

Abstract
Receptor subunits for the neurocytokine ciliary neurotrophic factor (CNTF) share sequence similarity with the receptor for leptin, an adipocyte-derived cytokine involved in body weight homeostasis. We report here that CNTF and leptin activate a similar pattern of STAT factors in neuronal cells, and that mRNAs for CNTF receptor subunits, similarly to the mRNA of leptin receptor, are localized in mouse hypothalamic nuclei involved in the regulation of energy balance. Systemic administration of CNTF or leptin led to rapid induction of the tis-11 primary response gene in the arcuate nucleus, suggesting that both cytokines can signal to hypothalamic satiety centers. Consistent with this idea, CNTF treatment of ob/ob mice, which lack functional leptin, was found to reduce the adiposity, hyperphagia, and hyperinsulinemia associated with leptin deficiency. Unlike leptin, CNTF also reduced obesity-related phenotypes in db/db mice, which lack functional leptin receptor, and in mice with diet-induced obesity, which are partially resistant to the actions of leptin. The identification of a cytokine-mediated anti-obesity mechanism that acts independently of the leptin system may help to develop strategies for the treatment of obesity associated with leptin resistance.
Ciliary neurotrophic factor (CNTF) is a pluripotent neurocytokine expressed by glial cells in peripheral nerve and in the central nervous system, which stimulates gene expression, cell survival, or differentiation in a variety of neuronal and nonneuronal cells (1, 2). In addition to preventing neuronal degeneration in various lesion paradigms (1, 2), systemically administered CNTF causes fever, acute-phase response, weight loss, and anorexia in experimental animals (3–7) and humans (8, 9). The effects of CNTF on body weight change and food intake in mice are transient (6, 7), and their mechanism is unknown.
CNTF exerts its biological actions through the binding, sequential assembly, and activation of a multisubunit receptor complex composed of a ligand-specific subunit (CNTFRα) and two signal-transducing subunits, gp130 and leukemia inhibitory factor (LIF) receptor (LIFR) (2, 10). Binding of CNTF to CNTFRα triggers the subsequent association and heterodimerization of gp130 and LIFR, leading to the activation of a signaling cascade mediated by protein tyrosine kinases of the Jak family and STAT transcription activators (2, 10). The signal transducers gp130 and LIFR share sequence similarity (11) and signaling capabilities (12) with the recently identified receptor (OB-R; refs. 11, 13, 14) for leptin, an adipocyte-derived cytokine involved in body weight homeostasis (15–17). Mice lacking leptin (ob/ob mice) or functional OB-R (db/db mice) fail to restrain their food intake and become obese (11, 14, 18). The hyperphagia and adiposity of ob/ob mice can be reversed by systemic administration of leptin (15–17).
The objective of the present study was to investigate whether the weight- and appetite-reducing effects of CNTF may be mediated by a mechanism similar to that of leptin. To this end, we compared the CNTF- and leptin-induced activation of STAT factors in neuronal cells, the biological activities of CNTF and leptin in obese mice, and the expression and activation of the corresponding cytokine receptors in hypothalamic satiety centers. Our results reveal a striking analogy between the metabolic actions of a lipostatic hormone and a neurocytokine.
MATERIALS AND METHODS
Protein Production.
Recombinant human CNTF and DH-CNTF (Ser-166 → Asp/Gln-167 → His CNTF; ref. 19), were produced in Escherichia coli BL21 as previously described (20). The DNA coding sequence for human leptin was assembled by PCR using synthetic oligodeoxyribonucleotides according to the method of Stemmer et al. (21) and subcloned into the bacterial expression plasmid pRSET-5d (22). Human leptin was produced using the same protocol as for CNTF. Following purification by reverse-phase HPLC (20), all proteins migrated as single bands of the expected size on reducing SDS/PAGE gels stained with Coomassie blue, and they contained less than 5 ng of endotoxin per mg of protein, as determined by the Limulus amoebocyte assay (Sigma).
STAT Activation Assay.
The murine septal neuron–neuroblastoma hybrid cell line SN-56 (23) was maintained in complete culture medium (Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum, penicillin, glutamine, and sodium pyruvate). Cells were plated in 100-mm dishes and used 24 hr later, when semiconfluent. An expression vector containing the entire coding region (nucleotides 141–3,770) of human OB-R (13) was prepared as previously described (24) and was transfected into the cells by using Lipofectamine (GIBCO/BRL) according to the manufacturer’s instructions. After 24 hr, cells were distributed into 60-mm culture dishes containing complete culture medium, and after an additional 24 hr, they were deprived of serum for 4 hr before a 10-min treatment with cytokines, as specified in Results. The cells were then washed with ice-cold phosphate-buffered saline containing 50 mM NaF, collected by centrifugation, and frozen in liquid nitrogen. Total cell extracts were prepared as previously described (25). Binding of activated STAT factors to the high-affinity SIE m67 oligonucleotide (26) was determined by electromobility shift assays according to Sadowski and Gilman (27), using 10 μg of cell extract. The oligonucleotide probe was labeled by filling in 5′ protruding ends with the Klenow fragment of DNA polymerase in the presence of [α-32P]dATP and [α-32P]dCTP (3,000 Ci/mmol; 1 Ci = 37 GBq). Complexes were resolved on 5% polyacrylamide/2.5% glycerol/0.5× TBE (45 mM Tris borate/0.5 mM EDTA, pH 7.8) gels, which were then dried and subjected to autoradiography.
Animal Studies.
