

Abstract
2-Amino-alpha-carboline (AaC, 2-amino-9H-pyrido[2,3-b]indole) is a genotoxic carcinogen produced by cooking of protein-containing foods and combustion of biomaterial. Humans are chronically exposed to low levels of AaC through foods (grilled or pan-fried meats), drinking water, and smoke inhalation (cigarette/wood smoke, diesel exhaust). We report herein 17 metabolites of AaC formed in vivo in male Sprague-Dawley rats (from bile, urine, plasma) and in situ in rat hepatocytes and human HepG2 liver tumor cells. We confirmed several expected sites of AaC metabolism, but also observed novel metabolites. The novel metabolites include extensive N-acetylated AaC conjugates, multiple N-glucuronides, and at least one additional site of aromatic ring hydroxylation. The abundance of N-acetylated metabolites is noteworthy because this metabolic pathway is generally unrecognized for HAAs. Also noteworthy are metabolites that were not detected, i.e. no direct AaC N-sulfonation to form the sulfamate. These results, combined with earlier publications on the reactive (DNA-adduct forming) metabolites of AaC, indicate that both bioactivation and detoxification of AaC share the same metabolic pathways—namely oxidation, acetylation, and sulfonation. This may be an important factor attenuating the risk of carcinogenesis from AaC exposure; increased potential for bioactivation could be balanced by increased potential for detoxification.
Keywords: AaC; AalphaC; 2-amino-9H-pyrido[2,3-b]indole; heterocyclic aromatic amine; multiple reaction monitoring; environmental exposure; detoxification; excretion; metabolism; biomarker
Introduction
2-Amino-α-carboline (AaC, 2-amino-9H-pyrido[2,3-b]indole) is one of the five major heterocyclic aromatic amines (HAAs) present in the Western diet, and it is formed by pyrolysis of tryptophan during cooking. HAAs are naturally occurring genotoxic carcinogens which are not present in uncooked foods, but are readily produced under normal household cooking conditions. In addition to dietary exposure, AaC is found in combustion smokes of wood and cigarettes, in automobile exhaust, and in municipal water sources (1). Human exposure to HAAs including AaC is low but chronic (2). AaC is genotoxic in bacterial, insect and mammalian systems and is a suspected human carcinogen. For example, AaC induced preneoplastic lesions in the livers of male F344 rats (3), mutations in the colon of transgenic Big Blue™ mice (4), lymphomas and liver tumors in CDF1 mice (5), and increased the number and diameter of small intestinal tumors in C57BL/6J-Min/+ (multiple intestinal neoplasia) mice (6). However, despite many years of study, association of AaC exposure with human carcinogenesis remains probable but unproven (7) due to the difficulty of accurately estimating the low chronic human exposures.
The aim of this study was to use model systems (tissue homogenates, purified enzymes, cultured cells, and rodents in vivo) to develop methods for efficient detection, identification and quantification of stable excreted metabolites of the heterocyclic aromatic amine AaC. We report herein in vivo metabolism of AaC in male Sprague-Dawley rats and in situ metabolism by rat hepatocytes and human HepG2 cells. Our goal is to develop plausible biomarkers for future human molecular epidemiological and exposure studies for AaC which could clarify whether AaC is a human carcinogen at the low and chronic human exposure levels.
AaC requires biotransformation to achieve its mutagenic and carcinogenic potential. Indeed, we have published the pathways of AαC metabolism leading to reactive metabolites and DNA adduct formation, including N-oxidation, O-acetylation, and O-sulfonation catalyzed by cytochrome P450s, acetyltransferases, and sulfotransferases, respectively (8-9). We and others have characterized the major AαC-DNA adduct (10-12) and measured major and minor AαC-DNA adducts formed in vitro and in rats (12-13). We and others have also determined several intermediary metabolites of AαC that are not reactive (8, 14, 15).
Our results reported herein show that AaC is highly metabolized by oxidation and conjugation to stable, excreted metabolites. At least fourteen metabolites were observed in the rat bile and urine from the in vivo study, and structure determination by LC/MS/MS indicated ring-oxidation and extensive conjugation including acetylation, sulfonation, and glucuronidation. More extensive structure elucidation was conducted on the metabolites formed in rat hepatocyte and human hepG2 liver tumor cell incubates. In total, we confirmed expected sites of AaC metabolism, but also observed unexpected metabolites. The unexpected metabolites include extensive N-acetylated conjugates, double conjugates, and multiple N-glucuronide conjugates. We found at least three sites of aromatic ring hydroxylation, including a minor form undetected in the microsomal studies (8, 15). Also noteworthy is that no direct N-sulfonation to AaC-sulfamate was detected.
Materials and Methods
Caution
AaC is a suspected human carcinogen. Care must be taken in handling of both powder AaC and derivative solutions including proper use of laboratory gloves and personal protective devices. Radiolabeled chemicals must be handled by authorized users according to regulatory agencies.
Chemicals
AaC and radiolabeled [3,4,5-3H]AaC (100 mCi/mmol) were purchased from Toronto Research Chemicals (Ontario, Canada). Solvable™ aqueous based tissue solubilizer, and Ultima Flo-AP flow scintillation cocktail were obtained from PerkinElmer Life & Analytical Sciences. Other reagents were of the highest grade available commercially.
Animals
This study was conducted under institutional IACUC approval (No. AN00-05-016) following guidelines for the appropriate treatment of animals. Male Sprague-Dawley rats (250–300 g) were procured from Charles River Breeders (Wilmington, MA) for the in vivo study, or Harlan Technologies for the hepatocyte study. The rats were fed with standard laboratory chow (Teklad® diet) and water ad libitum before use. Ketaject and Xylaject were mixed prior to use for anesthesia.
