Culture.
Raji (CCL86, American Type Culture Collection) is a Burkitt lymphoma B cell line expressing high levels of all three isotypes of MHC class II proteins. HeLa (CCL2, American Type Culture Collection) is a cervical carcinoma cell line. BLS-2 is an Epstein–Barr virus-transformed B cell line derived from MHC class II-deficient patients (13). Raji and BLS-2 cells were grown in RPMI 1640 medium (GIBCO/BRL), supplemented with 10% bovine calf serum (HyClone), 2 mM l-glutamine, penicillin (100 units/ml), and streptomycin (100 units/ml). HeLa cells were grown in Dulbecco’s modified Eagle’s medium with the same additives used for Raji cells.
Plasmid Constructions.
All oligonucleotides were purchased from Life Technologies. The cDNA for CIITA was cloned by reverse transcription-coupled PCR using the polymerase pfu (Stratagene). Raji cell poly(A)+ RNA was made with the FastTrack mRNA isolation kit (Invitrogen). Reverse transcription was carried out with oligo(dT) and Superscript II reverse transcriptase (GIBCO/BRL). The cDNA thus obtained was used for PCRs. Two sets of primers were used to get overlapping 5′ and 3′ PCR products. The 5′ product was digested with EcoRI and RsrII and the 3′ product was digested with XhoI and RsrII. The primer pairs used were as follows: for the 5′ product, 5′ primer, 5′-ATAGAATTCATGCGTTGCCTGGCT CCACGC-3′, and 3′ primer, 5′-TGCCGGTCCGCACGTGCTGTC-3′; for the 3′ product primer, 5′ primer, 5′-CACAGCACGTGCGGACCGGCA-3′, and 3′ primer, 5′-ATACTCGAGTCTCAGGCTGATCCGTGAATC-3′. The digested products were ligated to pcDNA3 digested with EcoRI and XhoI. The CIITA expression plasmid thus obtained was tested for function by cell surface staining of stable transfectants of MHC class II-negative BLS-2 cells. The construct for making the fusion protein containing the N-terminal 210 amino acids of CIITA fused to the Gal4 DNA binding domain (amino acids 1–147) was obtained by ligating a PCR product (digested with EcoRI and BamHI) corresponding to that region of CIITA into the EcoRI–BamHI site of the vector pBXG1. The primer pair used to generate the PCR product was 5′ primer, 5′-ATAGAA TTCATGCGTTGCCTG GCTCCACGC-3′, and 3′ primer, 5′-CGGGATCCCATGGG AATCTGGTCGGTTT-3′.
The construct used for making the glutatione S-transferase (GST) CIITA activation domain fusion protein GST–CIITA-(17–148) was made by ligating a PCR product (digested with EcoRI and XhoI) into the EcoRI–XhoI site of the vector pGEX-5x-1 (Pharmacia). The primer pair was 5′ primer, 5′-CCGGAATTCCAAGGCAGCTCACAGTGT-3′, and 3′ primer, 5′-CCGCTCGAGTGGGAAGGGTCTTTTCTGACT-3′. The plasmids pBXG1 and pBXGal-VP16 were a gift of M. Ptashne (37). The TFIIB mutant plasmids pETIIBNM1 (R185E/R193/E) and pETIIBNM2 (K189E/K200E) were a gift of Michael Green (23). The wild-type TFIIB expression plasmid was a gift of M. Ptashne. The plasmids pETβTAFII32, pTβdTAFII40, pTβTAFII60, and pTβhTAFII70 were a gift of Robert Tjian (University of California, Berkeley). The plasmid containing hTAFII30 cDNA in pBluescript SK(−) was a gift of Pierre Chambon (38). This was used to generate a PCR product of the hTAFII30 coding sequence with 5′ EcoRI and 3′ XhoI adaptors for subcloning into EcoRI–XhoI site of the vector pCITE-3a (Novagen). The sequence of the primer pair is as follows: 5′ primer, 5′-ATAGAATTCATGAGCTGCAGCGGCTCCGGC-3′, and 3′ primer, 5′-ATACTCGAGTCAGGTGAAGTAGTGCGGCTTC-3′.