Experiments were performed using groups of male 10- to 18-week-old C57BL/6J ob/ob and C57BL/KS db/db mice, and 19-week-old AKR/J mice rendered obese by feeding a high-fat diet (28) starting at 12 weeks of age. Except where noted otherwise, animals were housed in individual cages with ad libitum access to water and either standard or high-fat (AKR mice) rodent chow, under a 12-hr light–dark cycle (lights on at 0730, off at 1930). They were accustomed to daily (900 hr) intraperitoneal injections of vehicle (0.9% saline/0.2 mg/ml endotoxin-free bovine serum albumin) for two days before the beginning of the treatment (day 0) with either vehicle or cytokines. Animals were weighed after injection and food intake was determined by recording the amount of chow remaining in food dishes. In pair-feeding experiments, vehicle-treated mice were either fed ad libitum or fed the amount of chow consumed by DH-CNTF-treated mice during the preceding 24-hr period, starting at day 1. Blood samples were taken from the retroorbital sinus 24 hr after the last injection (900), or 7 hr after the last injection and the removal of food (1600). Serum glucose was determined by the glucose oxidase method and serum insulin by radioimmunoassay (Amersham), using rat insulin as standard.
Locomotor activity was measured by scoring the number of times mice crossed the middle of their home cages during 3 hr of the dark cycle (2100–2400). Grooming behavior was assessed by focal observations in home cages (five observations of 1 min each during 30 min of the light cycle), using a rating scale from 0 to 3 (0, no activity; 1, weak; 2, normal; and 3, hyperactive). Conditioned taste aversion experiments were performed using a two-bottle paradigm with 0.1% saccharin as a novel taste (29).
Body Composition.
Carcasses were homogenized, and 2-g aliquots were lyophilized and then oven-dried at 90°C until weight was constant. Fat was then extracted with diethyl ether/ethanol (20:1, vol/vol) (30). Water and fat mass were calculated from the weight differences after dehydration and fat extraction, respectively. Lean mass was defined as the remaining amount of carcass.
Determination of tis-11 mRNA Levels by Reverse Transcription (RT)-PCR.
Hypothalamic RNA was extracted using a commercial kit (Biotecx), and the relative expression levels of the tis-11 primary response gene (31) and of β-actin (as control) were determined by RT-PCR. First-strand cDNA, prepared from 1 μg of RNA using (dT)17NN as primer, was amplified using primers derived from murine tis-11 (5′-ATGGATCTCTCTGCCTCTACGAGAGCCTC-3′ and 5′-TAGGGGCAGCGGCCCTGGAGGTAGAACTTG-3′) or murine β-actin (5′-TCATGAAGTGTGACGTTGACATCCGT-3′ and 5′-CCTAGAAGCACTTGCGGTGCACGATG-3′). Following PCR amplification for 30 cycles (1 min 94°C, 1 min 62°C, 2 min 72°C), products were resolved on a 1% agarose gel and visualized by ethidium bromide staining. Under these conditions, staining intensity was proportional to the amount of input cDNA.
In Situ Hybridization.
Serial coronal brain sections were prepared in the region containing the arcuate and paraventricular hypothalamic nuclei. In situ hybridization was performed according to previously described procedures (32), using 35S-labeled cRNA probes. Probes for murine OB-Rb, CNTFRα, LIFR, and tis-11 were obtained by RT-PCR amplification of mouse brain RNA using appropriate oligonucleotide primers, and corresponded to nucleotides 2850–3407 (GenBank accession no. U46135U46135), 246–856 (numbering according to the human sequence; GenBank accession no. M73238M73238), 2620–3217 (numbering according to the human sequence; GenBank accession number X61615X61615), and 1–950 (GenBank accession no. M58565M58565) of the respective coding sequences. The specificity of the probes was confirmed by DNA sequencing and Northern analysis using total mouse brain RNA.
RESULTS AND DISCUSSION
STAT Activation by CNTF and Leptin.
We directly compared the ability of CNTF and leptin to induce STAT factor activation in murine septal neuron-derived SN-56 cells, which express functional CNTF receptors (20). CNTF induced a pattern of STAT activation characteristic of gp130-signaling cytokines (33), with predominant DNA binding of STAT3 homodimers and, to a lesser degree, STAT1 homodimers and STAT1/STAT3 heterodimers (Fig. 1). Leptin produced no detectable signal (Fig. 1), consistent with the fact that SN-56 cells do not express OB-Rb, the signaling-competent long-form splice variant of leptin receptor (12, 24, 35) (as determined by RT-PCR analysis; data not shown). In SN-56 cells transfected with an expression vector for human OB-Rb (24), both cytokines triggered STAT activation in a dose-dependent and saturable manner, with EC50 values in the ng/ml range (A.D., A.D.M., and R.L., unpublished data). The pattern of leptin-induced STAT factors was similar to that induced by CNTF (Fig. 1), consistent with the sequence similarity, including the presence of consensus motifs for Jak kinase and STAT factor interaction sites, between OB-Rb and receptors of the gp130 family (11). Similar results were obtained in an OB-Rb-transfected hypothalamic cell line (ref. 24 and unpublished results).
Figure 1.

Effects of leptin and CNTF on STAT factor activation in SN-56 cells and SN-56 cells transfected with human OB-Rb. Cells were incubated for 10 min in the presence or absence of CNTF (C) or leptin (L) (100 ng/ml). Arrows denote the positions of migration of DNA-bound STAT3 homodimers (a), STAT1–STAT3 heterodimers (b), and STAT1 homodimers (c) (34).
Anti-Obesity Effects of CNTF.
To determine whether CNTF shares biological activities with leptin, cytokines were administered to genetically obese mice and mice with diet-induced obesity (DIO). In agreement with previous reports (15–17, 36), leptin treatment of mutant ob/ob mice, which do not express functional leptin, reversed the obesity and hyperphagia associated with leptin deficiency. Daily intraperitoneal administration of human CNTF (from 2 to 50 μg per mouse) to ob/ob mice also produced a progressive and dose-dependent decrease of body weight, as well as a rapid reduction in food intake (Fig. 2). At the highest dose tested (50 μg), CNTF caused a 16% decrease in body weight after 7 days, as compared with a 5% increase in vehicle-treated controls, and a 5-fold decrease in food intake. These effects were comparable in magnitude to those of a high dose (100 μg) of leptin (13% ± 2% and 95% ± 2% reductions in body weight and food intake, respectively; P < 0.0001 by Student’s t test; see also Table 1). The human CNTF superagonist variant DH-CNTF, which possesses increased biological activity on mouse neuronal cells (20), produced reductions in body weight (Table 1) and food intake (data not shown) at doses ≈ of those for CNTF. This result, together with the lack of activity of CNTF variants (20) with impaired receptor interaction (data not shown), suggest that the anti-obesity effects of CNTF are mediated through activation of the CNTF receptor complex.
of those for CNTF. This result, together with the lack of activity of CNTF variants (20) with impaired receptor interaction (data not shown), suggest that the anti-obesity effects of CNTF are mediated through activation of the CNTF receptor complex.
