

Abstract
We describe a method that directly relates tissue neuropathological analysis to medical imaging. Presently, only indirect and often tenuous relationships are made between imaging (such as MRI or x-ray computed tomography) and neuropathology. We present a biochemistry-based, quantitative neuropathological method that can help to precisely quantify information provided by in vivo proton magnetic resonance spectroscopy (1HMRS), an emerging medical imaging technique. This method, high resolution magic angle spinning (HRMAS) 1HMRS, is rapid and requires only small amounts of unprocessed samples. Unlike chemical extraction or other forms of tissue processing, this method analyzes tissue directly, thus minimizing artifacts. We demonstrate the utility of this method by assessing neuronal damage using multiple tissue samples from differently affected brain regions in a case of Pick disease, a human neurodegenerative disorder. Among different regions, we found an excellent correlation between neuronal loss shown by traditional neurohistopathology and decrease of the neuronal marker N-acetylaspartate measured by HRMAS 1HMRS. This result demonstrates for the first time, to our knowledge, a direct, quantitative link between a decrease in N-acetylaspartate and neuronal loss in a human neurodegenerative disease. As a quantitative method, HRMAS 1HMRS has potential applications in experimental and clinical neuropathologic investigations. It should also provide a rational basis for the interpretation of in vivo 1HMRS studies of human neurological disorders.
Modern imaging methods such as x-ray computed tomography and MRI have revolutionized the detection of abnormalities of the brain. Although sensitive for lesion detection, x-ray computed tomography and MRI are often nonspecific in defining the underlying pathology. In many cases, diagnosis must await microscopic evaluation of tissue obtained by biopsy or autopsy. It has been hypothesized that greater diagnostic specificity may be achieved by magnetic resonance spectroscopy (MRS) because it can measure changes in neurochemistry that accompany specific diseases. Thus, many investigators and clinicians have applied MRS in vivo for clinical diagnoses (1–7), although a rational, quantitative basis for the interpretation of brain MR spectra is not yet established.
Currently, the understanding of brain MR spectra relies on ex vivo analysis of chemical extracts of tissue samples (8, 9). Unfortunately, these procedures may introduce artifacts. Direct proton MRS (1HMRS) analysis of brain tissue using standard high resolution 1HMRS methods is compromised by poor spectral resolution (10). Magic angle spinning (MAS) can reduce MR spectral line-widths; hence, high resolution MAS (HRMAS) 1HMRS of brain may be ideal for elucidating in vivo MRS observation because it can produce high resolution spectra of unprocessed brain (11).
In liquids, molecules do not experience significant motion restriction and tumble at rates faster than the MR time scale. Thus, spectral broadening effects due to molecular interactions are averaged, resulting in narrow spectral lines. In tissue, restriction of molecular motions and magnetic susceptibility results in spectral broadening that is not effectively averaged. As a consequence, tissues may be considered to have certain characteristics of solid molecular interactions. In solids, interactions such as dipole couplings and chemical shift anisotropy produce spectral broadening with an angular dependence of (3 cos2 θ − 1), where θ is the angle between the static magnetic field and the internuclear vector. It is known that if a sample is spun mechanically, at a rate faster than the spectral broadening originating from these interactions, and at the “magic angle” of 54°44′ (which meets the criterion of 3 cos2 (54°44′) − 1 = 0), the contribution from these interactions to the MR spectral broadening can be significantly reduced (12–15). In biological studies, MAS 1HMR has been demonstrated to be useful in the noninvasive analyses of plant material and lipids (16–20). Applying MAS on human/animal tissue should reduce the effect of residual molecular interactions and the influence of magnetic susceptibility on spectral broadening. Adipose tissue is another factor that prevents highly resolved proton spectra to be obtained. Recently, we have shown that enhanced spectral resolution of fatty lymphatic tissue can be achieved using a modified 1HMR technique that includes MAS, and a MAS rotor synchronized T2 (spin–spin relaxation time) filter. By using this technique, malignant cancerous rat lymph nodes could be distinguished from normal lymph nodes (11).
In the present study, we evaluated the quality of HRMAS spectra of unprocessed brain samples and tested the hypothesis that regional decrease of brain N-acetylaspartate (NAA) concentrations correlate directly with histopathological evidence of cortical neuronal loss due to Pick disease. Currently, Pick disease, a neurodegenerative disorder that accounts for approximately 5% of all dementias (21, 22), can be confidently diagnosed only by histopathologic analysis of postmortem brain showing marked neuronal loss, with or without characteristic neuronal inclusions, in the frontal and/or temporal lobes. In this study, we analyzed multiple postmortem brain samples of differently affected cortical regions obtained from a 66-year-old patient who died with Pick disease. An excellent correlation between regional neuron loss and regional NAA decrease was found. Furthermore, because MR analysis of intact tissue directly reflects the in vivo status of a diseased brain, this method should benefit the design and implementation of new in vivo MR techniques for early detection and diagnosis of neurological diseases.
