

Abstract
Activation of myocardial A1 adenosine receptors (A1AR) protects the heart from ischemic injury. In this study transgenic mice were created using the cardiac-specific α-myosin heavy chain promoter and rat A1AR cDNA. Heart membranes from two transgene positive lines displayed ≈1,000-fold overexpression of A1AR (6,574 ± 965 and 10,691 ± 1,002 fmol per mg of protein vs. 8 ± 5 fmol per mg of protein in control hearts). Compared with control hearts, transgenic Langendorff-perfused hearts had a significantly lower intrinsic heart rate (248 beats per min vs. 318 beats per min, P < 0.05), lower developed tension (1.2 g vs. 1.6 g, P < 0.05), and similar coronary resistance. The difference in developed tension was eliminated by pacing. Injury of control hearts during global ischemia, indexed by time-to-ischemic contracture, was accelerated by blocking adenosine receptors with 50 μM 8-(p-sulfophenyl) theophylline but was unaffected by addition of 20 nM N6-cyclopentyladenosine, an A1AR agonist. Thus A1ARs in ischemic myocardium are presumably saturated by endogenous adenosine. Overexpressing myocardial A1ARs increased time-to-ischemic contracture and improved functional recovery during reperfusion. The data indicate that A1AR activation by endogenous adenosine affords protection during ischemia, but that the response is limited by A1AR number in murine myocardium. Overexpression of A1AR affords additional protection. These data support the concept that genetic manipulation of A1AR expression may improve myocardial tolerance to ischemia.
The heart possesses intrinsic protective mechanisms to provide tolerance to injurious stimuli such as ischemia reperfusion. Recent research has focused on understanding and harnessing such endogenous cardioprotective mechanisms. The autacoid adenosine has been proposed to function as an endogenous cardioprotectant (1). Exogenous or endogenous activation of myocardial A1 adenosine receptor (A1AR) protects the heart from injury during global ischemia (2) and improves bioenergetic and mechanical recovery in reperfused myocardium (3–6). A brief period of ischemia, or brief exposure to adenosine, protects the heart from damaging effects of subsequent ischemic episodes: a phenomenon known as “preconditioning” (7). Adenosine appears to be of central importance as a mediator of ischemic preconditioning in a range of species (7–10). There is evidence that more than one of the four adenosine receptor subtypes may contribute to myocardial protection, but the A1AR appears to be primarily responsible for ischemic preconditioning (11). Additionally, A1AR activation has been shown to provide protection from ischemic injury in other tissues (12, 13), providing evidence that A1AR activation may have broad protective effects in other organ systems.
Despite evidence implicating endogenous adenosine as a mediator of cardioprotection during ischemic episodes, controversy remains regarding the ability of adenosinergic therapy to reduce ischemic or post-ischemic injury (e.g., ref. 14). Given that ischemia elicits a large increase in interstitial adenosine (10, 15), one possibility that has received little attention is that it may prove difficult to pharmacologically enhance an intrinsic response or mechanism that is normally maximally active during ischemia reperfusion. An alternative approach that could prove more effective is to increase the number of functional receptors present. There is evidence suggesting that “up-regulation” of A1AR can reduce ischemic injury (16). Transgenic manipulation provides the opportunity for substantial modification of receptor number. Transgenic models of G-protein-coupled receptors have been used in assessment of cardiac function and provide unique opportunities for study of receptor signaling (17). Recently, overexpression of heat shock protein and glutathione peroxidase have been shown to improve myocardial responses to ischemia reperfusion (18–20).
We hypothesized that a specific increase in myocardial A1AR density would enhance tolerance to ischemia. To test this hypothesis we used the cardiac-specific α-myosin heavy chain (α-MHC) promoter (21) to overexpress A1AR cDNA in a murine model. We have characterized two lines expressing the transgene and documented the response of hearts from these animals to global normothermic ischemia and reperfusion. The results indicate that overexpression of myocardial A1AR protects the heart from ischemic damage.
MATERIALS AND METHODS
Transgenic Construct.
The EcoRI XhoI fragment of the rat A1 cDNA (22) was subcloned into the SalI site of a construct containing the α-MHC promoter with a MEF-2 mutation (23) and the human growth hormone polyadenylylation signal. This promoter results in high-level expression in the heart of mature animals along with some aortic expression (23). The A1AR cDNA promoter construct was digested with NotI and purified for injection into the pronuclei of single-cell fertilized mouse embryos (24).
Transgene Detection.
Mice were screened for the presence of the transgene by Southern analysis. Mouse genomic DNA is digested with EcoRI and probed with an α-MHC promoter fragment. EcoRI digestion of the native α-MHC promoter results in a 2.6-kb fragment, and digestion of the transgenic α-MHC A1 construct results in a 1.6-kb fragment. Thus, transgenic mice demonstrated a 1.6-kb fragment from the transgenic promoter plus the 2.6-kb fragment from the native promoter.
Message Determination.
Total RNA was isolated using the method of Chomczynski and Sacchi (25). Northern analysis of RNA was performed by electrophoresis under denaturing conditions in a Mops/formaldehyde/1.2% agarose gel and transferred to a charged nylon membrane (Zetaprobe, Bio-Rad) by capillary action in high salt (20× standard saline phosphate/EDTA). Membranes then were UV-crosslinked, and A1AR cDNA was labeled by random priming with hybridization and washes at 65°C according to the method of Church and Gilbert (26).
Membrane Preparation.