Figure 2.

Effects of CNTF and leptin on body weight (Left) and food intake (Right) in genetically obese mice (ob and db) and mice with (DIO). Mice received daily intraperitoneal injections of either vehicle or cytokines (amounts in μg per mouse), starting at day 0. Body weight is expressed as percent of the original weight on day −2 and represents the mean ± SEM (n = 3 for ob/ob and db/db, n = 5 for DIO mice). Baseline weights for each group of vehicle-treated animals were (in g): ob/ob, 49.3 ± 0.3; db/db, 39.1 ± 2.5; DIO, 42.6 ± 0.8. Statistical significance was determined by repeated measures ANOVA. For all groups, P values for the effects of treatment, time, and time × treatment were P < 0.05, P < 0.0001, and P < 0.01, respectively.
Table 1.
Effects of cytokines and pair-feeding on body weight, serum insulin, and glucose in obese mice
| Treatment | Weight change, g | Serum glucose, mM | Serum insulin, ng/ml |
|---|---|---|---|
| Experiment 1 (ob/ob, 7 days) | |||
| Vehicle | +1.6 ± 0.1 | ND | 63.3 ± 12.7 |
| Leptin | −6.5 ± 0.4** | ND | 8.1 ± 9.1* |
| CNTF | −8.2 ± 0.1** | ND | 4.3 ± 1.0* |
| DH-CNTF | −7.7 ± 0.8** | ND | 3.2 ± 2.9* |
| Experiment 2 (ob/ob, 4 days) | |||
| Vehicle | +0.5 ± 0.5 | ND | 72.5 ± 25.7 |
| DH-CNTF | −8.4 ± 0.5**§ | ND | 8.1 ± 0.2*† |
| Pair-fed | −7.0 ± 0.5** | ND | 11.1 ± 0.4* |
| Experiment 3 (db/db, 7 days) | |||
| Vehicle | +0.2 ± 0.4 | 23.3 ± 0.8 | 9.1 ± 4.2 |
| Leptin | −0.8 ± 0.5 | 28.7 ± 0.8* | 9.7 ± 2.6 |
| CNTF | −6.8 ± 0.5** | 8.4 ± 1.7** | 8.2 ± 2.1 |
| Experiment 4 (dg/db, 4 days) | |||
| Vehicle | 0.0 ± 0.3 | 30.1 ± 2.0 | ND |
| DH-CNTF | −6.8 ± 0.4**§ | 12.3 ± 1.9**§ | ND |
| Pair-fed | −5.3 ± 0.4** | 24.8 ± 5.4 | ND |
Mice were treated with vehicle, leptin (100 μg), CNTF (50 μg), or DH-CNTF (10 μg). Blood samples were taken 24 hr after the last injection (experiments 1 and 3), or 7 hr after the last injection and the removal of food (experiments 2 and 4). Data represent mean values ± SEM from three to six animals per treatment group. ND, not determined. ∗, P < 0.05 vs. vehicle; ∗∗, P < 0.001 vs. vehicle; §, P < 0.05 vs. pair-fed; †, P < 0.001 vs. pair-fed (Student’s t test).
The db/db mouse mutant is resistant to the action of leptin (15–17, 36), due to a mutation in the gene coding for OB-R resulting in the production of a receptor splice variant with a truncated intracytoplasmic domain (11, 14). In contrast, treatment of db/db mice with CNTF led to dose- and time-dependent weight loss and suppression of food intake (Fig. 2). The superagonist DH-CNTF elicited comparable effects at ≈ the dose of CNTF (see Table 1). Other weight-loss-inducing cytokines such as tumor necrosis factor and interleukin 1 have been reported to enhance leptin expression and release from adipose tissue (37), suggesting that their metabolic effects are mediated, at least in part, through the leptin system. The results obtained in ob/ob and db/db mice, together with the inability of CNTF to activate signaling through OB-Rb and to bind to the mutant db/db OB-R (A.D., I.G., A.D.M., and R.L., unpublished results) demonstrate that CNTF does not act by stimulation of leptin release or direct activation of leptin receptors.
the dose of CNTF (see Table 1). Other weight-loss-inducing cytokines such as tumor necrosis factor and interleukin 1 have been reported to enhance leptin expression and release from adipose tissue (37), suggesting that their metabolic effects are mediated, at least in part, through the leptin system. The results obtained in ob/ob and db/db mice, together with the inability of CNTF to activate signaling through OB-Rb and to bind to the mutant db/db OB-R (A.D., I.G., A.D.M., and R.L., unpublished results) demonstrate that CNTF does not act by stimulation of leptin release or direct activation of leptin receptors.
AKR mice rendered obese by feeding a high-fat diet (DIO mice) have been reported to be less sensitive than ob/ob mice to the weight- and appetite-reducing actions of leptin (17). This finding, together with the observation that plasma levels of leptin are higher in DIO mice than in lean littermates, led to the proposal that diet-induced obesity is associated with leptin resistance (38, 39). As depicted in Fig. 2, a 5-day treatment of DIO mice with a 100 μg daily dose of leptin caused modest decreases in body weight (7% ± 1%; P < 0.05 vs. vehicle) and food intake (27% ± 2%; P < 0.05). In contrast, CNTF (50 μg) and DH-CNTF (10 μg) elicited potent reductions in body weight (19% ± 1% and 24% ± 1%, respectively; P < 0.0001) and food intake (76% ± 4% and 73% ± 7%, respectively; P < 0.0005) in DIO mice. The finding that CNTF can reverse the obesity of db/db as well as DIO mice could help to develop strategies for the treatment of human obesity, which was postulated to be associated with leptin resistance (38, 40, 41).