EXPERIMENTAL PROCEDURES
Collection of Brain Tissues and Neuropathology.
Human brain tissue was obtained through the Massachusetts General Hospital Brain Bank, which has approval from the Massachusetts General Hospital Subcommittee on Human Studies to distribute samples for scientific study.
Pick disease brain tissue (15 samples) from four different regions of cerebral cortex of the left hemisphere was dissected and snap frozen 14 h after the patient’s death and kept at approximately −80°C (23). Fresh monkey brain tissue was supplied by the New England Regional Primate Research Center and stored at approximately −80°C. Sample collection, dissection, and transportation were all performed on a dry-ice surface. Before each HRMAS 1HMRS measurement, the sample was warmed on a −10°C preparation surface and then transferred to a 7-mm MAS NMR rotor, where it was held between two kel-f combined rotational and multiple pulse inserts (Bruker, Billerica, MA). Samples weighed between 23 and 60 mg.
The right hemisphere of the Pick brain tissue was fixed in 10% formalin before being examined pathologically. Fixed brain tissue from similar regions were processed, microtome-sectioned (6 μm), and stained with the routine luxol fast blue/hematoxylin and eosin method before microscopic examination. For neuronal counts of each cortical region, three microscopic fields at ×250 magnification (area = 0.454 mm2) were evaluated for the number of nondegenerated pyramidal neurons in cortical layer III (the external pyramidal layer).
HRMAS 1HMRS of Unprocessed Brain Tissue.
HRMAS experiments were performed at 2°C unless otherwise specified on an MSL400 NMR spectrometer (proton frequency at 400.13 MHz) using a BD-MAS probe (Bruker). Temperature was controlled by a VT-1000 unit in combination with a MAS-DB pneumatic unit (Bruker). The MAS rate used in this study was stabilized at 2.500 ± 0.001 kHz. Spectra presented here were acquired using a rotor synchronized Carr–Purcell–Meibom–Gill pulse sequence, [90-(τ-180-τ)n-acquisition], as a T2 filter, to remove the effect of lipids on spectral broadening and to suppress the resonance from tissue water (11). The inter-pulse delay (τ = 2π/ωr = 400 μs) was synchronized to the rotor rotation, i.e., τ was a multiple of sample spinning speed in time units, where ωr/2π represented the MAS speed in kilohertz. The value for n was 750 (2nτ = 600 ms). The 90° pulse length was adjusted for each sample individually and varied from 9.6 to 10.6 μs. The number of transients was 512 with an acquisition time of 1,016 ms (16,000 complex points). A repetition time of 3.0 s and a spectral width of 8,064.52 Hz (20.15 ppm) were employed. The total acquisition time was less than 31 min. Tissue degradation studies for human and monkey brains were carried out at both 20°C and 2°C. With an acquisition of 31 min for each spectrum, the entire sequence of degradation spectra was collected continuously for more than 24 h. T2 measurements were performed using the same Carr–Purcell–Meibom–Gill sequence, varying n from 200 to 900. Before Fourier transformation and phasing, all free induction decays were subjected to 1-Hz apodization. Trimethylsilane at 0.00 ppm was used as an external chemical shift reference, from which the internal reference of the lactate doublet was determined to be 1.32 and 1.34 ppm.
Solution 1HMRS of Brain Extracts and Unprocessed Tissue.
Brain tissue (600 mg) was pulverized with a mortar cooled in liquid nitrogen. The tissue was then extracted using a methanol/chloroform extraction procedure (9). After centrifuging at 1,800 × g for 10 min, the three layers (aqueous, protein, and organic) were separated. The aqueous layer was lyophilized, dissolved in deuterium oxide, and lyophilized again. The final powder was then dissolved in deuterium oxide for spectroscopic study. Proton MR spectra were acquired at 20°C on the above-mentioned NMR spectrometer using a 5-mm probe (Bruker). Spectral acquisition parameters included a one-pulse experiment, a repetition time of 15 s, a spectral width of 4 kHz, and 32,000 complex points, and 64 transients were averaged. Monkey brain tissue, after being subjected to 2.5-kHz MAS in the MAS probe, was transferred to a 5-mm NMR tube and immersed in deuterated phosphate buffer solution. Glasswool was used at the bottom of the tube to adjust the sample to the coil active region. 1HMR spectra were collected at 20°C with a standard 5-mm probe, and with the same experimental parameters used to obtain the monkey brain HRMAS spectra, except for the 90° pulse was 20 μs with the 5-mm probe.
