

Abstract
Mutation analysis was performed in four apparently unrelated Dutch families with pantothenate kinase-associated neurodegeneration, formerly known as Hallervorden-Spatz syndrome. A novel 3-bp deletion encompassing the nucleotides GAG at positions 1,142 to 1,144 of exon 5 of the PANK2 gene was found in all patients. One patient was compound heterozygous; she also carried a novel nonsense mutation (Ser68Stop). The other patients were homozygous for the 1142_1144delGAG mutation. The 1142_1144delGAG mutation was also found in a German patient of unknown descent. We used polymorphic microsatellite markers flanking the PANK2 gene (spanning a region of approximately 8 cM) for haplotype analyses in all these families. A conserved haplotype of 1.5 cM was found for the 1142_1144delGAG mutation carriers. All the Dutch families originated from the same geographical region within the Netherlands. The results indicate a founder effect and suggest that the 1142_1144delGAG mutation probably originated from one common ancestor. It was estimated that this mutation arose at the beginning of the ninth century, approximately 38 generations ago.
Keywords: Hallervorden-Spatz syndrome, PANK2, Mutation, Founder, Haplotype
Introduction
Pantothenate kinase-associated neurodegeneration (PKAN; MIM 234200) is a rare autosomal recessive neurodegenerative disorder characterised by iron accumulation in the brain [1-3]. Its prevalence is estimated at 1 to 3 per million. Clinical features are progressive dementia and extrapyramidal dysfunction with dystonia, rigidity, dysarthria or choreoathetosis. Additional pigmentary retinopathy, acanthocytosis or hypoprebetalipoproteinemia can be detected in some cases [4, 5]. The onset of symptoms is classically during the first or second decade of life but may occur later in atypical cases. The diagnosis is based upon clinical findings and a typical pattern of iron accumulation in the globus pallidus visible on magnetic resonance imaging, also known as the ‘eye-of-the-tiger’ sign [6]. PKAN is caused by mutations in the human pantothenate kinase type 2 gene (PANK2; MIM 606157) located on chromosome 20p13 [7]. Pantothenate kinase type 2 plays a key role in mitochondrial coenzyme A biosynthesis [8-10]. It has been hypothesised that a deficiency of this enzyme causes brain iron and cysteine accumulation resulting in oxidative stress, membrane damage, cell death and subsequent neurodegeneration in PKAN patients. We recently started diagnostic DNA analysis of the PANK2 gene in the Dutch patients with PKAN and found a novel 3-bp deletion within a conserved haplotype of 1.5 cM in four apparently unrelated families.
Patients and methods
Patients and families were referred to our clinic for DNA diagnosis and genetic counselling. Diagnosis of Hallervorden-Spatz syndrome was established according to previously published criteria [3]. Essential features for diagnosis were a disease onset within the first two decades of life, progression of clinical signs and symptoms, extrapyramidal dysfunction and evidence of iron accumulation within the basal ganglia on magnetic resonance imaging (MRI). Corroborative features were corticospinal tract involvement, progressive intellectual impairment, retinal degeneration or a family history consistent with an autosomal recessive inheritance. The clinical features of the Dutch patients enlisted are summarised in Table 1. T2-weighted MRI scans, when available, showed the characteristic ‘eye-of-the-tiger’ sign in all affected individuals (Fig. 1).
Table 1.