In vivo model
Male Sprague-Dawley rats (0.4 kg) were anesthetized ip with a mixture of ketamine (100 mg/kg body wt) and xylazine (10 mg/kg body wt). After establishing the effect of anesthesia, the external urethra was ligated to prevent leakage of urine. The abdomen was opened surgically, and the bile duct was quickly cannulated with Becton Dickinson PE # 10 tubing and stabilized with stay sutures. The abdomen was closed by mattress suture. The bile flowed as yellow fluid and was collected in 1.5 mL centrifuge tubes. The jugular vein was cannulated with an angio-catheter # 24 and stay sutured. To confirm that the catheter was in the vein, it was flushed with a small volume (0.1 mL) of purified water. A single iv bolus dose containing 1 mg [3,4,5-3H]AαC in 0.1 mL DMSO (17.5 μCi per animal, ∼0.04 mCi/kg, 2.5 mg AaC/kg) was then injected via the jugular vein. For blood collection, the femoral vein was cannulated in three animals with an angio-catheter # 24 and stay sutured. Two (∼0.5 mL) blood samples were taken via the femoral vein, one and two hours after the injection of AαC. The animals were continuously observed for general condition and kept sedated throughout the bile collection by repeated anesthesia (half original dose every 2 h). Temperature was recorded periodically and warm fluid (0.5 mL) was supplemented every 30 min. Bile was collected on ice in 30 min fractions. After bile was collected for 3–7 h, the contents of the ligated bladder was collected by syringe, and the animal was euthanized by bleeding the portal vein and the abdominal aorta into 8 mL Vacutainer tubes (PST™, Becton Dickinson). Samples were obtained from a total of seven rats treated with [3,4,5-3H]AαC.
Cell system models
The cell systems used in this study were chosen as representative human (hepG2) and rat (hepatocyte) cell types for liver metabolism. Rat hepatocytes were obtained as primary culture from untreated male Sprague-Dawley rats. Hepatocytes were prepared just prior to use by washing the liver with modified Hank's Balanced salt solution (HBSS, without calcium, with EGTA). Hepatocytes were released by collagenase perfusion with Williams medium E (‘media’). Isolated hepatocytes were washed three times with media and cultured in insulin-supplemented media (1% v/v). The viability of the hepatocytes was assessed by Trypan blue exclusion, and the viability of cells used was 75-95%. Three million viable cells in 5 mL of culture medium were placed onto 60 mm Petri dish plates. The cultures were maintained at 37 °C in humidified air containing 5% CO2. Culture medium was supplemented (10% v/v) with heat inactivated Fetal Bovine Serum to favor attachment during the first four hours after plating. After the attachment, the supplemented media was replaced with serum-free media. During culture, media was supplemented with penicillin-streptomycin (1% v/v) to prevent contamination. Hepatocytes in primary culture (24 h after isolation) were incubated with [3,4,5-3H]AaC (0.1 mM, 0.01% DMSO) for 48 hours. After incubation, media and cells were collected together and an equal volume of acetonitrile added. After the precipitated materials were removed by centrifugation, the supernatant was concentrated and analyzed by HPLC or LC/MS/MS without further purification.
Human HepG2 liver tumor cells were obtained from ATCC and were cultured according to ATCC guidelines (HepG2/C3A, ATCC #CRL-10741). Cells in culture were incubated with [3,4,5-3H]AaC (0.05 or 0.1 mM) in growth media for 24 or 48 h. After incubation, media and cells were collected together and an equal volume of acetonitrile added. After the precipitated materials were removed by centrifugation, the supernatant was concentrated and analyzed by HPLC or LC/MS/MS without further purification.
Preparation of metabolite standards by in vitro incubation
Rat liver microsomal and cytosolic fractions prepared in our laboratory (16-17) were used for in vitro production of AaC metabolites. AaC (1 mM) was added to a solution containing rat liver microsomal fraction (1.5 mg/mL), 0.1 M potassium phosphate (pH 7.4), 2 mM EDTA, 2 mM ascorbic acid, and NADPH generating solution (NRS, comprised of 5 mM glucose-6-phosphate, 1 unit/mL glucose-6-phospate dehydrogenase, 1 mM NADP). The NRS was incubated for 7–10 min at 37 °C just prior to use. AaC was incubated at 37 °C for 1.5 h in a 1 mL assay. After 1.5 h of incubation with microsomes, some reactions were supplemented with 1 mg/mL rat liver cytosol, with or without 0.02 mM PAPS (3′-phosphoadenosine-5′-phosphosulfate), and incubated for total of 3 h. In other reactions, 1 mM UDPGA (uridine diphosphate-glucuronic acid) was added to the microsomal incubation. Both microsomal and cytosolic reactions were stopped after total of 1.5 or 3 h by addition of acetonitrile (1:1 v/v), and precipitated protein was pelleted. The supernatant was analyzed by HPLC or LC/MS/MS without further purification.
Sample preparation and purification of bile, urine, and plasma
Bile was collected in fractions from a total of seven rats administered [3,4,5-3H]AαC. Each fraction of bile was centrifuged for 10 min at 20,000g to pellet particulate matter. A portion of each bile fraction (0.1 mL) was analyzed directly by liquid scintillation counting. A portion of all fractions from each animal were pooled for further purification by solid phase extraction (SPE) and HPLC. Urine collected was centrifuged at 20,000g for 10 min to pellet particulate matter. A portion of urine (0.1 mL) was analyzed directly by liquid scintillation counting. A portion of the remaining urine sample of each animal was subjected to SPE and subsequent HPLC. The remaining urine volume was directly subjected to HPLC analysis after precipitation of protein by two volumes of acetonitrile. The blood collected in Vacutainer tubes was centrifuged to separate the plasma. A portion of plasma (0.1 mL) was analyzed directly by liquid scintillation counting. To the remaining plasma sample was added an equal volume of acidified acetonitrile (acetonitrile:acetic acid 25:1) to precipitate protein and clarify the solution. After centrifugation (5 min, 20,000g) the supernatant was analyzed by HPLC. Some plasma supernate samples were further purified by SPE before HPLC analysis.
Solid-phase extraction (SPE) permitted separation of some interfering materials and concentration prior to HPLC analysis. Bile, urine or clarified plasma sample to be analyzed was loaded on to a conditioned hydrophilic-lipophilic balance SPE cartridge (Water Oasis™ HLB, 6 cm3). Samples were eluted with 5 mL of 20 mM ammonium formate (pH 5.0), followed by a stepwise gradient of 20 to 100% acetonitrile in ammonium formate (step increases of 20%). A portion (0.1 mL) from each fraction was subjected to liquid scintillation counting, and fractions with significant radiolabel were subjected to HPLC analysis. The fractions collected from SPE were dried in vacuo and reconstituted (1-1.5 mL, SPE elution solvent) prior to HPLC analysis.