RESULTS
The Effect of MAS.
Fig. 1 demonstrates the effect of MAS on the 1HMR spectral resolution of brain tissue. Spectrum a in Fig. 1, acquired after spectrum b, was obtained with the same monkey brain tissue used to acquire the MAS spectrum (spectrum b), and was recorded with the same experimental protocol by which spectrum b was acquired. Both spectra were acquired at 20°C with water suppression. This figure shows the enhancement in spectral resolution obtainable with MAS. Fig. 2 compares the quality of 1HMRS using HRMAS with solution MR spectra of brain extracts. In this figure, an HRMAS 1HMR spectrum of unprocessed Pick brain tissue from the minimally degenerated posterior portion of the superior temporal gyrus region is shown (spectrum a). An adjacent sample from the same region was extracted using the methanol/chloroform method (9), and a routine solution 1HMRS was acquired (Fig. 2, spectrum b). The spectra shown in Fig. 2 have similar spectral resolutions, on the order of 1 Hz.
Figure 1.

Proton MR spectra (400 MHz) of monkey brain tissue at 20°C. Spectrum a, static; spectrum b, HRMAS at 2.5 kHz.
Figure 2.

Proton MR spectra (400 MHz) of human brain tissue collected from superior temporal gyrus histologically determined to be mildly affected with Pick disease. Spectrum a, intact tissue T2-weighted HRMAS at 2.5 kHz, at 2°C; spectrum b, tissue extraction solution at 20°C. Selected metabolite resonances were labeled on spectrum a (Lac:, lactate; GABA: γ-aminobutyric acid; Acet, acetate; Cre, creatine; Chol, choline; PCh, phosphorylcholine; Tau, taurine; Inos, inositol; alcohol conta: alcohol contamination). The resonances at 1.18 ppm (triplet) and 3.65 ppm (quartet) were only seen in the tissue spectrum, and were due to contamination with small amount of alcohol sprayed on the surface of the postmortem brain before tissue freezing (9).
Neuropathology and Tissue Metabolism of Pick Disease.
Fig. 3 illustrates the histopathological changes Pick disease can produce in human brain. Fig. 3a was taken from a relatively normal region of the primary visual cortex of the occipital lobe, whereas Fig. 3b shows the severely degenerated rostral inferior temporal gyrus with prominent neuronal loss and marked astrocytosis.
Figure 3.

(a) Photomicrograph taken from superficial occipital visual cortex, showing normal population of neurons and unremarkable neuropil. (×100.) (b) Photomicrograph taken from superficial cortex (layers 1 and 2) of the rostral inferior temporal gyrus, showing marked neuronal loss and severe astrogliosis. (×100.)
The numbers of apparently surviving pyramidal neurons in the cortical layer from selected cortical regions of Pick disease brain were compared with a neurologically normal elderly brain (from a 100-year old person). Results are shown in Table 1. Decreased neuronal counts are noted in all regions. Nevertheless, the severely degenerated inferior temporal gyrus cortex in Pick disease has lost the majority of its layer III pyramidal neurons compared with the primary visual cortex of the same brain and the same region of a normal elderly control.
Table 1.
Neuronal counts in cortical layer III of various cortical regions
| Cortical region examined | Pick disease brain
|
Elderly control
|
Difference in mean, % | ||||
|---|---|---|---|---|---|---|---|
| Counts | M | SE | Counts | M | SE | ||
| Primary visual cortex | 78, 84, 90 | 84 | 3.5 | 118, 102, 122 | 114 | 6.1 | 26.3 |
| Superior temporal gyrus | 50, 57, 46 | 51 | 3.2 | 56, 58, 68 | 61 | 3.7 | 16.4 |
| Superior frontal gyrus | 70, 63, 57 | 63 | 3.8 | 68, 78, 76 | 74 | 3.1 | 14.9 |
| Inferior temporal gyrus | 25, 32, 36 | 31 | 3.2 | 79, 78, 84 | 80 | 1.9 | 61.3 |
Counts were taken from a ×250 microscopic field (or 0.454 mm2). The control was fron a neurologically normal brain from a 100-year-old person. M, mean.