The altered specificity TBP coding sequence containing expression plasmid pCGNTBPAS, the c-fos (−56)[4xgal] reporter, the pCGNTBPAS with the mutation combinations (H1′+H2+S3′/S4′) and (H1′+S3/S4+S3′S4′) were a gift of W. P. Tansey and W. Herr (Cold Spring Harbor Laboratory). Additional mutations were made in the TBPAS coding region by using the Sculptor in vitro mutagenesis system (Amersham). Single-stranded DNA was obtained from pCGNTBPAS using helper phage (Stratagene). As the noncoding strand was recovered from the phage, mutagenic oligonucleotides formed parts of the coding strand and carried the desired base changes. The sequences of the mutagenic primers are as follows (base changes are shown in lowercase type): L283K, 5′-TATGAGCCAGAGaaATTTCCTGGTTT-3′; K232T/Y233A, 5′-ACTGGCAGCAAGAAcAgcTGCTAGAGTTGTAC-3′. The plasmid p62(TFIIH) was a gift of Ronald Drapkin and Danny Reinberg (University of Medicine and Dentistry of New Jersey, Piscataway). The plasmids pSV-HIV1-tat, pHKGSp1, and pGal5-HIV2 CAT were a gift of Justin Blau and David Bentely (Amgen).
GST Pull-Down Assays.
pGEX5X-1 GST expression plasmids were transformed into bacteria (HB101). Overnight cultures were used to inoculate larger cultures at 37°C and isoproppyl β-d-galactoside was added (to 1 mM, final concentration) and the bacteria were allowed to grow for an additional 4 hr. Cells were harvested by centrifugation, resuspended in the binding buffer (20 mM Tris/150 mM NaCl/1 mM dithiothreitol/1 mM phenylmethylsulfonyl fluoride), and freeze–thawed three times. Sonication was performed subsequently, and insoluble material was separated by centrifugation. The proteins GST and GST–CIITA-(17–148) were purified by binding to glutathione-agarose (Sigma) at 4°C for 1 hr, washing thoroughly with the binding buffer, and eluting with 10 mM reduced glutathione. Labeled (35S) proteins to be tested for binding to GST–CIITA-(17–148) were translated in vitro by using the transcription/translation-coupled reticulocyte lysate system (Promega). For a GST interaction assay, 2 μg of GST–CIITA-(17–148) or the control protein (GST) was bound to glutathione beads and incubated at 4°C for 1 hr with shaking. Excess protein was washed away with the binding buffer. The beads were resuspended in binding buffer containing 0.2% Nonidet P-40, and labeled in vitro translated protein was added. This was followed by incubation with shaking at 4°C for 2 hr and repeated washings with the binding buffer containing 0.2% Nonidet P-40. The beads with bound proteins were boiled in SDS/PAGE loading buffer and analyzed by Coomassie staining and autoradiography.
Altered Specificity TBP Assays.
Altered specificity TBP assays were performed essentially as described by Tansey et al. (36). Transient transfection in HeLa cells was carried out by the calcium phosphate coprecipitation method (39). The plasmids common for each transfection were 2 μg of c-fos (−56)[4xGAL] reporter, 224 ng of pCGNhTBPAS, 2 μg of pCMV βgal, and various amounts of pBluescript to a total DNA concentration of 25 μg. The amount of pCGNhTBPAS DNA to be used was determined empirically by titration to be in the linear range and below the level at which squelching is observed. Quantitative Western blot analyses of protein extracts from transfected cells with the anti-hemagglutinin antibody 12CA5 (Babco, Richmond, CA) was employed to ensure that equivalent amounts of different mutant proteins were being made for the amounts of DNA used in each transfection. Single and double point mutations were found not to affect the levels of protein expression within the cell. The amount of pBXG1-CIITA and pBXG1-VP16 DNA necessary to achieve high levels of activation was determined empirically to be 320 ng. Approximately 7.5 × 106 cells were taken on each plate 16–20 hr prior to transfection. Two hours before transfection, the medium was changed. The calcium phosphate–DNA precipitate was added to the cells and incubated at 37°C with 5% CO2/95% air for 16 hr. The cells were washed twice with phosphate-buffered saline, fresh medium was added, and incubation continued for another 48 hr. Cells were harvested and chloramphenicol acetyltransferase (CAT) activities were determined as reported (40). Transfection with each plasmid was performed at least twice in triplicate and the quantitative analysis was done with phosphoimaging equipment (Fuji). All transfections included 2 μg of pCMVβGal (41) as an internal control for transfection efficiency. β-Galactosidase activity was measured spectrophotometrically.
HIV-1 Tat Synergy Assay.
HeLa cells were transfected transiently as described above with 8 μg of the reporter pGal5-HIV2CAT and 1.5 μg of Gal4 expression plasmid encoding activator proteins. pSV-HIV-tat (1.5 μg) was included in samples where synergy of transactivation was being tested. pCMVβGal (2 μg) was used in each transfection as an internal control for transfection efficiency and β-galactosidase activity was measured spectrophotometrically.
RESULTS AND DISCUSSION
CIITA Interacts with TFIIB.