Clinical features in Dutch PKAN patients
| Features | Subjecta |
|||||
|---|---|---|---|---|---|---|
| FI V:2 | FI V:3 | FII II:4 | FIII II:3 | FIV II:1 | FIV II:3 | |
| Gender | F | M | M | F | M | F |
| Age at onset (years) | 12 | 9 | 5 | 6 | 11 | 10 |
| Age at diagnosis (years) | 18 | 9 | 8 | 13 | 13 | |
| Age at loss of ambulation (years) | 22 | 12 | 10 | 8 | 16 | 18 |
| Age at death (years) | 15 | 18 | 12 | 17 | ||
| Current age (years) | 33 | 27 | ||||
| Extrapyramidal dysfunction | ||||||
| Dystonia | + | + | + | + | + | + |
| Rigidity | + | + | + | + | + | + |
| Choreoathetosis | − | + | − | − | + | + |
| Gait disturbance | + | + | + | + | + | + |
| Intellectual impairment | + | + | + | + | + | − |
| Emotional lability | + | − | − | − | + | + |
| Retinal degeneration | − | NA | + | + | − | + |
| Hypoprebetalipoproteinemia | − | NA | NA | NA | − | NA |
| Acanthocytosis | + | + | − | − | + | − |
| Eye-of-the-tiger sign on MRI | + | NA | + | + | + | + |
NA Information not available
Numbering corresponds to the numbering of families (FI to FIV) and the identification within family trees (see Fig. 3)
Fig. 1.

Axial and coronal sections of T2-weighted MRI of subject FIV II:3 illustrating the characteristic ‘eye-of-the-tiger’ sign (arrow): a symmetrical area of high signal intensity within the globus pallidus surrounded by an area of low signal intensity
After informed consent, genomic DNA was extracted from peripheral blood samples obtained from affected and unaffected family members from the four Dutch families and one German family (HS72), using standard techniques. Exons 1C to 7 of the PANK2 gene, including the intron/exon boundaries, were amplified by polymerase chain reaction (PCR) [7]. The PCR-amplified fragments were sequenced in forward and reverse directions using BigDye Terminator version 3.1 cycle sequencing kit on an automated capillary sequencer (ABI 3100 Genetic Analyser, Applied Biosystems, Foster City, CA, USA). Sequence alignment and mutation detection were performed using Mutation Surveyor software (version 2.2, SoftGenetics Inc., State College, PA, USA). Nucleotides and amino acid residues were numbered according GenBank accession number BK000010 [7]. Polymorphic microsatellite markers D20S113, D20S198, D20S193, D20S473, D20S116, D20S482 and D20S895 flanking the PANK2 gene were analysed to construct haplotypes.
The age of the 3-bp deletion was estimated using previously described methods [11, 12]. By comparing marker allele frequencies in mutation carrying and normal chromosomes, the linkage equilibrium index between each marker and the mutation was calculated as δ=(Pm−Pn/1−Pn), where Pm is the frequency of the marker allele on the mutation-bearing chromosomes and Pn is the frequency of the same allele on normal chromosomes. Genetic distances between each marker and the PANK2 gene, and thereby the recombination fractions θ, were estimated from physical distances. The appropriate genetic to physical distance ratio was calculated by linear regression analysis of physical vs genetic position encompassing a region of approximately 8 cM on chromosome 20p13. Sex-averaged genetic distances were derived from the deCODE high-resolution recombination map [13]. The resulting conversion factor was 2.5 cM/Mb. The algorithm of Risch et al. [14] [g=ln δ/ln(1−θ)] was then applied to estimate the age (in generations) of the mutation. Estimates based on different markers were averaged to obtain an overall age estimate.
Results
We identified two novel mutations involving exon 1C and exon 5 of the PANK2 gene in the Dutch PKAN patients (Fig. 2). Three patients were homozygous for a 3-bp deletion involving nucleotides GAG at positions 1,142 to 1,144 of the PANK2 gene. As a result of this deletion, arginine at position 371 and glutamic acid at position 372 of the Pank2 protein are substituted for a glutamine (delArg371-Glu372insGln). The same mutation was found heterozygous in an additional Dutch patient (subject FIII II:3). She carried a second novel mutation (233C>A), generating a premature stop codon (Ser68Stop) on the other allele. The 1142_1144delGAG mutation was not present in 50 control chromosomes. Hayflick and colleagues [15] identified the 1142_1144delGAG mutation once in a (not previously published) German patient of unknown ancestry (family HS72). This particular patient was compound heterozygous and carried a missense mutation (Thr418Met) on the other PANK2 allele. The Thr418Met mutation is the second most common mutation identified in PKAN patients [15, 16].