Tissue HRMAS 1HMRS from the corresponding regions of the left hemisphere are shown in Fig. 4. The rostral inferior temporal gyrus spectrum in Fig. 4 (spectrum b) contained substantially fewer metabolites compared with the primary visual cortex spectrum (spectrum a). In this case of Pick disease, the superior temporal and frontal gyri were found to be histopathologically less damaged (see Table 1). These observations correlated well with the HRMAS 1HMRS measurements listed in Table 2. Metabolite concentrations in Table 2 were calculated, using tissue water as an internal standard, according to the following equation:
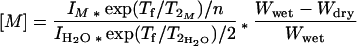 |
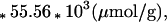 |
1 |
where I represents measured intensities for metabolites and tissue water, Tf (600 ms) was the length of T2 filter and n is the number of protons giving rise to the resonance. The percentage of tissue water in each brain sample was measured by weighing tissue before (Wwet) and after (Wdry) lyophilization. The mean value for the percent water content was 80.4 ± 1.8%.
Figure 4.

Comparison of HRMAS 1HMR spectra of brain tissue from primary visual cortex region, relatively unaffected by Pick disease (spectrum a), and the severely affected rostral inferior temporal gyrus region (spectrum b) at 2°C. Spectra were scaled according to the concentration of creatine (3.03 ppm, 8.48 μmol/g for spectrum a and 4.96 μmol/g for spectrum b) for better visualization.
Table 2.
Chemical shifts, concentration, and SE of metabolites of Pick disease brain
| Met | Chemical shift, ppm | PVC (n = 2)
|
ITG (n = 5)
|
STG (n = 4)
|
SFG (n = 4)
|
||||
|---|---|---|---|---|---|---|---|---|---|
| Conc., μmol/g | SE | Conc., μmol/g | SE | Conc., μmol/g | SE | Conc., μmol/g | SE | ||
| NAA | 2.01 (s) | 8.38 | 0.91 | 1.21*** | 0.14 | 3.98* | 0.94 | 2.98** | 0.50 |
| Acet | 1.92 (s) | 2.96 | 0.54 | 1.07** | 0.13 | 1.85* | 0.11 | 1.81 | 0.42 |
| Lac | 4.12 (q) | 35.35 | 2.09 | 15.90*** | 1.35 | 20.11* | 2.74 | 18.14** | 1.64 |
| 1.33 (d) | 38.14 | 2.99 | 20.94** | 2.33 | 24.84* | 1.41 | 24.94* | 1.96 | |
| Cre | 3.93 (s) | ||||||||
| 3.03 (s) | 8.48 | 0.99 | 4.96** | 0.41 | 7.69 | 0.66 | 6.86 | 1.09 | |
| Ala | 1.48 (d) | 3.20 | 0.51 | 1.94* | 0.22 | 3.45 | 0.12 | 2.92 | 0.71 |
| Gly | 3.56 (s) | ||||||||
| Cho | 3.20 (s) | 2.43 | 0.33 | 0.82** | 0.12 | 1.41* | 0.27 | 1.47 | 0.33 |
| PCh | 3.22 (s) | 2.33 | 0.43 | 1.76 | 0.28 | 4.15 | 0.66 | 2.80 | 0.57 |
| Glu | 2.34 (m) | 4.62 | 0.11 | 2.03*** | 0.32 | 4.69 | 0.88 | 3.93 | 0.61 |
| Gln | 2.44 (m) | ||||||||
| Asp | 2.85 (d), 2.79 (d), 2.68 (m) | ||||||||
| Tau | 3.41 (t) | ||||||||
| Inos | 3.54 (d) | 18.19 | 1.62 | 11.98 | 1.99 | 18.54 | 4.14 | 16.09 | 3.58 |
| 3.52 (d) | |||||||||
| 4.05 (t) | 29.05 | 7.01 | 15.09* | 2.18 | 26.52 | 4.34 | 23.46 | 5.75 | |
| GABA | 3.00 (m), 2.29 (m), 1.90 (m) | ||||||||
| Val | 1.05 (d), 0.95 (d) | ||||||||
Met, metabolites; PVC, primary visual cortex; ITG, inferior temporal gyrus; STG, superior temporal gyrus; SFG, superior frontal gyrus; Conc., concentration; s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. See the Fig. 2 legend for definitions of the metabolite abbreviations. The concentrations for TG, STG, and SFG samples are significant relative to the concentrations for PVC. ∗, P < 0.05; ∗∗, P < 0.005; ∗∗∗, P < 0.0005.