HPLC with UV and radiolabel analyses
HPLC separations with photodiode array and radiolabel detections were conducted on a Hitachi D-7000 system, equipped with Waters Symmetry C18 column, in-line vacuum degasser, L7100 4-solvent low pressure mixing dual pump, L7200 thermostatted sequential autosampler, L7455 photodiode array detector, Packard Radiomatic 505TR Flow Scintillation Analyzer (0.1 mL flow cell, Ultima Flo-AP scintillation cocktail), and a fraction collector.
Bile, urine and plasma samples (0.1 mL) were analyzed on a 3 × 150 mm column with 1.0 mL/min elution. The linear elution gradient in 20 mM ammonium formate (pH 5.0) was 0-7 min: 5-7% acetonitrile (v/v), 7-45 min: 7-50% acetonitrile, 50-55 min: 50-100% acetonitrile, 55-60 min steady at 100% acetonitrile. For radiolabel quantitation, the complete flow was passed through both the photodiode array and the flow scintillation detector. When eluent was collected for separate LC/MS/MS analysis, additional volume of the samples containing significant radiolabeled metabolite was separately injected and passed through only the photodiode array detector. Collected fractions were pooled and freeze-dried. Concentrated fractions from each major radiolabel-containing peak were analyzed by LC/MS/MS on the Quattro Ultima system as described below.
Cell incubate samples (supernatant from lysed cells plus media) were analyzed on a 2.1 × 50 mm column with 0.2 mL/min elution. The linear elution gradient in 20 mM ammonium formate (pH 5.0) was 0-40 min: 6-30% acetonitrile (v/v), and 40-50 min: 30-100% acetonitrile. For radiolabel quantitation, the complete flow was passed through both the photodiode array and the flow scintillation detector. For LC/MS/MS analysis, additional volume of each cell incubate sample was separately analyzed on the API2000 system as described below. When flow fractions were collected and concentrated for NMR analysis, the elution gradient was modified (1.0 mL/min; 0-50 min: 6-20% acetonitrile (v/v), 50-60 min: 20-100% acetonitrile) and the 3 × 150 mm column was used.
LC/MS/MS analyses
LC/MS/MS analysis of partially purified bile and urine samples was conducted by Dr. Daniel R. Doerge at the National Center for Toxicological Research (NCTR, Jefferson, Arkansas). Distinct LC elution conditions were used, including a linear elution gradient 0-45 min: 5-50% acetonitrile (v/v) in 20 mM ammonium acetate (pH 5.0). A Quattro Ultima triple stage quadropole mass spectrometer (Waters, Milford, MA), equipped with an electrospray (ES) source, was used with an ion source temperature of 120 °C, a desolvation gas temperature of 400 °C, and a constant cone voltage of 35 V. Full scan measurements (MS) were recorded over the range of m/z 100-600 for positive ions. For MS/MS measurements, a collision cell gas (Ar) pressure of 2-4 × 10-3 mbar was used to acquire either positive or negative ions in constant neutral loss, precursor ion, or product ion modes. This analysis focused on detecting the sulfate and glucuronide metabolites; the samples were screened for neutral loss of sulfate (80 mass units) or glucuronide (176 mass units). Parents molecules that lost 80 or 176 mass units were identified, and parents were screened for those consistent with potential AaC metabolite structures.
LC/MS/MS analysis on the cell incubates was conducted at the University of Rhode Island on an Applied Biosystems API2000 system equipped with turbo ion spray source and utilizing IDA and Metabolite ID software (Analyst 1.2 version). LC elution conditions were identical to those described above for radiolabel analysis of the cell incubate samples. The instrument conditions were as follows: curtain gas (N2), 45 psi; GS1, 40 psi; GS2, 45 psi; heated gas temperature, 300 °C; and ion spray voltage 5000 V. Full scan measurements (MS) were recorded over the range of m/z 100-600 for positive ions. For MS/MS collision-induced dissociation, the collision energy was varied, with nitrogen as the collision gas. Positive ions were acquired in either constant neutral loss, precursor ion, or product ion modes. Multiple reaction monitoring (MRM) was used for all final analyses incorporating appropriate m/z pairs for each metabolite.
NMR analysis
NMR was conducted on a Bruker AM300. Samples from rat hepatocyte incubations were collected as single peaks eluting from HPLC as described above, and dried in vacuo. Samples were reconstituted in deuterated methanol with tetramethylsilane as the reference resonance. NMR was utilized to clarify radiolabel substitution position on the aromatic ring of the commercial [3H]AaC, and to determine the substitution position of three purified metabolites (M3, M7, M10).
Hydrolysis of sulfate and glucuronide conjugates
After purification, selected samples were subjected to sulfatase or β-glucuronidase treatment. Samples (0.1 mL) were mixed with β-glucuronidase (375 U) or sulfatase (0.5 U) and incubated at 37 °C for 12 hours. Protein was precipitated with two volumes of acetonitrile and removed by centrifugation. The supernatant was dried in vacuo and reconstituted in 0.1 mL. Control samples were incubated in absence of enzyme. Hydrolysis products were subsequently analyzed by HPLC (UV, radiolabel) or LC/MS/MS.
Results
We collected bile, urine and plasma from seven adult male Sprague-Dawley rats treated intravenously with [3,4,5-3H]AaC. Excretion of radiolabel occurred at a reproducible and steady rate proportional with bile flow, and after four hours 15-20% of the dose had been excreted into the bile of each rat. Less than 1% of dose remained in plasma four hours after iv administration, indicating that AaC was extensively removed from circulation. Urine collected over 4 hours contained 10-15% of the dose administered.
Metabolites were separated by HPLC with or without prior solid phase extraction. The solid-phase extraction was beneficial only for the bile samples, removing strongly retained bile components prior to HPLC injection. As shown in Figures 1 and 2, flow scintillation analysis revealed at least 14 distinct metabolites in bile and urine (M1-M14). Many of these metabolites retained detectable UV absorbance at 340 nm (Figures 1B, 2B) even though their UV spectra were not identical to AaC. Radiolabel detection was used for relative quantification as summarized in Table 1. In bile, the metabolites eluted in two predominant fractions, M6 and M8, making up 60% of the total bile metabolites. In the urine, M6, M10 and M11 predominated, making up nearly 50% of the total urinary metabolites. Several bile metabolites were not detected in the urine (M1, M2, M5, M9, M13, M14). The major urinary and biliary metabolites were identified as N-acetyl-AaC-sulfate (M6 and M11), N-acetyl-AaC-O-glucuronide (M8), and AaC-sulfate (M10) by LC/MS/MS on the Quattro Ultima system. M6 is noted as M6a,b because two closely eluting components were distinguishable in the urine, both exhibiting the 322/242 MS/MS fragment pair. More data was needed distinguish the regioisomers and for conclusive structure identification of the less predominant metabolites. As described below, we used the cellular incubates to provide additional data providing conclusive match of structural identity with quantitation (Table 1) for the biliary and urinary AaC metabolites.