Fig. 2.
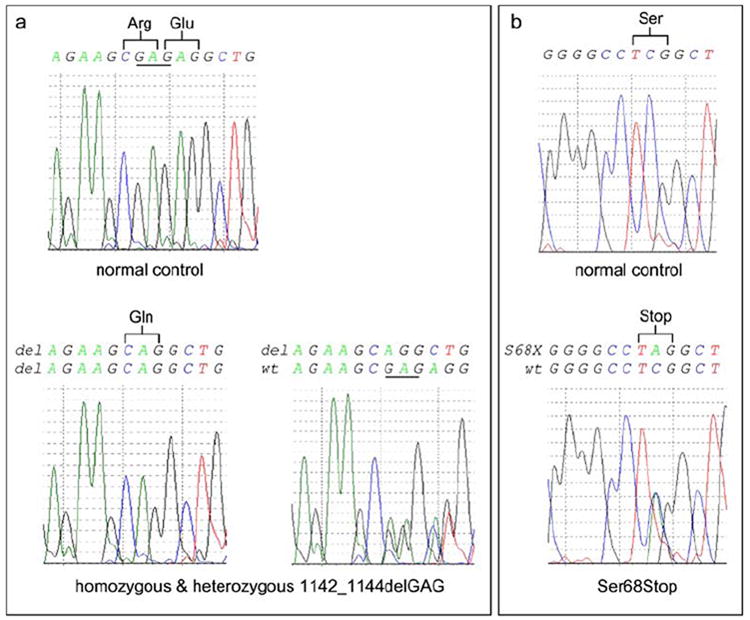
Mutations in the Dutch PKAN patients. A novel deletion of nucleotides 1,142 to 1,144 in exon 5 of the PANK2 gene was found in all enlisted Dutch patients (a). As a result of this mutation, arginine (Arg) at position 371 and glutamic acid (Glu) at position 372 of the Pank2 protein are substituted for glutamine (Gln). Three patients (subjects FI V:2, FII II:4 and FIV II:3) were found homozygous for this mutation. Another patient (subject FIII II:3) was found compound heterozygous for the 1142_1144delGAG mutation and a nonsense mutation (Ser68Stop) in exon 1C (b)
Haplotype analyses were performed in all these patients and their family members (Fig. 3). A conserved haplotype of 1.5 cM surrounding the PANK2 gene was found in all carriers of the 1142_1144delGAG mutation. The preserved alleles at these loci probably reflect the original ancestral disease-associated haplotype and indicate a founder effect. Table 2 shows the estimated ages of the 1142_1144delGAG mutation. The mean overall age estimate is 38 generations [95% confidence interval (CI) 24–51 generations].
Fig. 3.
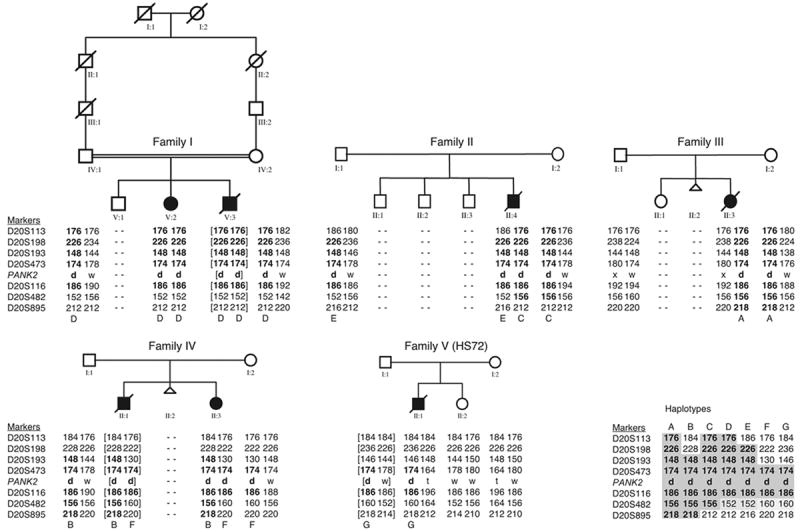
PKAN family pedigrees with disease-associated haplotypes. Filled symbols indicate affected individuals, and open symbols indicate unaffected individuals. The parents of family I are consanguineous. Family V (HS72) represents the German family (see text). Allele sizes (bp) for the microsatellite markers are shown. The letters A to G indicate the different haplotypes associated with the 1142_1144delGAG mutation. An alignment of these haplotypes is shown at the bottom right corner. The letters d, w, x and t indicate the 1142_1144delGAG mutation, the wild-type allele, the Ser68Stop and Thr418Met mutations, respectively. Shared haplotypes are given in boldface numbers. Derived haplotypes are presented between brackets
Table 2.