Fig. 5 depicts the correlations between the concentrations of NAA and NAA + Acet with neuronal counts measured in variously affected regions of Pick disease brain. The correlations appear linear. The correlation coefficients are 0.78 and 0.77 for [NAA + Acet] and [NAA], respectively, (P < 0.0001 for both), suggesting that the concentration of NAA is directly related to the number of neurons. Notably, the lines do not pass through the origin.
Figure 5.

The correlation between the population of surviving cortical neurons and the [NAA + Acet] (•) and [NAA] (○) concentrations measured in different Pick brain regions. The concentrations are corrected according to their measured T2 values. See text for details.
Changes of Metabolite T2 Due to Pick Disease.
Table 3 summarizes the alteration in tissue metabolite T2, spin–spin relaxation time, in each disease-affected brain region. We found prolongation of T2 for most of the metabolites in the diseased regions compared with the more normal region of primary visual cortex.
Table 3.
Spin–spin relaxation time T2 (ms) and SE of metabolites in regions of Pick disease brain
| Met | CS, ppm | PVC (n = 2)
|
ITG (n = 5)
|
STG (n = 4)
|
SFG (n = 4)
|
||||
|---|---|---|---|---|---|---|---|---|---|
| ms | SE | ms | SE | ms | SE | ms | SE | ||
| NAA | 2.01 (s) | 302.9 | 23.9 | 463.4** | 15.9 | 373.9* | 18.7 | 353.2 | 15.9 |
| Acet | 1.92 (s) | 409.3 | 29.0 | 738.3** | 43.7 | 565.3 | 61.7 | 539.8* | 40.0 |
| Lac | 4.12 (1) | 250.4 | 16.5 | 298.6** | 9.6 | 289.6* | 8.0 | 278.2* | 4.2 |
| 1.33 (d) | 285.0 | 20.8 | 411.9*** | 9.3 | 370.6** | 11.0 | 338.4* | 10.1 | |
| Cre | 3.03 (s) | 324.8 | 19.9 | 596.2*** | 17.8 | 509.7* | 29.2 | 433.8* | 31.5 |
| Ala | 1.48 (d) | 313.0 | 16.6 | 532.4*** | 26.5 | 388.0** | 15.9 | 384.7* | 27.0 |
| Cho | 3.20 (s) | 467.7 | 8.5 | 619.9 | 40.3 | 517.7 | 25.1 | 491.7 | 6.0 |
| PCh | 3.22 (s) | 327.8 | 7.4 | 374.3 | 13.9 | 353.0 | 13.5 | 334.0 | 10.3 |
| Glu | 2.34 (m) | 291.1 | 21.1 | 400.7** | 13.9 | 342.4 | 19.9 | 323.5 | 19.0 |
| Inos | 3.54 (d) | 284.1 | 26.9 | 351.5* | 14.4 | 336.0 | 11.4 | 311.6 | 9.3 |
| 4.05 (t) | 276.8 | 2.5 | 309.9 | 9.4 | 295.9 | 14.6 | 292.3 | 10.6 | |
| Water | 5.00 (s) | 91.2 | 1.4 | 122.6*** | 4.0 | 111.6 | 9.6 | 98.8 | 4.0 |
See the Table 2 legend for definitions of abbreviations and footnotes.
DISCUSSION
MAS Produces High Spectral Resolution in Unprocessed Brain Tissue.
Figs. 1 and 2 both demonstrate that HRMAS 1HMRS of unprocessed brain tissue can produce spectra of a quality heretofore observed only with liquid samples of brain extracts. In Fig. 1, the nonspinning MR spectrum (spectrum a) was measured with the same sample, the same protocol, and after the acquisition of the spinning spectrum (spectrum b). This fact illustrates that the observed enhancement in spectral resolution seen in spectrum b (Fig. 1) is the direct effect of MAS and is not the result of tissue rupture that might be caused by the mechanical force of spinning.
The Difference Between HRMAS and Solution NMR.