Duration and Specificity of the CNTF Effects.
A 25-day treatment of db/db mice with DH-CNTF led to a progressive and steady decrease in body weight, which by day 10 reached a level corresponding to that of strain-matched lean mice (Fig. 3). In parallel, DH-CNTF elicited an ≈50% decrease in food intake, which persisted throughout the treatment. In contrast, the CNTF superagonist elicited only transient effects in strain-matched wild-type mice. Thus, DH-CNTF rapidly depressed both food intake and the rate of body weight change in lean mice, but these effects subsided after approximately 5 and 10 days of treatment, respectively (Fig. 3). Interestingly, leptin also elicits transient effects in lean mice (15, 42).
Figure 3.

Duration of DH-CNTF effects on body weight and food intake in obese vs. lean mice. C57BL/KS db/db mice (circles), or age- and sex-matched C57BL/KS +/+ mice (squares), housed in groups of five, received daily intraperitoneal injections of either vehicle (empty symbols) or 10 μg of DH-CNTF (filled symbols) for 25 days. From day 26, all mice were treated with vehicle. Food intake is the number of grams consumed per group divided by five.
A possible explanation for the observed differences between obese and lean animals is that CNTF, similarly to leptin (15, 16), predominantly depletes adipose mass, such that the extent and duration of its effect would depend on the size of fat depots. Indeed, DH-CNTF specifically reduced the percentage of body fat in ob/ob and db/db mice, while increasing that of body water and lean mass as compared with vehicle-treated controls (Table 2). The absolute weight loss induced by DH-CNTF could be accounted for by a predominant loss of body fat (60–70% of lost mass), accompanied by a smaller reduction in water mass (see absolute weights in Table 2). Leptin produces similar effects in ob/ob mice (15, 16). Thus, in obese mice, CNTF shares specific anti-adiposity effects with leptin, rather than acting as a “cachectic” cytokine. Why does CNTF lead to a reduction in muscle (4) or protein (7) mass in lean, but not in obese, animals? A plausible explanation for this apparent discrepancy is that the predominant fat-depleting effect of the cytokine leads to a nearly total loss of body fat in lean animals (ref. 4 and unpublished results), which causes protein loss as a secondary event.
Table 2.
Effects of DH-CNTF treatment of obese mice on carcass composition
| Mice | Treatment | Body composition
|
|||||
|---|---|---|---|---|---|---|---|
| Water
|
Fat
|
Lean mass
|
|||||
| g | % | g | % | g | % | ||
| ob/ob | Vehicle | 18.1 ± 0.9 | 38.0 ± 1.5 | 22.1 ± 1.2 | 46.3 ± 1.7 | 7.5 ± 0.2 | 15.8 ± 0.5 |
| DH-CNTF | 15.5 ± 0.4* | 42.8 ± 1.7* | 14.1 ± 1.6** | 38.7 ± 3.0* | 6.7 ± 0.5 | 18.5 ± 1.6 | |
| db/db | Vehicle | 16.5 ± 0.6 | 38.8 ± 1.3 | 19.1 ± 1.5 | 44.6 ± 2.2 | 7.1 ± 0.4 | 16.6 ± 1.0 |
| DH-CNTF | 13.3 ± 0.5** | 41.4 ± 1.3 | 12.5 ± 0.8** | 38.1 ± 1.4* | 6.7 ± 0.2 | 20.6 ± 0.7** | |
Mice were treated for 10 days with either vehicle or 10 μg of DH-CNTF. Results are the mean ± SEM (n = 5). ∗, P < 0.05; ∗∗, P < 0.01 vs. vehicle by Student’s t test.
We next ascertained that CNTF does not induce toxicity, malaise, or illness. Irreversible toxicity was ruled out by the finding that body weight and food intake rapidly returned to pretreatment levels following interruption of cytokine administration, in both db/db (Fig. 3) and ob/ob mice (data not shown). Locomotor activity was not significantly altered by a 3-day treatment of db/db mice with DH-CNTF (10 μg) as compared with vehicle-treated controls (activity scores: 43 ± 6 and 49 ± 6, respectively; n = 5). Likewise, DH-CNTF treatment did not alter grooming behavior (activity scores: 1.2 ± 0.6 and 1.0 ± 0.4, for DH-CNTF- and vehicle-treated, respectively) and did not induce any form of stereotypic behavior. The behavioral effects of longer treatments or of higher doses remain to be analyzed. The possibility that the cytokine causes taste aversion was examined in DIO mice by using a two-bottle paradigm with 0.1% saccharin as a novel taste (29). Similarly to leptin, which was reported to reduce water intake in ob/ob mice (16), DH-CNTF (10 μg) caused a decrease in water intake of DIO mice 2 days after conditioning (1.8 ± 0.1 ml vs. 2.8 ± 0.2 ml in vehicle-treated controls; n = 9; P < 0.001). However, DH-CNTF did not cause taste aversion (saccharin intake 49 ± 2% of total fluid vs. 51 ± 4% in controls). These results indicate that the satiety effect of DH-CNTF is not due to cytokine-induced sickness behavior.
Reversal of Obesity-Associated Metabolic Defects by CNTF.