Estimation of the age of the 1142_1144delGAG PANK2 mutation
| Marker | Allele size | Distance from PANK2 |
θ | Pm | Pn | δ | g | |
|---|---|---|---|---|---|---|---|---|
| (bp) | (Mb) | (cM) | ||||||
| D20S113 | 176 | 1.86 | 4.66 | 0.047 | 0.63 | 0.60 | 0.063 | 57.9 |
| D20S198 | 226 | 1.26 | 3.16 | 0.032 | 0.63 | 0.40 | 0.375 | 30.5 |
| D20S193 | 148 | 0.59 | 1.48 | 0.015 | 0.75 | 0.30 | 0.643 | 29.6 |
| D20S473 | 174 | 0.44 | 1.10 | 0.011 | 1.00 | 0.30 | 1.000 | – |
| D20S116 | 186 | 0.16 | 0.40 | 0.004 | 1.00 | 0.30 | 1.000 | – |
| D20S482 | 156 | 0.62 | 1.55 | 0.016 | 0.38 | 0.70 | – | – |
| D20S895 | 218 | 1.20 | 3.01 | 0.030 | 0.38 | 0.00 | 0.375 | 32.1 |
θ Recombination fraction, Pm marker allele frequency on mutation-bearing chromosomes, Pn marker allele frequency on normal chromosomes, δ linkage disequilibrium, g estimated age in number of generations
Discussion
Since the discovery of the PANK2 gene, many different mutations have been identified in PKAN patients [7, 15-17]. These include missense mutations, nonsense mutations, splice-site mutations and frameshift mutations. Frameshift mutations may result from small insertions, deletions or duplications. In the present study, a novel 3-bp deletion was identified in the Dutch PKAN patients. In contrast to most of the previously identified small deletions, the 1142_1144delGAG mutation is in frame with the coding sequence of the wild-type gene and does not lead to a truncated Pank2 protein. As a consequence of this mutation, the residues arginine at position 371 and glutamic acid at position 372 are substituted for a glutamine. These amino acid changes occur within the catalytic domain of the protein. The substituted arginine (Arg371) is a conserved amino acid among different eukaryotic pantothenate kinase genes, including the human and mouse PANK1, PANK2 and PANK3 genes and the Drosophila homologue fumble [7, 18]. It is not known whether the 1142_1144delGAG mutation will influence kinase activity or could affect mRNA or protein processing, stability and lifetime. Thus far, many (mostly single) amino acid substitutions have been reported in PKAN patients, including patients with a classic early presentation of the disease [15, 17]. Previous studies have shown that such amino acid substitutions indeed may cause protein instability or reduce enzymatic activity [10]. There is evidence suggesting that human Pank2 may function as a homodimer, as is true for the bacterial enzyme [10, 19]. In theory, amino acid substitutions could also affect protein functioning by disturbing such a dimer formation. Further functional studies are needed to clarify the exact mechanisms by which the 1142_1144delGAG mutation could lead to aberrant Pank2 function.