There are quantitative and qualitative differences between the spectra of intact tissue and extracts, as shown in Fig. 2. We are particularly interested in differences in the NAA resonance, a metabolite found exclusively in neurons in adult brain (24–29) and proposed as a marker for neuronal loss or dysfunction. In the unprocessed brain spectrum (Fig. 2, spectrum a), there is a prominent resonance due to the methyl moiety of Acet at 1.92 ppm. Our HRMAS 1HMRS studies on degradation of human and monkey brain show that the increase in Acet is mostly due to the degradation of NAA (Fig. 6). Although Acet may also be produced by other neurochemical degradation processes, this observation is in agreement with results reported in a study of rabbit brain (8). Therefore, the sum of NAA and Acet may represent an upper limit, and NAA alone may be a lower limit of the total NAA concentration before death. As is clear in Fig. 2, the brain extraction procedure that we employed results in loss of Acet. The NAA concentration of a brain extract is very likely lower than the upper limit of the tissue spectrum. A lower limit cannot be estimated without knowledge of the postmortem decomposition of NAA, the loss of Acet during extraction, and the efficiency of the extraction procedure for NAA. Results shown in Fig. 6 suggest that the longer the postmortem time period, the larger the error of this underestimation. In practice, one may need to evaluate these two limits whenever human tissue is the focus of studies because the time period between tissue death and tissue preservation always exists in such cases, even with tissue obtained from surgery.
Figure 6.

Degradation of normal human brain tissue at 20°C measured at 2.5 kHz spinning. The concentrations of NAA (○) and Acet (•) are normalized by the total spectral intensity in the region of 0.5–4.5 ppm.
Another aspect related to tissue degradation is that intact brain tissue undergoing MR measurements at room temperature will still be subjected to neurochemical degradation, as shown in Fig. 6, unlike tissue extraction, where metabolic reactions are terminated at the point when the tissue is frozen in liquid nitrogen and subsequently extracted. In both human and monkey brain studies, we found a 4-fold reduction in rates of NAA decrease and Acet increase when the experimental temperature was reduced from 20°C to 2°C. This reduction in degradation rates resulted in less than 5% of signal changes for the most rapidly degrading metabolites during the duration of NMR measurements (2 h). Thus, brain tissue studied at low temperature may effectively prevent tissue degradation during NMR measurements. The extra spectral broadening at such low temperature is overcome using MAS.
Another major difference between the unprocessed brain and extract spectra is the relative intensities of the choline containing metabolites at ≈3.2 ppm. These metabolites give rise to a prominent and characteristic resonance observed in 1HMRS spectra of mammalian brain in vivo, and have been found to be abnormal in several neurological diseases. We note that, compared with the extract spectrum, the choline and phosphocholine peaks in the spinning spectrum are substantially more intense relative to the intensity of the creatine resonance (at 3.03 ppm). This difference is maintained after correction for T2 effects. The creatine/phosphocreatine resonance is generally thought to be less affected by chemical extraction. Our creatine HRMAS concentration of 8.48 μmol/g in the relatively normal primary visual cortex of Pick disease brain indeed shows a close agreement with the commonly accepted value of 7.6 μmol/g from extracts (30). However, comparing our HRMAS concentrations for choline and phosphocholine with those from extracts, we observed a 3-fold difference in concentration for the sum of both metabolites, 4.76 μmol/g from HRMAS vs. 1.6 μmol/g from extracts (30). Thus, the extraction procedure may only partially release choline-containing metabolites, possibly due to close association with cell membranes. With HRMAS, we can increase the observability of the these molecules, resulting in better representation of their in vivo MRS-observable concentrations in brain. Several other differences between the tissue and extract spectra were detected. Clearly, direct analyses of unprocessed brain by HRMAS is superior to brain extract MRS for understanding changes observed by in vivo brain MRS.
Pick Disease Neuronal Loss and NAA Decrease.
Our major goal was to establish a correlation between histopathological evidence of neuronal loss and a decrease in NAA concentration as determined by HRMAS 1HMRS. Although qualitatively apparent on microscopic examination (Fig. 3), we performed a quantitative evaluation of neuronal loss. We counted surviving pyramidal neurons per unit surface area (×250 microscopic field or 0.454 mm2) specifically located within cortical layer III and found significant differences between the Pick disease brain and the normal elderly control brain as well as among different areas in the former. It should be noted that due to the “shrinkage effect” of neuropil loss, the actual degree of neuronal loss in the inferior temporal cortex of Pick brain likely exceeds the 61% loss relative to the normal control (see Table 1). The NAA concentration in unprocessed tissue measured by HRMAS 1HMRS, in our opinion, is not subject to some of these complicating factors of histological evaluation and may provide a more reliable estimation of the remaining functional neuronal population.