Among the mutations chosen for study was the TBP mutant L189K, one of the three mutants found to be defective specifically for activated transcription mediated by the VP16 activation domain but not for basal transcription (34) in an in vitro transcription assay with yeast TBP. The mutation involved a conserved TBP residue, and therefore, it was generated in the human TBP. In addition, an established in vivo altered specificity TBP (TBPAS) transfection assay in HeLa cells was used (36, 42). The altered specificity TBP assay uses a TBP derivative with triple amino acid substitution in its DNA binding region. TBPAS has a relaxed DNA binding specificity and can recognize an altered TGTAAA box in addition to the canonical TATAAA box. When used in conjunction with a reporter containing TGTAAA box, the combination allows one to bypass the contribution to transcriptional activity from the endogenous TBP (42). The mutation in the larger human TBP that correspond to the yeast mutation L189K is L283K (Fig. 1A). The CIITA activation domain fused to the Gal4 DNA binding domain that can activate transcription from a reporter plasmid with a wild-type TBPAS was totally defective in activating transcription through the TBP mutant L283K (Fig. 2).
Figure 1.

Structure of TBP indicating the position of the mutations. The diagram was obtained with the program rasmol (52) with coordinates of the yeast TBP (53). For a comparison of the numbering of the residues of yeast and human TBP see ref. 54. The mutations H2, H1′, S3/S4, and S3′/S4′ are alanine-scan mutations described by Tansey et al. (36). H2 is a triple mutation (R231A, R235A, and R239A). H1′ is a double mutation (R269A and E271A). S3/S4 is a double mutant (E206A and R208A). S3′/S4′ is a double mutant (K297A and R299A). L283K is a mutation equivalent to the yeast mutation L189K (34) and K232T/Y233A is equivalent to the yeast mutation N2–1 (35). The use of the mutant L283K is based on the assumption that it is function for basal transcription as has been demonstrated for the yeast counterpart.
Figure 2.
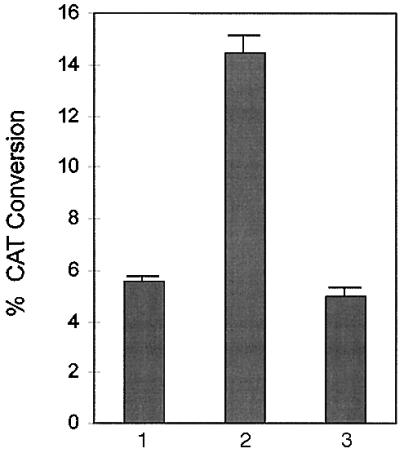
Altered specificity assay in HeLa cells to test the effect of the TBP mutation L283K in CIITA-mediated transactivation. Histograms represent the following transfections. Lane 1; transfections to check the levels of CIITA-mediated transactivation due to endogenous TBP. Each sample had 2 μg of c-fos(−56)TGTAAAA(4xgal reporter) construct and 320 ng of pBXG1CIITA(1–210) effector construct. Lane 2; transfections in triplicate that contained 224 ng of the altered specificity TBP construct pCGNhTBPAS-WT in addition to the reporter and the effector constructs. Lane 3; transfections that contained 224 ng of the pCGNhTBPAS with the mutation L283K in addition to the reporter and the effector constructs. For reference, the CIITA activation domain in this assay had about 20% of the activity of the VP16 activation domain, e.g., 8.9% CAT conversion with CIITA and 43.5% with VP16.
We can draw a parallel with the mechanism suggested for the defect caused in the VP16-mediated transactivation due to this mutation (34). Under certain special conditions, yeast TBP L189K was defective in forming the TBP–TFIIB–DNA complex but could form a TBP–TFIIB–TFIIA–DNA complex stable for electrophoresis (34). It was suggested that the mutant TBP–TFIIB complex had reduced DNA binding potential that could be sufficiently compensated for basal transcription to take place by the inclusion of TFIIA but not for activated transcription by the VP16 activation domain. Also, yeast TBP L189K had been shown to be markedly deficient in recruiting TFIIB into the initiation complex (34). This is consistent with the observation that TBP L189 is involved in interaction with the N-terminal domain of TFIIB (43) and a mutation in this residue could destabilize this interaction. The precise mechanism of how this mutation specifically affects activator-dependent transcription is not understood but has been suggested to arise from the necessity that both domains of TFIIB bind DNA and the simultaneous requirement that the VP16 activation domain contact both domains of TFIIB. The mutation L189K probably destabilizes the interaction of the N-terminal domain of TFIIB with TBP, thereby preventing formation of a stable TBP–TFIIB–DNA complex that would allow both domains of TFIIB to interact with the activation domain of VP16.