A variable clinical presentation is seen in patients homozygous for the 1142_1144delGAG mutation. A disease onset at the age of approximately 10 years, with a slow progression and survival beyond the age of 30 years, as well as an earlier onset with a fast and severe progression leading to an early death are observed (Table 1). All male patients homozygous for the 1142_1144delGAG mutation showed a severe progression of disease symptoms and died before reaching the age of 20 years. In contrast, two female patients homozygous for the 1142_1144delGAG mutation had a slower disease progression and are still alive. These findings are consistent with intra-familial phenotypic variability, as is seen in other families [20]. The number of cases in this study was too small to investigate the effect of gender on the course of the disease in patients homozygous for the 1142_1144delGAG mutation. The observed gender differences could just be coincidental.
One girl (subject FIII II:3) was found to be compound heterozygous for the 1142_1144delGAG mutation and a novel Ser68Stop mutation. She had a severe progression of disease symptoms and an early death (Table 1). The Ser68Stop mutation is located in exon 1C after the putative mitochondrial target sequences [7, 8]. The expected product would be a truncated non-functional protein that lacks the complete catalytic domain. Nonsense mutations like these are found more frequently in cases with an early onset and severe progression of the disease [15]. The clinical features of the patient carrying the Ser68Stop mutation are consistent with this finding. Although she was referred to our hospital for her first neurological evaluation at the age of 6 years, the parents reported that, retrospectively, the first symptoms already started to develop at the age of 3 years. After a rapid progression of the disease, she died at 12 years of age, 4 years after the clinical diagnosis was established.
We estimated that the age of the 1142_1144delGAG mutation is approximately 38 generations. Assuming an intergenerational time of 30 years [21], our estimate dates the first appearance of this mutation 1,126 years ago. With an average year of birth of the mutation-carrying parents of 1946, the 1142_1144delGAG mutation can be dated to approximately 820 years a.d. (95% CI 419–1,220 years a.d.). This calculated age of the appearance of the 1142_1144delGAG mutation naturally only provides a very rough estimate since its value is influenced by many factors, including the choice of the founder marker allele, estimated recombination fractions, differences in population growth over past generations and sampling variation.
All the Dutch parents heterozygous for the 1142_1144delGAG mutation originated from the same geographical area in the northern part of the Netherlands. They were all born in a province called Friesland. These families were not aware of any interfamilial relationship, and consanguinity was only known for the parents of family I. Genealogical studies based on birth records and marriage certificates, tracing all ancestors for at least six generations, did not reveal any common ancestor between the families. A somewhat closer relationship between some of the carriers, however, seems apparent from the location of birthplaces and the haplotype branching tree (Fig. 4). Unfortunately, no information on birthplace or ancestral origins of the 1142_1144delGAG mutation-carrying parent from the German patient was available. So far, the 1142_1144delGAG mutation has not been reported in PKAN patients from other European countries [16, 22-24].
Fig. 4.