In addition to its weight- and appetite-regulating actions, CNTF reversed the hyperglycemia and hyperinsulinemia associated with the ob or db mutations. Mice bearing the ob mutation on the C57BL/6 background exhibit strong hyperinsulinemia (with nearly normal glucose levels after the age of 2–3 months) (43), which can be corrected by leptin treatment (15, 16, 36). Treatment of ob/ob mice with CNTF or DH-CNTF also led to strong reductions in serum insulin levels (Table 1, experiments 1 and 2). The db/db mutant on the C57BL/KS background is characterized by severe hyperglycemia (with nearly normal insulin levels after the age of 2–3 months) (44). As previously reported (15, 16, 36), leptin was unable to reverse hyperglycemia in db/db mice. In contrast, CNTF and DH-CNTF led to 2- to 3-fold reductions in both fed and fasted serum glucose levels, without affecting the already low levels of insulin (Table 1, experiments 3 and 4). CNTF was previously shown to cause hypoglycemia in lean mice (6). The DH-CNTF-induced reductions in body weight, serum insulin, and serum glucose exceeded those induced by pair-feeding of ob/ob or db/db mice to the food intake of cytokine-treated animals (Table 1, experiments 2 and 4). These results show that the effects of CNTF, similarly to those of leptin (15, 42, 45) are not solely due to decreased food intake.
Expression of CNTF Receptor Subunits in Brain Regions Involved in Body Weight Control.
Part of the fat-depleting activity of CNTF could be related to its ability to inhibit lipoprotein lipase in cultured adipocytes (46), an effect mediated by soluble CNTFRα (2, 46) and shared with other gp130-signaling cytokines (47). OB-Rb is expressed in adipose tissue (14), raising the possibility that leptin might also exert a direct action on fat cells. Yet gp130-signaling cytokines have only modest effects, if any, on lipolysis or lipogenesis in cultured adipocytes, and it has been pointed out that inhibition of lipoprotein lipase activity alone is unlikely to lead to fat cell depletion (47). Moreover, such a mechanism would not explain the potent appetite-reducing effects of CNTF and leptin.
A more likely explanation for the overlapping biological activities of leptin and CNTF is that the cytokines stimulate common effector pathways in brain areas involved in the regulation of energy intake and expenditure. OB-Rb is predominantly expressed in such regions, including the arcuate, ventromedial, and paraventricular hypothalamic nuclei (48, 49). To determine whether hypothalamic satiety centers could also be targets for CNTF, in situ hybridization was performed using cRNA probes for murine OB-Rb, CNTFRα, and LIFR. As shown in Fig. 4, the arcuate and paraventricular nuclei of the mouse hypothalamus express mRNAs for leptin and CNTF receptor subunits. Further studies are in progress to determine whether the receptors for leptin and CNTF are expressed by neurons [as already reported for CNTFRα in the rat central nervous system (50, 51), including the paraventricular nucleus (51)] or glia, and whether they are coexpressed by the same cells.
Figure 4.
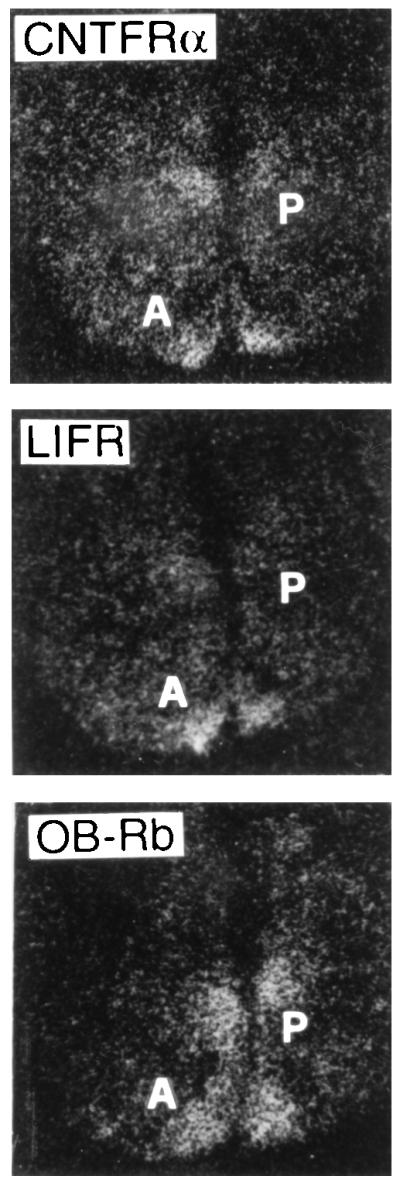
Expression of receptor subunits for leptin (OB-Rb) and CNTF (CNTFRα and LIFR) in mouse hypothalamus. A, arcuate nucleus; P, paraventricular nucleus. (×100.)
Activation of Hypothalamic Cells by CNTF and Leptin.
In agreement with the existence of a cytokine signaling pathway to central satiety centers, systemically administered leptin activates early signaling responses in mouse hypothalamus (52, 53). If the mechanism of action of CNTF is similar to that of leptin, hypothalamic responses should be detectable also after peripheral administration of CNTF. The tis-11 primary response gene (31), which is rapidly induced by CNTF and other STAT3-dependent cytokines (50), was used as a marker for cellular activation. Hypothalamic tis-11 mRNA of ob mice was found to be significantly elevated 1 hr after intraperitoneal injection of leptin or DH-CNTF as compared with vehicle-treated controls (Fig. 5A). In situ hybridization revealed a similar pattern of tis-11 staining for leptin and DH-CNTF; the arcuate nucleus was a major site of activation by both cytokines (Fig. 5B).
Figure 5.