This complex mechanism has important implications for how the CIITA activation domain is affected by a mutation in human TBP, TBP L283K. The fact that this TBP mutant is inactive with CIITA (Fig. 2) strongly suggests a direct involvement of TFIIB in CIITA-mediated transactivation, and therefore, the ability of CIITA activation domain to interact with TFIIB was tested. Binding experiments performed with a bacterially produced protein containing GST fused to amino acids 17–148 of CIITA and with in vitro-translated TFIIB showed that, indeed, there was an interaction between the two proteins (Fig. 3). Moreover, it was found that two double point mutants of TFIIB, R185E/R193E and K189E/K200E, which have been shown to be defective in their interaction with the VP16 activation domain and also to be defective in supporting activated transcription mediated by the VP16 activation domain (23), did not interact with the CIITA activation domain. Thus TFIIB could be an important target for CIITA in the transcription machinery. TFIIB has been shown to interact with activators other than those of the acidic type such as CTF1 (44) but it is unclear at present how general this interaction is for transcriptional activation.
Figure 3.

Specific interaction between immobilized CIITA activation domain with TFIIB. [35S]Methionine-labeled TFIIB and TFIIB mutants were incubated with glutathione-agarose bound to either the control GST protein or a GST–CIITA-(17–148) fusion protein. After repeated washings, the agarose beads with bound proteins were boiled in SDS/PAGE buffer and analyzed by SDS/PAGE and autoradiography. Lanes 1–3 are inputs for the [35S]methionine-labeled TFIIB molecules K189E/K200E, R185E/R193E, and TFIIB-WT, respectively, without binding to beads or washing. Lanes 4–9 represent the results of the GST interaction assays. The reason for the slightly faster migration of TFIIB-WT released from the glutathione bead compared with the migration of the same protein as input (lane 3) is not clear.
TBP–TAF Complex Interaction Is Also Essential for CIITA-Mediated Transactivation.
In addition to TFIIB, a complex of proteins called the TAFs are associated with TBP, and different members of this complex may serve the function of bridging the activation domain of a given activator with TBP (25). The question arises whether TFIIB (which can associate with both TBP and CIITA and, therefore, act as a bridge) is solely required or whether CIITA-mediated transactivation depends also on other molecules, such as the TAFs, that are thought to perform a similar bridging function. The system of TBP alanine-scan mutants (36) and altered specificity TBP transcription assay was employed to address this question. These mutant TBP molecules carry several amino acid changes across the surface of TBP. Also, since these mutants were characterized for their binding to TFIIB, the effects of disrupting the TBP–TFIIB interaction could be distinguished from that of disrupting TBP–TAF interaction in transcription activation by CIITA. According to our present knowledge, human TAFII250 binds directly to TBP and forms a scaffold for the assembly of TFIID (TBP–TAF complex) (25), and mutations in TBP that disrupt its binding to TAFII250 would prevent the formation of a complete TBP–TAF complex. Such mutant TBP molecules should be impaired in supporting activated transcription if TAFs were the sole molecules to bridge the activation domain with TBP, but they may or may not be impaired if another molecule such as TFIIB could perform the bridging function. However, the TBP mutant (H1′+H2+S3′/S4′) (ref. 36; H and S represent the helices and strands of TBP, respectively; Fig. 1B), which is severely impaired in its interaction with TAFII250 and produces consistent effects on transcription regardless of the nature of the activation domain, was totally nonfunctional in mediating activated transcription through CIITA activation domain (Fig. 4). Therefore, the integrity of TBP–TAF complex appears to be absolutely essential for CIITA-mediated transcription. This occurs despite the fact that TFIIB in view of its interaction with both TBP and CIITA activation domain could theoretically serve the bridging function mentioned above. Thus interaction with TFIIB and the integrity of the TBP–TAF complex are both required for CIITA mediated transcription, and therefore, functions in addition to bridging of the activation domain and TBP are probably being performed by TFIIB, TAFs, or both.
Figure 4.
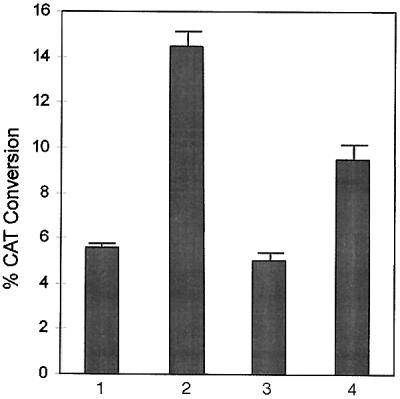
Altered specificity TBP assay in HeLa cells to test the effect of TBP mutations H1′+H2+S3′/S4′ and H1′+S3/S4+S3′/S4′ on the transactivation potential of CIITA activation domain. Histograms represent the following transfections. Lane 1; transfections to check the levels of CIITA-mediated activation due to endogenous TBP. c-fos(-56)TGTAAAA(4xgal) reporter construct (2 μg) and 320 ng pf PBGX1CIITA(1–210) effector construct was used for each transfection. The same amounts of the reporter and effector plasmids were used for the remaining transfections. Lane 2; transfections to check the levels of CIITA-mediated activation achieved by the addition of 224 ng of pCGNhTBPAS-WT, the expression plasmid for the altered specificity TBP. Also shown are transfections with the mutant altered specificity TBP molecules H1′+H2+S3′/S4′ (560 ng, lane 3) and H1′+S3/S4+S3′/S4′ (560 ng, lane 4), respectively. These amounts of the two combinatorial TBP mutants were previously determined (36) by quantitative Western blotting to result in expression levels equivalent to that obtained with 224 ng of TBPAS-WT plasmid.