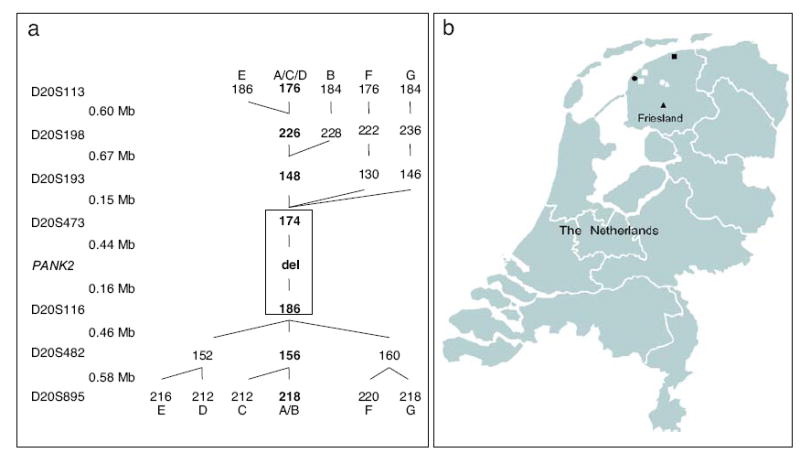
Haplotype branching tree (a). The markers used are shown with their relative physical distances. The observed haplotypes are indicated by different letters (from A to G), with the shared haplotypes in boldface numbers. The conserved haplotype region shared by all carriers of the 1142_1144delGAG mutation is boxed. Geographic origins of the Dutch families with the 1142_1144delGAG PANK2 mutation (b). The places of birth are depicted for each parent heterozygous for the 1142_1144delGAG mutation. The various haplotypes are indicated by different symbols (•=A, ○=B, ■=C, ◻=D, ▲=E, △=F). As shown, all the Dutch families were of Friesian origin
The presence of the 1142_1144delGAG mutation in the province Friesland could be explained by the demographic and settlement history of the region. The first human settlements in the province date from approximately 600 to 400 years b.c. Between 400 b.c. and the start of the Christian era, these first Friesians colonized the north and northwestern parts of the Netherlands. They developed their own language and identity. Frequent floods and the fall of the Roman Empire caused a desertion of the region between 250 and 400 years a.d. It is believed that the Friesians migrated to Germany and central England. They returned (together with a mixture of other Germanic tribes originating from Denmark and the northern provinces of Germany) to recolonize the area during the fifth century. These new Friesians subsequently repopulated the Dutch coasts between the rivers ‘Sincfal’ (Belgium) and ‘Weser’ (Germany). During the eighth century, however, the armies of Charles the Great forced them to retreat. After the domination of the Franks, the Vikings from Denmark invaded the area for a period of time during the end of the ninth century. Since we have roughly estimated the time of appearance of the 1142_1144delGAG mutation during this period, the Franks or the Vikings might have introduced this mutation into the Friesian population. Alternatively, it could have arisen de novo. Friesland remained more or less autonomous throughout the rest of the Middle Ages, until the sixteenth century when it became part of the Republic of the Seven United Netherlands. It appears that the Friesian population remained relatively stable during this period. Past geographical borders like rivers and seas, in addition to a linguistic and political separation, seem to have limited migration. This could explain a limited spreading of the 1142_1144delGAG mutation after its first appearance. The presence of a stable founder population in Friesland has been suggested before [25]. Within such a founder population, patients with a genetic disorder are likely to have a higher frequency of genes identical by descent [26]. A founder effect within the Friesian population has, for instance, also been reported for a rare form of congenital adrenal hyperplasia [27].
In summary, the results of the present study indicate that the newly identified 1142_1144delGAG PANK2 mutation found in the Dutch PKAN patients represents a founder mutation descended from one common ancestor, probably of Friesian origin.
Acknowledgments
We thank all patients, families and referring physicians for their participation. Additionally, we thank M. de Raad for her technical assistance regarding the haplotype analyses. All experiments comply with the current laws of the Netherlands. Electronic data and URLs: http://www.ncbi.nlm.nih.gov/Omim (for genetic disease details), http://www.ncbi.nlm.nih.gov/entrez/ (for polymorphic microsatellite markers and nucleotide information), http://www.decode.com (for genetic distance).