Figure 1.
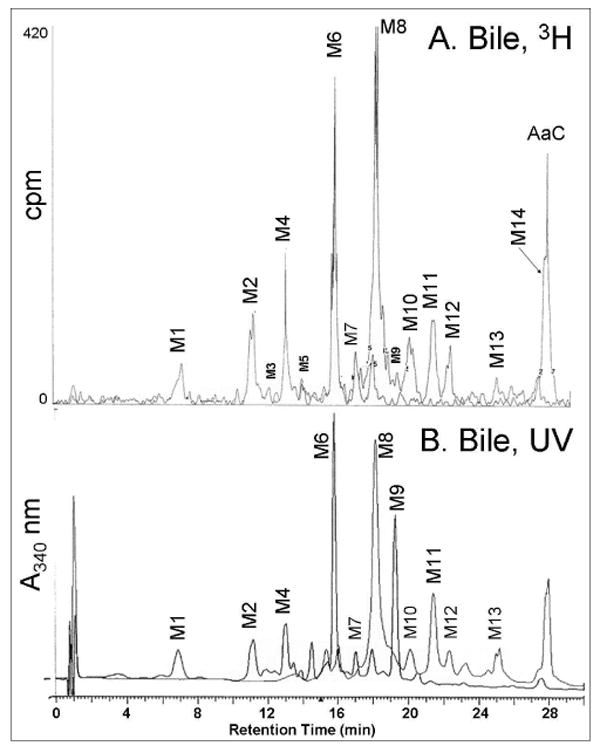
HPLC separation and in-line detection of rat bile [3,4,5-3H]AaC metabolites. (A) Flow scintillation radio-chromatogram used for quantitation, and (B) UV/vis profile at 340 nm. Metabolite identifications (Table 1) were made by LC/MS/MS, NMR, and UV analyses.
Figure 2.
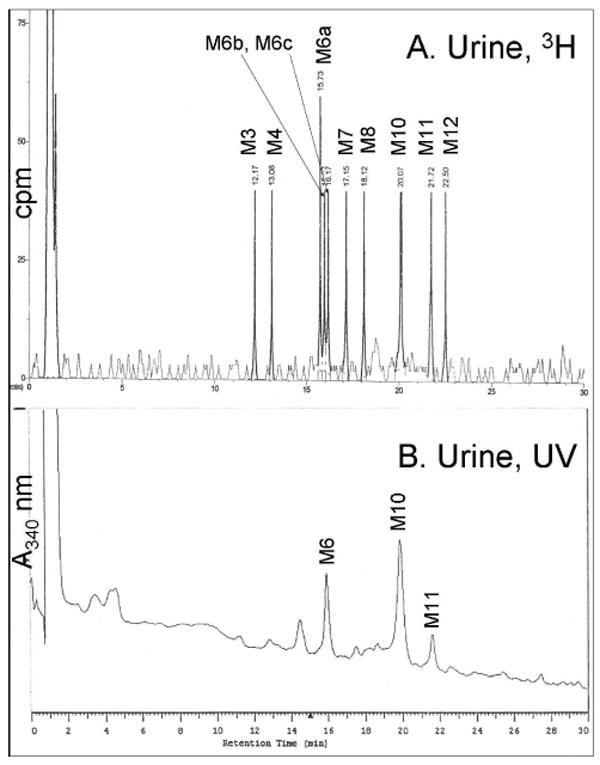
HPLC separation and in-line detection of rat urine [3,4,5-3H]AaC metabolites. (A) Flow scintillation radio-chromatogram used for quantitation, and (B) UV/vis profile at 340 nm. Metabolite identifications (Table 1) were made by LC/MS/MS, NMR, and UV analyses.
Table 1.
Quantitation of AaC metabolites present in rat bile, rat urine, rat hepatocyte incubates, and human HepG2 cell incubates.
| In Vivo | Cellular incubate | Metabolite name b | |||
|---|---|---|---|---|---|
| Metabolite number | Rat Bile
%a |
Rat Urine
%a |
Rat Hepatocyte
%a |
Human HepG2
%a |
|
| M1 | 2 | 0 | <1 | 0 | N-acetyl-AaC-C6-O-glucuronide (#1)c |
| M2 | 6 | 0 | 0 | 0 | AaC-C6-O-glucuronide (#1) |
| M3 | 0.5 | 8 | 11 | 36 | AaC-C6-sulfate |
| M4 | 6 | 7 | 3 | 0 | AaC-N-glucuronide (#1) |
| M6a,b | 20 | 19 | 6 | 19 | N-acetyl-AaC-C6-sulfate (#1) |
| M5 | 0.5 | 0 | 0 | 0 | AaC-N-glucuronide (#2) |
| M6c | 0 | 10 | 0 | 0 | AaC-C3-O-glucuronide (#2) |
| M8 | 40 | 9 | 0 | 0 | N-acetyl-AaC-C3-O-glucuronide (#2) |
| M7 | 2 | 10 | 14 | 0 | AaC-N-glucuronide (#3) |
| 0 | 0 | <1 | <1 | N-acetyl-AaC-C?d-sulfate (#2) | |
| M9 | 0.5 | 0 | <1 | <1 | AaC-C?-sulfate |
| M10 | 5 | 16 | 24 | 25 | AaC-C3-sulfate |
| M11 | 6 | 14 | 4 | 2 | N-acetyl-AaC-C3-sulfate (#3) |
| M12 | 4 | 8 | 0 | 0 | unknown |
| M13 | 0.5 | 0 | 0 | 0 | unknown |
| parent | nqe | 0 | nq | nq | AaC |
| M14 | 7 | 0 | 38 | 18 | N-acetyl-AaC |
Calculated as percent of total radiolabeled metabolites in each system.
Metabolite identifications were made by LC/MS/MS; regioisomers were determined by NMR and UV analyses.
Numbers in parentheses indicate multiple regioisomers in order of elution from HPLC.
Question mark indicates unidentified regioisomer.