Direct interaction of the CIITA activation domain with different TAFs of human (h) and Drosophila (d) origin was also tested. The CIITA activation domain interacted with dTAFII40 and dTAFII60 and their human homologues hTAFII32 and hTAFII70 (Fig. 5). The CIITA activation domain did not interact with hTAFII30 (Fig. 5a), the TAF reported to be specifically involved in interacting with estrogen receptor activation domain and mediating estrogen receptor-dependent transcription (38). dTAFII40 and its human counterpart hTAFII32 physically interact with and have been shown to mediate VP16-dependent transactivation (45, 46) and dTAFII40 and dTAFII60 can physically interact with and mediate transactivation through the p53 activation domain (26). Thus, CIITA interacts with specific polypeptides in the TAF complex shown to be involved in mediating activation. hTAFII32, hTAFII70, or both may be involved in mediating CIITA interactions with the TAF complex, which as shown above is required to interact with TBP via TAFII250 to activate transcription.
Figure 5.

Specific interaction between immobilized CIITA activation domain and TAFs. [35S]Methionine-labeled Drosophila and human TAFs were incubated with glutathione-agarose beads bound to either the control GST protein or a GST–CIITA-(17–148) fusion protein. After extensive washings, the agarose beads were boiled in SDS/PAGE buffer and bound proteins were analyzed by SDS/PAGE and autoradiography. (a) Binding of the CIITA activation domain was tested with labeled dTAFII40 (lanes 1–3) or hTAFII32 (lanes 4–6). The interactions of the [35S]methionine-translated proteins with GST (lanes 3 and 6) is insignificant compared with that with GST–CIITA (lanes 2 and 5). (b) Binding of CIITA activation domain to dTAFII60 (lanes 1–3) and hTAFII70 (lanes 4–6).
A VP16 Activation Domain-Specific Defective Combinatorial TBP Mutant Is Partially Defective in CIITA-Mediated Activation.
The TBP mutant (H1′+S3/S4+S3′/S4′; Fig. 1B) predominantly affects activation by the VP16 activation domain and much less activation by the proline-rich CTF1 and the glutamine-rich Oct-2 activation domains (36). The use of this TBP mutant leads to a 55% loss of CIITA-mediated transactivation (Fig. 4). The strength of the interaction between this mutant TBP and TAFII250 is reduced but is perhaps sufficiently strong for activation, given the fact that the response of some activators to this mutant TBP is largely unaffected (36). Therefore, functioning of TBP is probably altered in a way detrimental to only a particular class of activators. A clear understanding of the basis of the effect of this mutation may shed light on the mechanism of activation by CIITA or by acidic activation domains in general. Another TBP mutant namely, N2-1 (K138T/Y139A in yeast and K232T/Y233A in human TBP; Fig. 1A) reported to be defective for responding to acidic activators in yeast due to a defect in association with TFIIA (35) was also tested. CIITA and VP16 mediated activation as measured by altered specificity TBP assay were reduced by 33% and 39% respectively (data not shown) suggesting that maximal activation through CIITA and VP16 requires TBP-TFIIA interaction.
CIITA Activation Domain Is Capable of Stimulating Transcription Elongation.
The ability of CIITA activation domain to stimulate transcription elongation was tested next. Numerous examples document the existence of rate limiting steps after initiation of transcription (48, 49). Promoter-proximal pausing appears to be quite widespread, and therefore, the potential of an activator to stimulate transcription elongation is an important aspect of its function (50). In a broad sense, all nucleotide addition steps after initiation could be termed elongation. The ability or otherwise of an activation domain to act synergistically with HIV-1 Tat protein has been used to test whether the CIITA activation domain could stimulate elongation (51). Like the VP16 activation domain, the CIITA activation domain could not act synergistically with HIV-1 Tat (Fig. 6a). Under the same conditions the SP1 activation domain showed synergy with HIV-1 Tat. Since, in this assay, the only Gal4 fusion protein that can exhibit synergy is the one where the Gal4 fusion protein and HIV-1 Tat stimulate distinct steps of transcription (initiation and elongation), the results suggest that like VP16 the CIITA activation domain can stimulate elongation on its own and, hence, cannot act synergistically with HIV-1 Tat.