References
- 1.Dooling EC, Schoene WC, Richardson EP. Hallervorden-Spatz syndrome. Arch Neurol. 1974;30:70–83. doi: 10.1001/archneur.1974.00490310072012. [DOI] [PubMed] [Google Scholar]
- 2.Gordon N. Pantothenate kinase-associated neurodegeneration (Hallervorden-Spatz syndrome) Eur J Paediatr Neurol. 2005;6:243–247. doi: 10.1053/ejpn.2002.0606. [DOI] [PubMed] [Google Scholar]
- 3.Swaiman KF. Hallervorden-Spatz syndrome. Pediatr Neurol. 2001;25:102–108. doi: 10.1016/s0887-8994(01)00253-3. [DOI] [PubMed] [Google Scholar]
- 4.Ching KHL, Westaway SK, Gitschier J, Higgins JJ, Hayflick SJ. HARP syndrome is allelic with pantothenate kinase-associated neurodegeneration. Neurology. 2002;58:1673–1674. doi: 10.1212/wnl.58.11.1673. [DOI] [PubMed] [Google Scholar]
- 5.Houlden H, Lincoln S, Farrer M, Cleland PG, Hardy J, Orrell RW. Compound heterozygous PANK2 mutations confirm HARP and Hallervorden-Spatz syndromes are allelic. Neurology. 2003;61:1423–1426. doi: 10.1212/01.wnl.0000094120.09977.92. [DOI] [PubMed] [Google Scholar]
- 6.Sethi KD, Adams RJ, Loring DW, Elgammal T. Hallervorden-Spatz syndrome—clinical and magnetic resonance imaging correlations. Ann Neurol. 1988;24:692–694. doi: 10.1002/ana.410240519. [DOI] [PubMed] [Google Scholar]
- 7.Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet. 2001;28:345–349. doi: 10.1038/ng572. [DOI] [PubMed] [Google Scholar]
- 8.Hortnagel K, Prokisch H, Meitinger T. An isoform of hPANK2, deficient in pantothenate kinase-associated neurodegeneration, localizes to mitochondria. Hum Mol Genet. 2003;12:321–327. doi: 10.1093/hmg/ddg026. [DOI] [PubMed] [Google Scholar]
- 9.Johnson MA, Kuo YM, Westaway SK, Parker SM, Ching KHL, Gitschier J, Hayflick SJ. Mitochondrial localization of human PANK2 and hypotheses of secondary iron accumulation in pantothenate kinase-associated neurodegeneration. Ann N Y Acad Sci. 2004;1012:282–298. doi: 10.1196/annals.1306.023. [DOI] [PubMed] [Google Scholar]
- 10.Kotzbauer PT, Truax AC, Trojanowski JQ, Lee VMY. Altered neuronal mitochondrial coenzyme a synthesis in neurodegeneration with brain iron accumulation caused by abnormal processing, stability, and catalytic activity of mutant pantothenate kinase 2. J Neurosci. 2005;25:689–698. doi: 10.1523/JNEUROSCI.4265-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bergman A, Einbeigi Z, Olofsson U, Taib Z, Wallgren A, Karlsson P, Wahlstrom J, Martinsson T, Nordling M. The Western Swedish BRCA1 founder mutation 3171ins5; a 3.7 cM conserved haplotype of today is a reminiscence of a 1500-year-old mutation. Eur J Hum Genet. 2001;9:787–793. doi: 10.1038/sj.ejhg.5200704. [DOI] [PubMed] [Google Scholar]
- 12.Karpati M, Gazit E, Goldman B, Frisch A, Colombo R, Peleg L. Specific mutations in the HEXA gene among Iraqi Jewish Tay-Sachs disease carriers: dating of founder ancestor. Neurogenetics. 2004;5:35–40. doi: 10.1007/s10048-003-0166-8. [DOI] [PubMed] [Google Scholar]
- 13.Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, Shlien A, Palsson ST, Frigge ML, Thorgeirsson TE, Gulcher JR, Stefansson K. A high-resolution recombination map of the human genome. Nat Genet. 2002;31:241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- 14.Risch N, Deleon D, Ozelius L, Kramer P, Almasy L, Singer B, Fahn S, Breakefield X, Bressman S. Genetic analysis of idiopathic torsion dystonia in Ashkenazi Jews and their recent descent from a small founder population. Nat Genet. 1995;9:152–159. doi: 10.1038/ng0295-152. [DOI] [PubMed] [Google Scholar]