Not quantified.
To confirm that the novel N-acetyl-AaC metabolites were not simply an artifact caused by the use of exogenous ammonium acetate buffer for the LC/MS/MS experiments of the bile and urine samples, we were careful to eliminate exogenous acetate from all further experiments. All metabolite purification steps and MS/MS analyses from the rat hepatocyte and human HepG2 cellular incubations were conducted in the absence of exogenous acetate, instead using ammonium formate buffer (Figures 3, 4). In addition, the bile samples were reanalyzed on the API2000 using ammonium formate mobile phase. In the absence of exogenous acetate, the extensive N-acetylated AaC metabolites were consistently observed in the two cellular systems and bile fractions, indicating that these N-acetylated metabolites were not artifacts of the analysis. Moreover, N-acetyl metabolites were not observed in samples that should not have contained them, i.e., the microsomal and cytosolic incubation systems used for synthesis of sulfate and glucuronide metabolite standards. Thus, we conclude that the extensive N-acetylation of AaC is caused through enzymatic biotransformation rather than by artifact of the analysis conditions.
Figure 3.
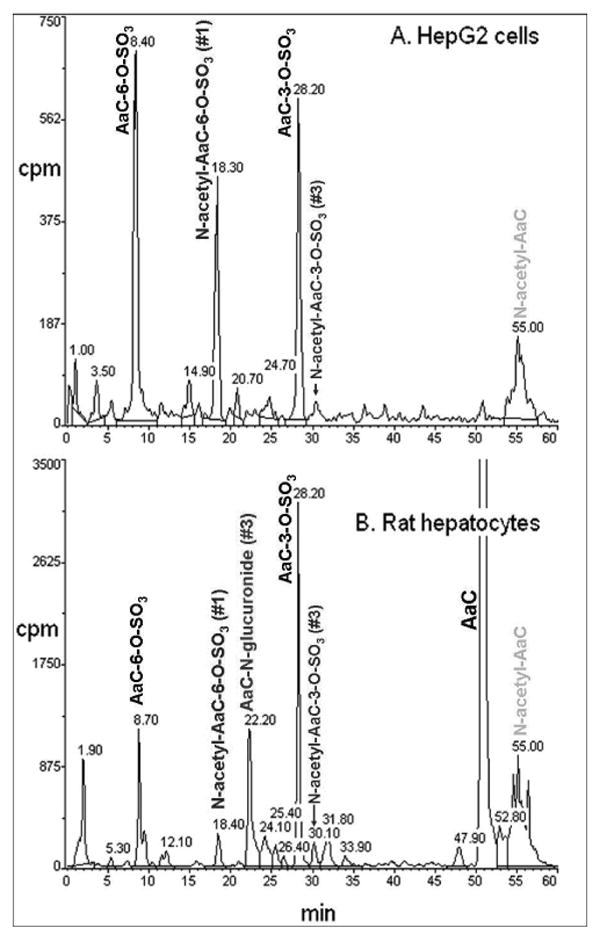
Flow scintillation radio-chromatograms used for quantitation of [3,4,5-3H]AaC metabolites in (A) HepG2 cell incubates and (B) rat hepatocyte incubates. Metabolite identifications were made by LC/MS/MS, NMR, and UV analyses.
Figure 4.
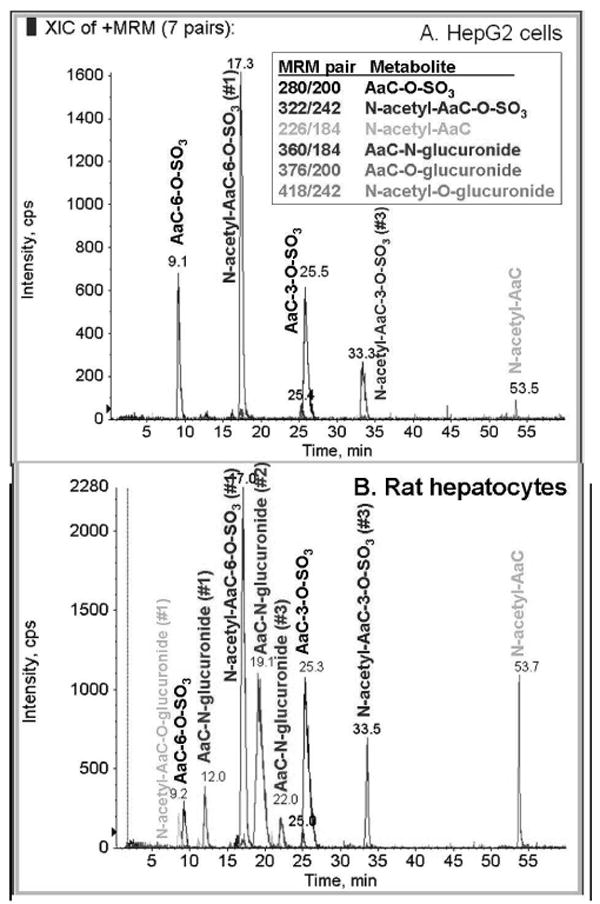
Overlay of six multiple reaction monitoring (MRM) extracted ion chromatograms (XIC) used for structure identification of [3,4,5-3H]AaC metabolites in (A) HepG2 cell incubates, and (B) rat hepatocyte incubates. Blue color indicates MRM XIC from 280/200 m/z pair, red indicates 322/242 m/z pair, blue-green indicates 226/184 m/z pair, pink indicates 360/184 m/z pair, gray indicates 376/200 m/z pair, and light blue indicates 418/242 m/z pair.
The radiolabel profile of the cellular incubates showed four major AaC metabolites for each cell system (Figure 3). LC/MS/MS with multiple reaction monitoring (Figure 4) identified these major metabolites as N-acetyl, N-glucuronosyl, and O-sulfonyl conjugates, and revealed additional minor AaC metabolites. In HepG2 cell incubates, the metabolites eluted in four predominant fractions, corresponding to M3, M6, M10, and M14, making up over 95% of the total HepG2 cellular metabolites. In rat hepatocyte incubates, M3, M7, M10 and M14 predominated, making up 87% of the total hepatocyte metabolites. In both HepG2 and rat hepatocyte cellular systems, AaC-C6-sulfate (M3), AaC-C3-sulfate (M10), and N-acetyl-AaC (M14) were major metabolites. The HepG2 cell system also formed N-acetyl-AaC-C6-sulfate (M6a), while rat hepatocytes formed a significant proportion of AaC-N-glucuronide (M7). It is not surprising that the HepG2 cellular incubates failed to form glucuronide metabolites because these cells are known to lack an active glucuronidation system.