As demonstrated in Fig. 5, the correlations between the concentrations of upper and lower limits of NAA ([NAA + Acet] and [NAA], respectively) and neuronal counts in different, variously affected regions of Pick disease brain are linear. These concentrations were quantified using tissue water as an internal standard and T2 measurements to correct for T2-filter effects. For the upper limit, the most severely affected brain region (rostral inferior temporal gyrus) had a [NAA + Acet] of ≈20% of the histopathologically relatively normal primary visual cortex (P < 0.0001). The less severely affected regions, the superior temporal gyrus and the superior frontal gyrus, had a [NAA + Acet] of 49% (P < 0.0174) and 58% (P < 0.0070) of the normal, respectively. For the lower limit of [NAA], we observed a similar degree of relative alteration in NAA concentration with respect to different brain regions. These linear correlations between NAA concentrations measured in unprocessed tissue and neuronal populations measured by histopathology suggest that this spectroscopic method can indeed be used to evaluate the population change of cortical neurons due to neuronal diseases. Extrapolating to zero [NAA], the estimated neuronal count remains above zero. We interpret this to mean that diminished NAA may also represent damage to remaining neurons. That is, although neurons are still visible, they may be dysfunctional and this is reflected in a decreased level of NAA. To the best of our knowledge, this is the first direct, quantitative demonstration of a correlation between neuronal loss and diminished NAA in a human neurological disease.
Pick Disease Increases T2 of Metabolites.
Finally, we examined the T2 relaxation times of each of these four brain regions. Table 2 reveals a relationship between T2 changes and disease severity. The more severely affected regions had a longer T2 for most metabolites, as well as tissue water. An increase in the T2 relaxation time of tissue water revealed by MR imaging has been observed in many brain diseases. This observation of T2 prolongation in metabolites has substantial significance in the interpretation of in vivo MR experiments. It suggests that constancy of metabolite T2 relaxations cannot be assumed, and in vivo estimates of metabolite concentrations must take this factor into account if a T2-weighted study is conducted.
CONCLUSION
Our results indicate that HRMAS 1HMRS is an effective new method for neurochemical investigation. It is rapid, quantitative, nondestructive, and relevant in vivo, which makes it suitable as a tool for clinical and experimental neuropathologic investigations. More important, it permits an understanding of the diagnostic information provided by in vivo MR spectroscopy, and thus can guide its development as a highly specific medical imaging method.
Acknowledgments
We gratefully acknowledge helpful discussions with Professors R. G. Griffin of Massachusetts Institute of Technology and E. T. Hedley-Whyte of Harvard Medical School. This work was supported in part by Public Health Service Grants NS34626 to R.G.G.; RR07000, RR00168, and NS30769 to A.L.; and RR00995 to R. G. Griffin and the Francis Bitter Magnet Laboratory at the Massachusetts Institute of Technology.
ABBREVIATIONS
- MR
magnetic resonance
- MAS
magic angle spinning
- HRMAS
high resolution MAS
- MRS
MR spectroscopy
- NAA
N-acetylaspartate
- Acet
acetate
References
- 1.Howe F A, Maxwell R J, Saunders D E, Brown M M, Griffiths J R. Magn Reson Q. 1993;9:31–59. [PubMed] [Google Scholar]
- 2.Vion-Dury J, Meyerhoff D J, Cozzone P J, Weiner M W. J Neurol. 1994;241:354–371. doi: 10.1007/BF02033352. [DOI] [PubMed] [Google Scholar]
- 3.Coetey A, Jarvik J G, Lenkinski R E, Grossman R I, Frank I, Delivoria-Papadopoulos M. Am J Neuroradiol. 1994;15:1853–1859. [PMC free article] [PubMed] [Google Scholar]