Figure 6.

Synergy between different activation domains and HIV-1 Tat. (a) HeLa cells were transfected with pGal5-HIV2 CAT, pCMVβgal, and different Gal4 fusion activation domains with or without pSV-HIV-1 Tat. CAT activities were normalized by the spectrophotometrically determined β-galactosidase activity. The relative percent CAT conversions plus and minus pSV-HIV-1 Tat (from which the histograms were derived were as follows: SP1, 4.9 and 0.13; VP16, 10.0 and 3.2; CIITA, 10.9 and 6.0. The 36-fold activation observed in the presence of HIV-1 Tat is lower than the levels reported in the literature (50-fold approximately) (51). (b) Binding of [35S]methionine-labeled p62 subunit of TFIIH to the activation domain of CIITA immobilized on glutathione beads. The experiments were performed as described in Figs. 3 and 5. The lanes represent 35S labeled p62 alone (lane 1) or its interaction with GST and GST-CIITA (lanes 2 and 3, respectively). The background interaction between GST and p62 has been noted (51).
Stimulation of elongation, analogous to that of initiation, is thought to involve interaction with general transcription factors. A correlation has been demonstrated between an activator’s ability to stimulate elongation and its binding to the multisubunit general factor TFIIH, specifically the p62 subunit of TFIIH (51). This could be a way of recruiting TFIIH that through its kinase activity could phosphorylate the polymerase II large subunit C-terminal domain and thereby stimulate elongation (51). The CIITA activation domain immobilized as a GST fusion protein bound the 35S-labeled p62 subunit of TFIIH (Fig. 6b). This is consistent with observations in the literature that activation domains that stimulate elongation such as VP16 and TAT-(1–48) can interact with p62 whereas those that could not stimulate elongation such as Sp1 and CTF could not interact with p62.
In conclusion, the results presented herein indicate that the CIITA activation domain stimulates transcription by facilitating the initiation and the elongation step of transcription. Whether an activator can interact with more than one general transcription factor simultaneously is unknown. A more likely possibility would be that it can interact sequentially with different individual general factors and facilitate steps in transcription that are temporally separated. The present results open multiple avenues of investigation to dissect CIITA mediated transcription activation. In addition to these results on the interaction and consequences thereof of the CIITA activation domain with the general transcription complex, in an accompanying paper (16), we further show that CIITA that is not a DNA binding protein interacts with DNA through the intermediacy of RFX5 that binds to the X element of the class II MHC promoters.
Acknowledgments
We are very grateful to Christopher Sears for his help in making figures. This work was supported by National Institutes of Health Grant CA47554 and by fellowships of the Irvington Foundation for Medical Research to S.K.M. and of the American Cancer Society to T.S.
ABBREVIATIONS
- MHC
major histocompatibility complex
- TBP
TATA box binding protein
- TAF
TBP-associated factor
- GST
glutathione S-transferase
- CAT
chloramphenicol acetyltransferase
References
- 1.Kappes D, Strominger J L. Annu Rev Biochem. 1988;57:991–1028. doi: 10.1146/annurev.bi.57.070188.005015. [DOI] [PubMed] [Google Scholar]
- 2.Bottazo G F, Dean B M, McNally J M, McKay E H, Swift P G, Gamble D R. N Engl J Med. 1985;313:353–360. doi: 10.1056/NEJM198508083130604. [DOI] [PubMed] [Google Scholar]