The regio-isomers were identified by two approaches: proton NMR and hydrolysis with either sulfatase or β-glucuronidase. Three of the major metabolites formed in the rat hepatocyte system (corresponding to M3, M7, M10) were purified and analyzed by NMR. As shown in Figure 5, proton NMR spectra of M10 and parent AaC were compared. In the M10 spectrum, the C3 proton resonance (6.42 ppm) of AaC disappeared, and the C4 proton doublet of AaC moved downfield (8.05 ppm to 8.15 ppm) to appear as a singlet. The sulfatase hydrolysis product of M10 exhibited the characteristic the UV spectra of AaC-C3-OH, and it co-eluted (HPLC) with AaC-C3-OH standard produced in the microsomal incubations. The NMR and hydrolysis/UV data indicate that M10 is substituted at the C3 of AaC. Combined with the MS MRM (280/200), these data conclusively identify M10 as AaC-C3-sulfate. The NMR spectrum of M3 (280/200) was compared to that of AaC in a manner similar to that described for M10. In the M3 spectrum the AaC C6 proton triplet resonance was absent, and alteration of splitting of protons on adjacent carbons indicated C6 substitution. The sulfatase hydrolysis product of M3 exhibited the characteristic the UV spectra of AaC-C6-OH and co-eluted (HPLC) with AaC-C6-OH standard produced in the microsomal incubations. These data conclusively identify M3 as AaC-C6-sulfate. We also obtained NMR spectrum for purified M7 (360/184) which showed no differences in the proton resonances as compared to AaC, as expected for an N-glucuronide conjugate. The position of this N-glucuronide was not established, but M7 represents one of three total N-glucuronides (with M4 and M5) which must include the N9, N1, and N2 nitrogens. The sulfate substitution position of N-acetyl-AaC-C6-sulfate (M6a,b) and N-acetyl-AaC-C3-sulfate (M11) were determined by similarity of their UV spectra to AaC-C6-sulfate and AaC-C3-sulfate, respectively. Lastly, the regio-isomer identifications of N-acetyl-AaC-O-glucuronide (M1, M8) and AaC-O-glucuronide (M2, M6c) were tentatively assigned by the observation that for all identified metabolites (AaC-OH, AaC-sulfate, N-acetyl-AaC-sulfate) the retention time for the 6-substituted metabolite was characteristically less than for the 3-substituted metabolite.
Figure 5.
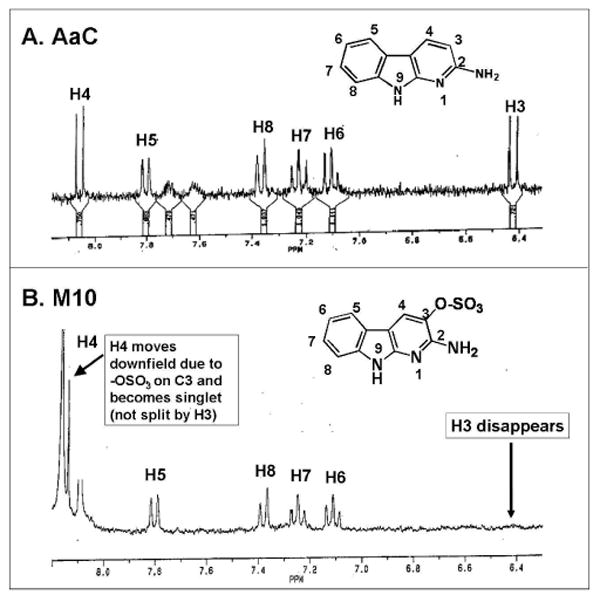
NMR spectra of (A) AaC and (B) M10 purified from human HepG2 incubations. M10 corresponds to metabolite in Figure 3 eluting at 28 min, and corresponds to metabolite in Figure 4 with MRM pair (280/200) eluting at 25.5 min. Aromatic proton resonance region (6.3-8.2 ppm) is shown.
Discussion
The goal of this study was to use model systems to develop methods for efficient detection, identification and quantification of stable and excreted metabolites of the genotoxic heterocyclic aromatic amine 2-amino-α-carboline (AaC). We designed our method to isolate only stable AaC metabolites because the reactive metabolites of AaC have been determined (8, 9, 15). A total of seventeen distinct AaC metabolites formed by in vivo and cellular systems were chromatographically separated, quantified by retention of radiolabel from the parent [3,4,5-3H]AaC, and identified structurally by combination of MS, MS/MS, NMR, UV and enzymatic hydrolysis experiments.
Many studies on the metabolism of various HAAs (reviewed in 18-19), have shown that multiple HAA metabolites are formed from combination of only a limited number of biotransformations typically including C- and N-oxidation, O- and N-glucuronidation, and O- and N-sulfonation. Moreover, we show herein that N-acetylation should be added to this list for some HAAs including AaC. Exact proportions of each metabolite have been shown to vary amongst different HAAs, species and systems (18-19). For AaC (Table 1), N-acetylation, N-glucuronidation, and ring-carbon oxidation with subsequent O-sulfonation or O-glucuronidation combined to make a total of at least 17 different stable AaC metabolites isolated from rat bile, rat urine, rat hepatocyte incubates, and human hepG2 cell incubates. These include the two major (AaC-C3-OSO3, AaC-C6-OSO3) and three minor (AaC-N-glucuronide, AaC-C3-O-glucuronide, AaC-C6-O-glucuronide) metabolites quantified from rat urine by Frederiksen and Frandsen (20).