- 4.Maier M. Br J Psychiatry. 1995;167:299–306. doi: 10.1192/bjp.167.3.299. [DOI] [PubMed] [Google Scholar]
- 5.Auld K L, Ashwal S, Holshouser B A, Tomasi L G, Perkin R M, Ross B D, Hinshaw D B., Jr Pediatr Neurol. 1995;12:323–334. doi: 10.1016/0887-8994(95)00062-k. [DOI] [PubMed] [Google Scholar]
- 6.Shonk T K, Moats R A, Gifford P, Michaelis T, Mandigo J C, Izumi J, Ross B D. Radiology. 1995;195:65–72. doi: 10.1148/radiology.195.1.7892497. [DOI] [PubMed] [Google Scholar]
- 7.Preul M C, Caramanos Z, Collins D L, Villemure J-G, Leblanc R, Olivier A, Pokrupa R, Arnold D L. Nat Med. 1996;2:323–325. doi: 10.1038/nm0396-323. [DOI] [PubMed] [Google Scholar]
- 8.Petroff O A C, Ogino T, Alger J R. J Neurochem. 1988;51:163–171. doi: 10.1111/j.1471-4159.1988.tb04850.x. [DOI] [PubMed] [Google Scholar]
- 9.Guimaraes A R, Schwartz P, Prakash M R, Carr C A, Berger U V, Jenkins B G, Coyle J T, Gonzalez R G. Neuroscience. 1995;96:1095–1101. doi: 10.1016/0306-4522(95)00300-8. [DOI] [PubMed] [Google Scholar]
- 10.Kuesel A C, Sutherland G R, Halliday W, Smith I C P. NMR Biomed. 1994;7:149–155. doi: 10.1002/nbm.1940070308. [DOI] [PubMed] [Google Scholar]
- 11.Cheng L L, Lean C L, Bogdanova A, Wright S C, Jr, Ackerman J L, Brady T J, Garrido L. Magn Reson Med. 1996;36:653–658. doi: 10.1002/mrm.1910360502. [DOI] [PubMed] [Google Scholar]
- 12.Andrew E R, Bradbury A, Eades R G. Nature (London) 1958;182:1695. [Google Scholar]
- 13.Lowe I J. Phys Rev Lett. 1959;2:285. [Google Scholar]
- 14.Maricq M M, Waugh J S. J Chem Phys. 1979;70:3300–3316. [Google Scholar]
- 15.VanderHart D L, Earl W L, Garroway A N. J Magn Reson. 1981;44:361. [Google Scholar]
- 16.Haberkron R A, Herzfeld J, Griffin R G. J Am Chem Soc. 1987;100:1296. [Google Scholar]
- 17.Forbes J, Husted C, Oldfield E. J Am Chem Soc. 1988;110:1059. [Google Scholar]
- 18.Rutar V. Food Chem. 1989;37:67–70. [Google Scholar]
- 19.Gross J D, Costa P R, Dubacq J P, Warschawski D E, Lirsac P N, Devaux P F, Griffin R G. J Magn Reson Ser B. 1995;106:187–190. doi: 10.1006/jmrb.1995.1031. [DOI] [PubMed] [Google Scholar]
- 20.Davis J H, Auger M, Hodges R S. Biophys J. 1995;69:1917–1932. doi: 10.1016/S0006-3495(95)80062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baldwin B, Forstl H. Br J Psychiatry. 1993;163:100–104. doi: 10.1192/bjp.163.1.100. [DOI] [PubMed] [Google Scholar]
- 22.Hansen L A. In: Alzheimer’s Disease. Terry R D, Katzman R, Bick K L, editors. New York: Raven; 1994. pp. 167–177. [Google Scholar]
- 23.Vonsattel J P G, Aizawa H, Ge P, DiFiglia M, McKee A C, MacDonald M, Gusella J F, Landwehrmeyer G B, Bird E D, Richardson E P, Hedley-White E T. J Neuropathol Exp Neurol. 1995;54:42–56. doi: 10.1097/00005072-199501000-00006. [DOI] [PubMed] [Google Scholar]
- 24.Tallan H, Moore S, Stein W. J Biol Chem. 1956;219:247–264. [PubMed] [Google Scholar]
- 25.Nadler J, Cooper J. J Neurochem. 1972;14:551–554. [Google Scholar]
- 26.Moffett J, Namboodiri M, Cangro C, Neale J. NeuroReport. 1991;2:131–134. doi: 10.1097/00001756-199103000-00005. [DOI] [PubMed] [Google Scholar]
- 27.Simmons M L, Frondoza C G, Coyle J T. Neuroscience. 1991;45:37–45. doi: 10.1016/0306-4522(91)90101-s. [DOI] [PubMed] [Google Scholar]
- 28.Miller D H, Austin S J, Connelly A, Youl B D, Gadian D G, McDonald W I. Lancet. 1991;337:58–59. doi: 10.1016/0140-6736(91)93383-k. [DOI] [PubMed] [Google Scholar]
- 29.Petroff O A C, Graham G D, Blamire A M, Al-Rayess M, Rothman D L, Fayad P B, Brass L M, Shulman R G, Prichard J W. Neurology. 1992;42:1349–1354. doi: 10.1212/wnl.42.7.1349. [DOI] [PubMed] [Google Scholar]
- 30.Jenkins B G, Chen I Y, Rosen B R. In: Mitochondria and Free Radicals in Neurodegenerative Diseases. Beal M F, Howell N, Bodis-Wolnet I, editors. New York: Wiley–Liss; 1997. , in press. [Google Scholar]