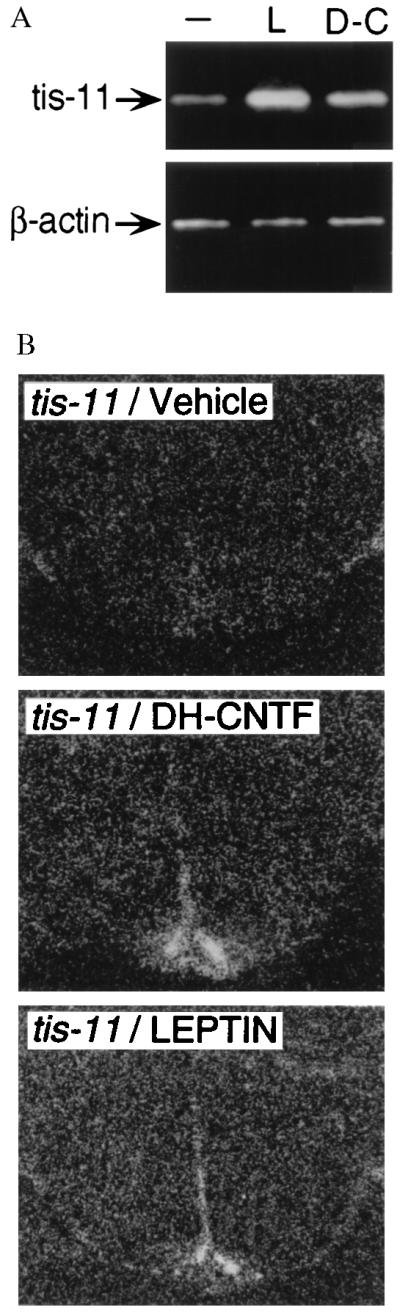
Effects of leptin and CNTF on tis-11 expression in mouse hypothalamus. Groups of three ob/ob mice received intraperitoneal injections of vehicle (−), leptin (L, 100 μg), or DH-CNTF (D-C, 10 μg) and were sacrificed 1 hr later by cervical dislocation. (A) Relative expression levels of tis-11 and β-actin (as control), as determined by RT-PCR. (B) In situ hybridization on coronal brain sections from vehicle- or cytokine-treated mice, using a 35S-labeled cRNA probe for murine tis-11. (×100.)
This result demonstrates that systemically administered CNTF and leptin can signal to a brain region that has been implicated as an important target of leptin action (36, 49). It cannot be excluded that the cytokines activate hypothalamic cells indirectly, for instance through peripheral mediators or via afferent nerves. Yet the rapidity of this effect, together with the expression of specific receptors for CNTF and leptin in the arcuate nucleus, argues for a direct action on the hypothalamus. Both CNTF (54) and leptin (55) can cross the blood–brain barrier. Cytokines may penetrate into the brain by means of specific transport systems, as reported for leptin (55). They may also gain access through circumventricular organs lying outside the blood–brain barrier, such as the median eminence, which is adjacent to the arcuate nucleus (56). Further studies are necessary to distinguish between these possibilities. In addition, it will be important to determine whether CNTF can alter the expression of neural mediators involved in energy homeostasis, such as neuropeptide Y, whose synthesis in the arcuate nucleus is down-regulated by leptin (36, 49).
Conclusions.
The present results are consistent with the idea that the shared biological activities of CNTF and leptin involve a related mechanism of action. In addition to their pharmacological significance, these findings raise the possibility that CNTF or related molecules, such as a putative second ligand for CNTFRα (57) or LIF [whose receptor is part of the CNTF receptor complex (2)], might play a physiological role in the regulation of energy balance. Even though null mutations in the CNTF or LIF genes (58, 59) have not been reported to be associated with metabolic defects, other gp130-signaling cytokines might play compensatory roles. In contrast to CNTF- or LIF-deficient animals, mice lacking CNTFRα (57) or LIFR (60) present severe neuronal deficits, suggesting the existence of additional receptor ligand(s). Since these mice die perinatally, the possible roles of such factors in adult animals remain unknown. Considering the glucose-lowering effects of CNTF, it is noteworthy, however, that the livers of LIFR-deficient mouse embryos contain increased glycogen stores, suggesting fetal hyperglycemia (60). Further studies, using CNTF receptor antagonists (20) or conditional receptor-knockouts, will help to assess the possible involvement of CNTFRα or LIFR ligands in the regulation of body weight and metabolism.
Finally, the present findings raise the intriguing possibility that leptin may share additional biological activities with CNTF, such as the ability to regulate neuronal cell survival or differentiation. A role for leptin in central nervous system function would be consistent with the expression of OB-Rb mRNA in rodent thalamus, cortex, and hippocampus (48).
Acknowledgments
We thank S. Germoni, M. Aquilina, and E. Dammassa for expert technical assistance, J. K. Blusztajn and B. Wainer for the gift of SN-56 cells, and M. Emili for artwork. This paper is dedicated to the memory of Yoav Citri.
ABBREVIATIONS
- CNTF
ciliary neurotrophic factor, CNTFRα, CNTF receptor α subunit
- DH-CNTF
Ser-166 → Asp/Gln-167 → His CNTF
- LIF
leukemia inhibitory factor
- LIFR
LIF receptor
- OB-R
leptin receptor
- RT-PCR
reverse transcription–PCR
- DIO
diet-induced obesity
References
- 1.Manthorpe M, Louis J C, Hagg T, Varon S. In: Neurotrophic Factors. Loughlin S E, Fallon J H, editors. San Diego: Academic; 1993. pp. 443–473. [Google Scholar]
- 2.Ip N Y, Yancopoulos G D. Annu Rev Neurosci. 1996;19:491–515. doi: 10.1146/annurev.ne.19.030196.002423. [DOI] [PubMed] [Google Scholar]
- 3.Shapiro L, Zhang X X, Rupp R G, Wolff S M, Dinarello C A. Proc Natl Acad Sci USA. 1993;90:8614–8618. doi: 10.1073/pnas.90.18.8614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henderson J T, Seniuk N A, Richardson P M, Gauldie J, Roder J C. J Clin Invest. 1994;93:2632–2638. doi: 10.1172/JCI117276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dittrich F, Thoenen H, Sendtner M. Ann Neurol. 1994;35:151–163. doi: 10.1002/ana.410350206. [DOI] [PubMed] [Google Scholar]