The major difference amongst the four model systems in this study of AaC metabolism was the proportion of metabolites formed from single, double, or triple sequential biotransformations. For 13 example, in the bile, triple reactions predominated (68% N-acetylation, plus ring-carbon oxidation, plus O-sulfonation or O-glucuronidation). In the urine, double and triple reactions were formed in similar proportions (44% ring-carbon oxidation, plus O-sulfonation or O-glucuronidation; 42% N-acetylation, plus ring-carbon oxidation, plus O-sulfonation or O-glucuronidation). In the hepG2 system, double reactions predominated (61% ring-carbon oxidation, with subsequent O-sulfonation or O-glucuronidation). In contrast, the rat hepatocytes utilized predominantly single reactions (55% N-acetylation or N-glucuronidation) with a smaller portion double biotransformations (35% ring-carbon oxidation plus O-sulfonation). Thus, the cellular systems produced fewer total AaC metabolites utilizing the same pathways as observed in vivo. The exception was lack of glucuronidation in the hepG2 cellular system. Even with these differences, all samples contained a significant proportion of N-acetyl-AaC-C6-sulfate (M6a), AaC-C3-sulfate (M10), and N-acetyl-AaC-C3-sulfate (M11). N-acetyl-AaC (M14) was formed in both cellular systems and was excreted into rat bile, but was not excreted into rat urine.
Seventy-five percent of the total quantified biliary (42% of urinary, 48% of rat hepatocyte, and 39% of human hepG2) metabolites were N-acetylated, either alone or in combination with ring oxidation and sulfonation or glucuronidation. Two lines of evidence show that these N-acetyl-AaC metabolites were formed by enzymatic biotransformation rather than as artifacts of the analysis conditions. First, these N-acetyl AaC metabolites were consistently detected from both the in vivo and cellular incubation systems even after removal of all exogenous sources of acetate. Second, N-acetyl metabolites were not observed in samples that should not have contained them (i.e., the microsomal and cytosolic incubation systems used for synthesis of sulfate and glucuronide metabolite standards) despite presence of exogenous acetate during LC/MS/MS of these samples. This evidence confirms that N-acetylation represents a large, previously unrecognized, proportion of total AaC metabolism in both human and rat model systems. Indeed, N-acetylation of AaC was not detected previously in the urine or feces of Wistar rats after oral gavage (20). It is not clear whether N-acetylated AaC metabolites did not occur under these conditions, or whether they were lost during the extensive sample processing by these authors.
At least two other carcinogenic HAAs undergo N-acetylation in both human and rat in vivo and in vitro systems (21-23). N-acetyl derivatives of 2-amino-6-methyldipyrido[1,2-a:3′,2′-d]imidazole (Glu-P-1) and 2-aminodipyrido[1,2-a:3′,2′-d] imidazole (Glu-P-2) were detected in significant levels human and rat urine and bile. This extensive N-acetylation of certain HAAs has not been generally recognized because other model HAAs, including PhIP and MeIQx, are not N-acetylated (18). Consideration of acetylation as an important detoxification/excretion pathway may be important for molecular epidemiological studies assessing exposure risk to HAAs such as AaC.
Human studies generally use urinary samples to estimate exposure to AaC and other heterocyclic aromatic amines (HAAs). Because of the very low human exposure levels, MS methods are generally used for detection and quantitation. However, most MS methods suffer from lack of specificity and from the tendency to monitor a single mass. The multiple reaction monitoring (MRM) MS method described herein proved to be both sensitive and specific and may be advantageous as a general methodological approach for biomarker studies. As shown by comparison of Figures 3 and 4, the MRM method was much more sensitive than flow scintillation analysis of the radiolabel, and allowed detection and structural identification of metabolites present at very low concentration. After establishment of the ionization efficiency for each metabolite in the presence of radiolabel, the MRM method could be used quantitatively without need for additional radiolabel. Moreover, for metabolite detection using MRM analysis, only minimal sample purification to remove protein and precipitates was necessary prior to LC injection. Selective detection of metabolites was achieved by prior experimental determination of appropriate MRM pairs. We believe this method will prove to be advantageous for human studies of environmental or dietary exposure.
Scheme 1.

Summary of 17 total AaC metabolite substitutions observed in vivo in rat bile and urine, in human hepG2 cell incubates, and rat hepatocyte incubates. At least six metabolites contained double conjugates.
Acknowledgments
We thank Dr. Fred F. Kadlubar (NCTR) for donation of a portion of radiolabeled AaC used in this study. We thank Dr. Daniel R. Doerge and colleagues at the National Center for Toxicological Research (NCTR, Jefferson, AR) for mass spectral analyses on the partially purified bile and urine samples. Further LC/MS/MS analyses on the cell incubates were conducted in the laboratory of Dr. Fatemeh Akhlaghi (University of Rhode Island) on an Applied Biosystems API 2000. Research was supported by (1) NIH/NCRR grant number P20 RR16457, (2) The Burroughs Wellcome Fund and the American Foundation for Pharmaceutical Education, and (3) the University of Rhode Island Foundation, Council for Research. These study sponsors had no role in study design; in the collection, analysis, nor interpretation of data; in the writing of the report; nor in the decision to submit the paper for publication. A portion of this work has been presented in abstract form: Drug Metab. Rev. 34(S1):190 (2002), and Chem. Res. Toxicol. 17(12):1776 (2004).