- 3.Mach B, Steimle V, Reith W. Immunol Rev. 1994;138:207–221. doi: 10.1111/j.1600-065x.1994.tb00853.x. [DOI] [PubMed] [Google Scholar]
- 4.Glimcher L H, Kara C J. Annu Rev Immunol. 1992;10:13–50. doi: 10.1146/annurev.iy.10.040192.000305. [DOI] [PubMed] [Google Scholar]
- 5.Lala P K, Johnson G R, Battye F L, Nossal G V J. J Immunol. 1979;122:334–341. [PubMed] [Google Scholar]
- 6.Halper J, Fu S M, Wang C Y, Winchester R, Kunkel H G. J Immunol. 1978;120:1480–1484. [PubMed] [Google Scholar]
- 7.King D P, Jones P P. J Immunol. 1983;131:315–318. [PubMed] [Google Scholar]
- 8.Liou H-C, Boothby M R, Finn P W, Davidon R, Nabavi N, Zelezink-Le N J, Ting J P Y, Glimcher L H. Science. 1990;247:1581–1584. doi: 10.1126/science.2321018. [DOI] [PubMed] [Google Scholar]
- 9.Sugawara M, Scholl T, Ponath P D, Strominger J L. Mol Cell Biol. 1994;14:8438–8450. doi: 10.1128/mcb.14.12.8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Z, Accola R S, Pious D, Zegers B J M, Strominger J L. EMBO J. 1988;7:1965–1972. doi: 10.1002/j.1460-2075.1988.tb03034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hume C R, Lee J S. Hum Immunol. 1989;26:228–309. doi: 10.1016/0198-8859(89)90007-4. [DOI] [PubMed] [Google Scholar]
- 12.Benichou B, Strominger J L. Proc Natl Acad Sci USA. 1991;88:4285–4288. doi: 10.1073/pnas.88.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seidl C, Saraiya C, Ofterweil Z, Fu Y P, Lee J S. J Immunol. 1992;148:1576–1584. [PubMed] [Google Scholar]
- 14.Steimle V, Otten L A, Zuffery M, Mach B. Cell. 1993;75:135–146. [PubMed] [Google Scholar]
- 15.Steimle V, Durand B, Barras E, Zufferey M, Hadam M, Mach B, Reith W. Genes Dev. 1995;9:1021–1032. doi: 10.1101/gad.9.9.1021. [DOI] [PubMed] [Google Scholar]
- 16.Scholl T, Mahanta S K, Strominger J L. Proc Natl Acad Sci USA. 1997;94:6320–6334. doi: 10.1073/pnas.94.12.6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reily J L, Westerheide S D, Price J A, Brown J A, Boss J M. Immunity. 1995;2:533–543. doi: 10.1016/1074-7613(95)90033-0. [DOI] [PubMed] [Google Scholar]
- 18.Zhou H, Glimcher L H. Immunity. 1995;2:545–553. doi: 10.1016/1074-7613(95)90034-9. [DOI] [PubMed] [Google Scholar]
- 19.Zawel L, Reinberg D. In: Progress in Nucleic Acid Research and Molecular Biology. Cohn W E, Moldave K, editors. San Diego: Academic; 1993. pp. 67–108. [DOI] [PubMed] [Google Scholar]
- 20.Triezenberg S J. Curr Opin Genet Dev. 1995;5:190–196. doi: 10.1016/0959-437x(95)80007-7. [DOI] [PubMed] [Google Scholar]
- 21.Ingles C J, Shales M, Cress W D, Triezenberg S J, Greenblatt J. Nature (London) 1991;351:588–590. doi: 10.1038/351588a0. [DOI] [PubMed] [Google Scholar]
- 22.Lieberman P M, Berk A J. Genes Dev. 1991;5:2441–2554. doi: 10.1101/gad.5.12b.2441. [DOI] [PubMed] [Google Scholar]
- 23.Roberts S G E, Ha Ilho, Maldonado E, Reinberg D, Green M. Nature (London) 1993;363:741–744. doi: 10.1038/363741a0. [DOI] [PubMed] [Google Scholar]
- 24.Goodrich J A, Hoey T, Thut C J, Admon A, Tjian R. Cell. 1993;75:519–530. doi: 10.1016/0092-8674(93)90386-5. [DOI] [PubMed] [Google Scholar]
- 25.Chen J -L, Attradi L D, Verrijer C P, Yokomori R, Tjian R. Cell. 1994;79:93–105. doi: 10.1016/0092-8674(94)90403-0. [DOI] [PubMed] [Google Scholar]
- 26.Thut C J, Chen J -L, Klemm R, Tjian R. Science. 1995;265:100–104. doi: 10.1126/science.7809597. [DOI] [PubMed] [Google Scholar]
- 27.Joliot V, Demma M, Prywes R. Nature (London) 1995;373:632–635. doi: 10.1038/373632a0. [DOI] [PubMed] [Google Scholar]
- 28.Xiao H, Pearson A, Coulombe B, Truant R, Zhang S, Regier J L, Triezenberg J, Reinberg D, Flores O, Ingles C J, Greenblatt J. Mol Cell Biol. 1994;14:7013–7024. doi: 10.1128/mcb.14.10.7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goodrich J, Tjian R. Cell. 1994;77:145–156. doi: 10.1016/0092-8674(94)90242-9. [DOI] [PubMed] [Google Scholar]