- 6.Fantuzzi G, Benigni F, Sironi M, Conni M, Carelli M, Cantoni L, Shapiro L, Dinarello C A, Sipe J D, Ghezzi P. Cytokine. 1995;7:150–156. doi: 10.1006/cyto.1995.1020. [DOI] [PubMed] [Google Scholar]
- 7.Espat N J, Auffenberg T, Rosenberg J J, Rogy M, Martin D, Fang C H, Hasselgren P O, Copeland E M, Moldawer L L. Am J Physiol. 1996;271:R185–R190. doi: 10.1152/ajpregu.1996.271.1.R185. [DOI] [PubMed] [Google Scholar]
- 8.Miller R G, Petajan J H, Bryan W W, Armon C, Barohn R J, Goodpasture J C, Hoagland R J, Parry G J, Ross M A, Stromatt S C the rhCNTF ALS Study Group. Ann Neurol. 1996;39:256–260. doi: 10.1002/ana.410390215. [DOI] [PubMed] [Google Scholar]
- 9.ALS CNTF Treatment Study Group. Neurology. 1996;46:1244–1249. doi: 10.1212/wnl.46.5.1244. [DOI] [PubMed] [Google Scholar]
- 10.Kishimoto T, Akira S, Narazaki M, Taga T. Blood. 1995;86:1243–1254. [PubMed] [Google Scholar]
- 11.Chen H, Charlat O, Tartaglia L A, Woolf E A, Weng X, Ellis S J, Lakey N D, Culpepper J, Moore K J, Breitbart R E, Duyk G M, Tepper R I, Morgenstern J P. Cell. 1996;84:491–495. doi: 10.1016/s0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- 12.Baumann H, Morella K K, White D W, Dembski M, Bailon P S, Kim H, Lai C-F, Tartaglia L A. Proc Natl Acad Sci USA. 1996;93:8374–8378. doi: 10.1073/pnas.93.16.8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tartaglia L A, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards G J, Campfield L A, Clark F T, Deeds J, Muir C, Sanker S, Moriarty A, Moore K J, Smutko J S, Mays G G, Woolf E A, Monroe C A, Tepper R I. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 14.Lee G-H, Proenca R, Montez J M, Carroll K M, Darvishzadeh J G, Lee J I, Friedman J M. Nature (London) 1996;379:632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 15.Halaas J L, Gajiwala K S, Maffei M, Cohen S L, Chait B T, Rabinowitz D, Lallone R L, Burley S K, Friedman J M. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 16.Pelleymounter M A, Cullen M J, Baker M B, Hecht R, Winters D, Boone T, Collins F. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 17.Campfield L A, Smith F J, Guisez Y, Devos R, Burn P. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman J M. Nature (London) 1994;372:425–431. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 19.Saggio I, Gloaguen I, Poiana G, Laufer R. EMBO J. 1995;14:3045–3054. doi: 10.1002/j.1460-2075.1995.tb07307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Marco A, Gloaguen I, Graziani R, Paonessa G, Saggio I, Hudson K R, Laufer R. Proc Natl Acad Sci USA. 1996;93:9247–9252. doi: 10.1073/pnas.93.17.9247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stemmer W P, Crameri A, Ha K D, Brennan T M, Heyneker H L. Gene. 1995;164:49–53. doi: 10.1016/0378-1119(95)00511-4. [DOI] [PubMed] [Google Scholar]
- 22.Schoepfer R. Gene. 1993;124:83–85. doi: 10.1016/0378-1119(93)90764-t. [DOI] [PubMed] [Google Scholar]
- 23.Lee H J, Hammond D N, Large T H, Wainer B H. Dev Brain Res. 1990;52:219–228. doi: 10.1016/0165-3806(90)90238-t. [DOI] [PubMed] [Google Scholar]
- 24.Rosenblum C I, Tota M, Cully D, Smith T, Collum R, Qureshi S, Hess J F, Phillips M S, Hey P J, Vongs A, Fong T M, Xu L, Chen H Y, Smith R G, Schindler C, Van der Ploeg L H T. Endocrinology. 1996;137:5178–5181. doi: 10.1210/endo.137.11.8895396. [DOI] [PubMed] [Google Scholar]
- 25.Demartis A, Bernassola F, Savino R, Melino G, Ciliberto G. Cancer Res. 1996;56:4213–4218. [PubMed] [Google Scholar]
- 26.Wagner B J, Hayes T E, Hoban C J, Cochran B H. EMBO J. 1990;9:4477–4484. doi: 10.1002/j.1460-2075.1990.tb07898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sadowski H B, Gilman M Z. Nature (London) 1993;362:79–83. doi: 10.1038/362079a0. [DOI] [PubMed] [Google Scholar]
- 28.West D B, Boozer C N, Moody D L, Atkinson R L. Am J Physiol. 1992;262:R1025–R1032. doi: 10.1152/ajpregu.1992.262.6.R1025. [DOI] [PubMed] [Google Scholar]
- 29.Langhans W, Harlacher R, Balkowski G, Scharrer E. Physiol Behav. 1990;47:805–813. doi: 10.1016/0031-9384(90)90001-k. [DOI] [PubMed] [Google Scholar]
- 30.Leshner A I, Litwin V A, Squibb R L. Physiol Behav. 1972;9:281–282. doi: 10.1016/0031-9384(72)90251-x. [DOI] [PubMed] [Google Scholar]
- 31.Varnum B C, Ma Q, Chi T, Fletcher B, Herschman H R. Mol Cell Biol. 1991;11:1754–1758. doi: 10.1128/mcb.11.3.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lazzaro D, Price M, De Felice M, Di Lauro R. Development (Cambridge, UK) 1991;113:1093–1104. doi: 10.1242/dev.113.4.1093. [DOI] [PubMed] [Google Scholar]