- 15.Hayflick SJ, Westaway SK, Levinson B, Zhou B, Johnson MA, Ching KHL, Gitschier J. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med. 2003;348:33–40. doi: 10.1056/NEJMoa020817. [DOI] [PubMed] [Google Scholar]
- 16.Pellecchia MT, Valente EM, Cif L, Salvi S, Albanese A, Scarano V, Bonuccelli U, Bentivoglio AR, D’Amico A, Marelli C, DiGiorgio A, Coubes P, Barone P, Dallapiccola B. The diverse phenotype and genotype of pantothenate kinase-associated neurodegeneration. Neurology. 2005;64:1810–1812. doi: 10.1212/01.WNL.0000161843.52641.EC. [DOI] [PubMed] [Google Scholar]
- 17.Thomas M, Hayflick SJ, Jankovic J. Clinical heterogeneity of neurodegeneration with brain iron accumulation (Hallervorden-Spatz syndrome) and pantothenate kinase-associated neurodegeneration. Mov Disord. 2004;19:36–42. doi: 10.1002/mds.10650. [DOI] [PubMed] [Google Scholar]
- 18.Afshar K, Gonczy P, DiNardo S, Wasserman SA. fumble encodes a pantothenate kinase homolog required for proper mitosis and meiosis in Drosophila melanogaster. Genetics. 2001;157:1267–1276. doi: 10.1093/genetics/157.3.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yun M, Park CG, Kim JY, Rock CO, Jackowski S, Park HW. Structural basis for the feedback regulation of Escherichia coli pantothenate kinase by coenzyme A. J Biol Chem. 2000;275:28093–28099. doi: 10.1074/jbc.M003190200. [DOI] [PubMed] [Google Scholar]
- 20.Gouider-Khouja N, Miladi N, Belal S, Hentati F. Intrafamilial phenotypic variability of Hallervorden-Spatz syndrome in a Tunisian family. Parkinsonism Relat Disord. 2000;6:175–179. doi: 10.1016/s1353-8020(99)00060-7. [DOI] [PubMed] [Google Scholar]
- 21.Tremblay M, Vezina H. New estimates of intergenerational time intervals for the calculation of age and origins of mutations. Am J Hum Genet. 2000;66:651–658. doi: 10.1086/302770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cossu G, Melis M, Floris G, Hayflick SJ, Spissu A. Hallervorden Spatz syndrome (pantothenate kinase associated neurodegeneration) in two Sardinian brothers with homozygous mutation in PANK2 gene. J Neurol. 2002;249:1599–1600. doi: 10.1007/s00415-002-0865-3. [DOI] [PubMed] [Google Scholar]
- 23.Castelnau P, Cif L, Valente EM, Vayssiere N, Hemm S, Gannau A, DiGiorgio A, Coubes P. Pallidal stimulation improves pantothenate kinase-associated neurodegeneration. Ann Neurol. 2005;57:738–741. doi: 10.1002/ana.20457. [DOI] [PubMed] [Google Scholar]
- 24.Molinuevo JL, Marti MJ, Blesa R, Tolosa E. Pure akinesia: an unusual phenotype of Hallervorden-Spatz syndrome. Mov Disord. 2003;18:1351–1353. doi: 10.1002/mds.10520. [DOI] [PubMed] [Google Scholar]
- 25.Sonneveld DJA, Schaapveld M, Sleijfer DT, Te Meerman GJ, Van der Graaf WTA, Sijmons RH, Koops HS, Hoekstra HJ. Geographic clustering of testicular cancer incidence in the northern part of the Netherlands. Br J Cancer. 1999;81:1262–1267. doi: 10.1038/sj.bjc.6690839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Te Meerman GJ, Van der Meulen MA, Sandkuijl LA. Perspectives of identity by descent (IBD) mapping in founder populations. Clin Exp Allergy. 1995;25:97–102. doi: 10.1111/j.1365-2222.1995.tb00433.x. [DOI] [PubMed] [Google Scholar]
- 27.Imai T, Yanase T, Waterman MR, Simpson ER, Pratt JJ. Canadian Mennonites and individuals residing in the Friesland region of the Netherlands share the same molecular basis of 17-alpha-hydroxylase deficiency. Hum Genet. 1992;89:95–96. doi: 10.1007/BF00207050. [DOI] [PubMed] [Google Scholar]