References
- 1.Kataoka H, Hayatsu T, Hietsch G, Steinkellner H, Sachiko N, Narimatsu S, Knasmüller S, Hayatsu H. Identification of mutagenic heterocyclic amines (IQ, Trp-P-1 and AαC in the water of the Danube River. Mutn Res. 2000;466:27–35. doi: 10.1016/s1383-5718(99)00235-1. [DOI] [PubMed] [Google Scholar]
- 2.Layton DW, Bogen KT, Knize MG, Hatch FT, Johnson VM, Felton JS. Cancer risk of heterocyclic amines in cooked foods: an analysis and implications for research. Carcinogenesis. 1995;16:39–52. doi: 10.1093/carcin/16.1.39. [DOI] [PubMed] [Google Scholar]
- 3.Hasegawa R, Yoshimura I, Imaida K, Ito N, Shirai T. Analysis of synergism in hepatocarcinogenesis based on preneoplastic foci induction by 10 heterocyclic amines in the rat. Japanese J Canc Res. 1996;87:1125–1133. doi: 10.1111/j.1349-7006.1996.tb03122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang XB, Felton JS, Tucker JD, Urlando C, Heddle JA. Intestinal mutagenicity of two carcinogenic food mutagens in transgenic mice: 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and amino(α)carboline. Carcinogenesis. 1996;17:2259–2265. doi: 10.1093/carcin/17.10.2259. [DOI] [PubMed] [Google Scholar]
- 5.Ohgaki H, Matsukura N, Morino K, Kawachi T, Sugimura T, Takayama S. Carcinogenicity in mice of mutagenic compounds from glutamic acid and soybean globulin pyrolysates. Carcinogenesis. 1984;5:815–819. doi: 10.1093/carcin/5.6.815. [DOI] [PubMed] [Google Scholar]
- 6.Steffensen IL, Paulsen JE, Alexander J. The food mutagen 2-amino-9H-pyrido[2,3-b]indole (AalphaC) but not its methylated form (MeAalphaC) increases intestinal tumorigenesis in neonatally exposed multiple intestinal neoplasia mice. Carcinogenesis. 2002;23:1373–1378. doi: 10.1093/carcin/23.8.1373. [DOI] [PubMed] [Google Scholar]
- 7.Augustsson K, Steineck G. Cancer risk based on epidemiological studies. In: Nagao M, Sugimura T, editors. Food Borne Carcinogens: Heterocyclic Amines. John Wiley & Sons Ltd.; London: 2000. pp. 332–347. [Google Scholar]
- 8.Raza H, King RS, Squires RB, Guengerich FP, Miller DW, Freeman JP, Lang NP, Kadlubar FF. Metabolism of 2-amino-α-carboline: A food-borne heterocyclic amine mutagen and carcinogen by human and rodent liver microsomes and by human cytochrome P4501A2. Drug Metab Dispos. 1996;24:395–400. [PubMed] [Google Scholar]
- 9.King RS, Teitel CH, Kadlubar FF. In vitro bioactivation of N-hydroxy-2-amino-α-carboline. Carcinogenesis. 2000;21:1347–1354. [PubMed] [Google Scholar]
- 10.Baranczewski P, Gustafsson JA, Moller L. DNA adduct formation of 14 heterocyclic aromatic amines in mouse tissue after oral administration and characterization of the DNA adduct formed by 2-amino-9H-pyrido[2,3-b]indole (AalphaC), analysed by 32P_HPLC. Biomarkers. 2004;9:243–257. doi: 10.1080/13547500400010056. [DOI] [PubMed] [Google Scholar]
- 11.Pfau W, Schulze C, Shirai T, Hasegawa R, Brockstedt U. Identification of the major hepatic DNA adduct formed by the food mutagen 2-amino-9H-pyrido[2,3-b]indole (A alpha C) Chem Res Toxicol. 1997;10:1192–1197. doi: 10.1021/tx9701182. [DOI] [PubMed] [Google Scholar]
- 12.Snyderwine EG, Sadrieh N, King RS, Schut HAJ. Formation of DNA adducts of the food-derived mutagen 2-amino(α)carboline and bioassay of mammary gland carcinogenicity in Sprague-Dawley rats. Food Chem Toxicol. 1998;36:1033–1040. doi: 10.1016/s0278-6915(98)00088-x. [DOI] [PubMed] [Google Scholar]
- 13.Majer BJ, Kassie F, Sasaki Y, Pfau W, Glatt H, Meinl W, Darroudi F, Knasmuller S. Investigation of the genotoxic effects of 2-amino-9H-pyrido[2,3-b]indole in different organs of rodents and in human derived cells. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;802:167–173. doi: 10.1016/j.jchromb.2003.10.042. [DOI] [PubMed] [Google Scholar]
- 14.King RS, Teitel CH, Shaddock JG, Casciano DA, Kadlubar FF. Detoxication of carcinogenic aromatic and heterocyclic amines by enzymatic reduction of the N-hydroxy derivative. Cancer Letters. 1999;143:167–171. doi: 10.1016/s0304-3835(99)00119-6. [DOI] [PubMed] [Google Scholar]
- 15.Frederiksen H, Frandsen H. In vitro metabolism of two heterocyclic amines, 2-amino-9H-pyrido[2,3-b]indole (A(alpha)C) and 2-amino-3-methyl-9H-pyrido[2,3-b]indole (MeA(alpha)C) in human and rat hepatic microsomes. Pharmacol Toxicol. 2002;90:127–134. doi: 10.1034/j.1600-0773.2002.900303.x. [DOI] [PubMed] [Google Scholar]
- 16.Banoglu E, Jha GG, King RS. Hepatic microsomal metabolism of indole to indoxyl, a precursor of indoxyl sulfate. Eur J Drug Metab Pharmacokin. 2001;26:235–240. doi: 10.1007/BF03226377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banoglu E, King RS. Sulfation of indoxyl by human and rat aryl (phenol) sulfotransferases to form indoxyl sulfate. Eur J Drug Metab Pharmacokin. 2002;27:135–140. doi: 10.1007/BF03190428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King RS, Kadlubar FF, Turesky RJ. In vivo metabolism. In: Nagao M, Sugimura T, editors. Food Borne Carcinogens: Heterocyclic Amines. John Wiley & Sons Ltd.; London: 2000. pp. 90–111. [Google Scholar]
- 19.Yamazoe Y, Nagata K. In vitro metabolism. In: Nagao M, Sugimura T, editors. Food Borne Carcinogens: Heterocyclic Amines. John Wiley & Sons Ltd.; London: 2000. pp. 74–89. [Google Scholar]
- 20.Frederiksen H, Frandsen H. Excretion of metabolites in urine and faeces from rats dosed with the heterocyclic amine, 2-amino-9H-pyrido[2,3-b]indole (AalphaC) Food Chem Toxicol. 2004;42:879–885. doi: 10.1016/j.fct.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 21.Kanai Y, Manabe S, Wada O. In vitro and in vivo N-acetylation of carcinogenic glutamic acid pyrolysis products in humans. Carcinogenesis. 1988;9:2179–2184. doi: 10.1093/carcin/9.12.2179. [DOI] [PubMed] [Google Scholar]
- 22.Kanai Y, Manabe S, Wada O. Detection of N-acetyl derivative of 2-amino-6-methyl-dipyrido[1,2-a:3′,2′-d]imidazole (Glu-P-1) in Glu-P-1-injected rats. Mutat Res. 1988;207:63–67. doi: 10.1016/0165-7992(88)90043-7. [DOI] [PubMed] [Google Scholar]
- 23.Hein DW, Doll MA, Rustan TD, Gray K, Feng Y, Ferguson RJ, Grant DM. Metabolic activation and deactivation of arylamine carcinogens by recombinant human NAT1 and polymorphic NAT2 acetyltransferases. Carcinogenesis. 1993;14:1633–1638. doi: 10.1093/carcin/14.8.1633. [DOI] [PubMed] [Google Scholar]