- 33.Schindler C, Darnell J E. Annu Rev Biochem. 1995;64:621–651. doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- 34.Zhong Z, Wen Z, Darnell J E. Science. 1994;264:95–98. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- 35.Ghilardi N, Ziegler S, Wiestner A, Stoffel R, Heim M H, Skoda R C. Proc Natl Acad Sci USA. 1996;93:6231–6235. doi: 10.1073/pnas.93.13.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stephens T W, Basinski M, Bristow P K, Bue-Valleskey J M, Burgett S G, Craft L, Hale J, Hoffmann J, Hsiung H M, Kriauciunas A, MacKellar W, Rosteck P R J, Schoner B, Smith D, Tinsley F C, Zhang X-Y, Heiman M. Nature (London) 1995;377:530–532. doi: 10.1038/377530a0. [DOI] [PubMed] [Google Scholar]
- 37.Grunfeld C, Zhao C, Fuller J, Pollock A, Moser A, Friedman J, Feingold K R. J Clin Invest. 1996;97:2152–2157. doi: 10.1172/JCI118653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maffei M, Halaas J, Ravussin E, Pratley R E, Lee G H, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, Kern P A, Friedman J M. Nat Med. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 39.Frederich R C, Hamann A, Anderson S, Löllmann B, Lowell B B, Flier J S. Nat Med. 1995;1:1311–1314. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- 40.Hamilton B S, Paglia D, Kwan A Y M, Deitel M. Nat Med. 1995;1:953–956. doi: 10.1038/nm0995-953. [DOI] [PubMed] [Google Scholar]
- 41.Considine R V, Sinha M K, Heiman M L, Kriauciunas A, Stephens T W, Nyce M R, Ohannesian J P, Marco C C, McKee L J, Bauer T L, Caro J F. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 42.Levin N, Nelson C, Gurney A, Vandlen R, De Sauvage F. Proc Natl Acad Sci USA. 1996;93:1726–1730. doi: 10.1073/pnas.93.4.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coleman D L, Hummel K P. Diabetologia. 1973;9:287–293. doi: 10.1007/BF01221856. [DOI] [PubMed] [Google Scholar]
- 44.Coleman D L, Hummel K P. Diabetologia. 1967;3:238–248. doi: 10.1007/BF01222201. [DOI] [PubMed] [Google Scholar]
- 45.Schwartz M W, Baskin D G, Bukowski T R, Kuijper J L, Foster D, Lasser G, Prunkard D E, Porte D, Jr, Woods S C, Seeley R J, Weigle D S. Diabetes. 1996;45:531–535. doi: 10.2337/diab.45.4.531. [DOI] [PubMed] [Google Scholar]
- 46.Gimble J M, Wanker F, Wang C-S, Bass H, Wu X, Kelly K, Yancopoulos G D, Hill M R. J Cell Biochem. 1994;54:122–133. doi: 10.1002/jcb.240540113. [DOI] [PubMed] [Google Scholar]
- 47.Marshall M K, Doerrler W, Feingold K R, Grunfeld C. Endocrinology. 1994;135:141–147. doi: 10.1210/endo.135.1.8013346. [DOI] [PubMed] [Google Scholar]
- 48.Mercer J G, Hoggard N, Williams L M, Lawrence C B, Hannah L T, Trayhurn P. FEBS Lett. 1996;387:113–116. doi: 10.1016/0014-5793(96)00473-5. [DOI] [PubMed] [Google Scholar]
- 49.Schwartz M W, Seeley R J, Campfield L A, Burn P, Baskin D G. J Clin Invest. 1996;98:1101–1106. doi: 10.1172/JCI118891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ip N Y, McClain J, Barrezueta N X, Aldrich T H, Pan L, Li Y, Wiegand S J, Friedman B, Davis S, Yancopoulos G D. Neuron. 1993;10:89–102. doi: 10.1016/0896-6273(93)90245-m. [DOI] [PubMed] [Google Scholar]
- 51.MacLennan A J, Vinson E N, Marks L, McLaurin D L, Pfeifer M, Lee N. J Neurosci. 1996;16:621–630. doi: 10.1523/JNEUROSCI.16-02-00621.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Woods A J, Stock M J. Nature (London) 1996;381:745–740. doi: 10.1038/381745a0. [DOI] [PubMed] [Google Scholar]
- 53.Vaisse C, Halaas J L, Horvath C M, Darnell J E, Jr, Stoffel M, Friedman J M. Nat Genet. 1996;14:95–97. doi: 10.1038/ng0996-95. [DOI] [PubMed] [Google Scholar]
- 54.Poduslo J F, Curran G L. Mol Brain Res. 1996;36:280–286. doi: 10.1016/0169-328x(95)00250-v. [DOI] [PubMed] [Google Scholar]
- 55.Banks W A, Kastin A J, Huang W, Jaspan J B, Maness L M. Peptides. 1996;17:305–311. doi: 10.1016/0196-9781(96)00025-3. [DOI] [PubMed] [Google Scholar]
- 56.Johnson A K, Gross P M. FASEB J. 1993;7:678–686. doi: 10.1096/fasebj.7.8.8500693. [DOI] [PubMed] [Google Scholar]
- 57.DeChiara T M, Vejsada R, Poueymirou W T, Acheson A, Suri C, Conover J C, Friedman B, McClain J, Pan L, Stahl N, Ip N Y, Kato A, Yancopoulos G D. Cell. 1995;83:313–322. doi: 10.1016/0092-8674(95)90172-8. [DOI] [PubMed] [Google Scholar]
- 58.Takahashi R, Yokoji H, Misawa H, Hayashi M, Hu J, Deguchi T. Nat Genet. 1994;7:79–84. doi: 10.1038/ng0594-79. [DOI] [PubMed] [Google Scholar]
- 59.Sendtner M, Götz R, Holtmann B, Escary J-L, Masu Y, Carroll P, Wolf E, Brem G, Brulet P, Thoenen H. Curr Biol. 1996;6:686–694. doi: 10.1016/s0960-9822(09)00450-3. [DOI] [PubMed] [Google Scholar]
- 60.Ware C B, Horowitz M C, Renshaw B R, Hunt J S, Liggitt D, Koblar S A, Gliniak B C, McKenna H J, Papayannopoulou T, Thoma B, Cheng L, Donovan P J, Peschon J J, Bartlett P F, Willis C R, Wright B D, Carpenter M K, Davison B L, Gearing D P. Development (Cambridge, UK) 1995;121:1283–1299. doi: 10.1242/dev.121.5.1283. [DOI] [PubMed] [Google Scholar]