- 30.Zeidler M P, Yokomori K, Tjian R, Mlodzik M. Genes Dev. 1996;10:50–59. doi: 10.1101/gad.10.1.50. [DOI] [PubMed] [Google Scholar]
- 31.Berger S L, Pine B, Silverman N, Marcus G A, Agapite J, Regier J L, Triezenberg S J, Guarente L. Cell. 1992;70:251–265. doi: 10.1016/0092-8674(92)90100-q. [DOI] [PubMed] [Google Scholar]
- 32.Riely J L, Boss J M. J Immunol. 1993;151:6942–6953. [PubMed] [Google Scholar]
- 33.Kerr L D, Ransome L J, Wamsley P, Schmitt M J, Boyer T G, Zhou Q, Berk A, Verma I. Nature (London) 1993;365:412–419. doi: 10.1038/365412a0. [DOI] [PubMed] [Google Scholar]
- 34.Kim T K, Hashimoto S, Kelleher R J, III, Flanagan P M, Kornberg R D, Horikoshi M, Roeder R G. Nature (London) 1994;369:252–255. doi: 10.1038/369252a0. [DOI] [PubMed] [Google Scholar]
- 35.Stargell L A, Struhl K. Science. 1995;269:75–78. doi: 10.1126/science.7604282. [DOI] [PubMed] [Google Scholar]
- 36.Tansey W P, Ruppert S, Tjian R, Herr W. Genes Dev. 1994;8:2756–2769. doi: 10.1101/gad.8.22.2756. [DOI] [PubMed] [Google Scholar]
- 37.Sadowski I, Ma J, Triezenberg S, Ptashne M. Nature (London) 1988;335:563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- 38.Jacq X, Brou C, Lutz Y, Davidson I, Chambon P, Tora L. Cell. 1994;79:107–117. doi: 10.1016/0092-8674(94)90404-9. [DOI] [PubMed] [Google Scholar]
- 39.Tanaka M, Lai J -S, Herr W. Cell. 1992;68:755–767. doi: 10.1016/0092-8674(92)90150-b. [DOI] [PubMed] [Google Scholar]
- 40.Gorman C M, Moffat L F, Howard B H. Mol Cell Biol. 1982;2:1044–1051. doi: 10.1128/mcb.2.9.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MacGregor G R, Caskey C T. Nucleic Acids Res. 1989;17:2365. doi: 10.1093/nar/17.6.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strubin M, Struhl K. Cell. 1992;68:721–730. doi: 10.1016/0092-8674(92)90147-5. [DOI] [PubMed] [Google Scholar]
- 43.Nikolov D B, Chen H, Halay E D, Usheva A A, Hisatake K, Lee D K, Roeder B G, Burley S K. Nature (London) 1995;377:119–128. doi: 10.1038/377119a0. [DOI] [PubMed] [Google Scholar]
- 44.Kim T K, Roeder R G. Proc Nat Acad Sci USA. 1994;91:4170–4174. doi: 10.1073/pnas.91.10.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goodrich J A, Hoey T, Thut C J, Admon A, Tjian R. Cell. 1993;75:519–530. doi: 10.1016/0092-8674(93)90386-5. [DOI] [PubMed] [Google Scholar]
- 46.Klemm R D, Goodrich J A, Zhou S, Tjian R. Proc Natl Acad Sci USA. 1995;92:5788–5792. doi: 10.1073/pnas.92.13.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yokomori K, Zeidler M P, Chen J -L, Verrijzer P, Mlodzik M, Tjian R. Genes Dev. 1994;8:2313–2323. doi: 10.1101/gad.8.19.2313. [DOI] [PubMed] [Google Scholar]
- 48.Krumm A, Meulia T, Brunvand M, Groudine M. Genes Dev. 1992;6:2201–2213. doi: 10.1101/gad.6.11.2201. [DOI] [PubMed] [Google Scholar]
- 49.Strobl L J, Eick D. EMBO J. 1992;11:3307–3314. doi: 10.1002/j.1460-2075.1992.tb05409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krumm A, Hickey L, Groudine M. Genes Dev. 1995;9:559–572. doi: 10.1101/gad.9.5.559. [DOI] [PubMed] [Google Scholar]
- 51.Blau J, Xiao H, McCracken S, O’Hare P, Greenblatt J, Benteley D. Mol Cell Biol. 1996;16:2044–2055. doi: 10.1128/mcb.16.5.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sayle R A, Milner-White E J. Trends Biochem Sci. 1995;20:374–376. doi: 10.1016/s0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]
- 53.Kim Y, Geiger J H, Hahn S, Sigler P. Nature (London) 1993;365:512–516. doi: 10.1038/365512a0. [DOI] [PubMed] [Google Scholar]
- 54.Lichtsteiner S, Tjian R. Proc Natl Acad Sci USA. 1993;90:9673–9677. doi: 10.1073/pnas.90.20.9673. [DOI] [PMC free article] [PubMed] [Google Scholar]