Hearts from transgenic positive and negative animals were homogenized in 10 vol of ice-cold buffer (10 mM EDTA/10 mM Hepes/0.1 mM benzamidine, pH 7.4). Homogenate was centrifuged at 48,000 × g for 10 min. The pellet was resuspended in 30 ml of buffer with EDTA reduced to 1 mM, recentrifuged, and washed twice more by resuspension/centrifugation. The final pellet was resuspended in 1 vol of the appropriate buffer for assays. Membrane suspensions were stored at −80°C. Protein was determined by the Lowry method using BSA for standards.
Ligand Binding.
A1AR density and equilibrium dissociation constants in heart membranes from control and transgene positive animals were determined by quantitation of specific binding of an adenosine A1 receptor antagonist, 8-cyclopentyl-1,3-[3H]dipropylxanthine ([3H]CPX, 0.1 to 7 nM), using standard techniques (27). Briefly, 100-μl aliquots of membrane (0.2–0.7 mg of protein in negative animals and 10–15 μg of protein in transgene positive animals) were incubated with adenosine deaminase (5 units/ml) in membrane buffer (50 mM Tris⋅HCl/5 mM MgCl2, pH 7.4) with radioligand. After a 2-hr incubation at 21°C, 3 ml of ice-cold rinse buffer (10 mM Tris⋅HCl/5 mM MgCl2, pH 7.4) was added to each sample. Membranes were collected onto Whatman GF/C glass fiber filters, which were washed three times for 10 sec with ice-cold buffer (10 mM Tris⋅HCl/5 mM MgCl2, pH 7.4). Radioactivity trapped on filters was counted. Nonspecific binding was determined by adding 10 μM N6-(phenylisopropyl)adenosine to displace specific binding of [3H]CPX.
The number of high-affinity agonist binding sites, a measure of G-protein-coupled receptors, was determined using the agonist radioligand 125I-ABA (28). Specific binding was fit to a single site-binding model using nonlinear least-squares curve fitting of the untransformed data to calculate receptor density and dissociation constants. To compare receptor density between transgenic and control hearts, Bmax was reported as fmol of receptor per mg of protein. Coupling was defined as the ratio of specific 125I-ABA/[3H]CPX binding.
The A1AR structure activity profile was determined by calculating Kis of competing drugs. To calculate the Ki, [3H]CPX or 125I-ABA was added to tubes at ≈50% of the Kd for the radiolabeled ligand, and competing ligand was added over a range of concentrations. IC50 values were calculated using a three-parameter logistic equation:
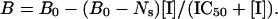 |
Ki values were precisely calculated from IC50, Bmax, the concentration of radioactive ligand, and the Kd (29).
Langendorff Perfused Heart Model.
Male and female mice (7–9 weeks, 21.8 ± 0.4 g body weight) were anesthetized with 50 mg per kg of sodium pentobarbital, a thoracotomy performed, and hearts excised into ice-cold perfusion fluid. The aorta was cannulated, and hearts retrogradely perfused at a pressure of 80 mmHg with modified Krebs buffer containing: 120 mM NaCl; 25 mM NaHCO3; 4.7 mM KCl; 1.2 mM KH2PO4; 1.25 mM CaCl2; 1.2 mM MgSO4; 15 mM glucose; and 0.05 mM EDTA. Buffer was equilibrated with 95% O2/5% CO2 at 37°C, giving a pH of 7.4. Hearts were bathed in perfusate in a water-jacketed bath maintained at 37°C. The left ventricle was vented with a polyethylene apical drain. Coronary perfusion was monitored via an ultrasonic flow-probe in the aortic perfusion line. To assess contractile function a stainless steel hook was attached to the ventricular apex and connected to a Grass FT03C strain gauge. Transducer position was adjusted to yield a diastolic tension of 1.0 g. Apicobasal displacement was continuously measured via a Gould RS3400 physiograph and the signal electronically processed to yield heart rate and +dP/dt. After 20-min of perfusion at intrinsic heart rate, hearts were switched to electrical pacing at 6 Hz (12 ms square wave, voltage 20% in excess of threshold) and allowed to stabilize for an additional 10 min before experimentation.
Ischemia-Reperfusion Studies.
Ischemia was produced by clamping the aortic cannula and simultaneously bubbling the bathing perfusate with 95% N2/5% CO2 to reduce PO2. Pacing was stopped during ischemia and resumed on reperfusion.
Two studies were performed. In the initial study time-to-ischemic contracture (TIC) was measured in control and transgenic hearts in the absence and presence of 50 μM 8-(p-sulfophenyl)theophylline (8-SPT). A group of control hearts also was pretreated with 20 nM N6-cyclopentyladenosine (CPA). The hearts were subjected to 30 min of global normothermic ischemia. TIC was defined as time between cessation of coronary flow and the point at which diastolic tension increased by 0.2 g (20% above basal tension). In the second study, control and transgenic hearts were subjected to 20 min of global normothermic ischemia followed by 30 min of reperfusion. Recovery of developed tension, diastolic tension, and coronary flow was assessed during reperfusion.
Data Analysis and Statistical Comparisons.
Baseline function in control and transgenic hearts was assessed via an unpaired t test, and TIC values were compared using a one-way ANOVA with Newman–Keuls post-hoc test for individual comparisons. Functional changes during ischemia-reperfusion were statistically analyzed by two-way ANOVA for repeated measures with Newman–Keuls post-hoc test. In all tests significance was accepted for P < 0.05.
RESULTS
Transgenic Animals.
Two positive transgenic lines were established and bred for analysis (lines 1 and 5). Fig. 1A is a Southern blot demonstrating the presence of the transgene in lines 1 and 5. Northern analysis revealed abundant message in hearts from lines 1 and 5 (Fig. 1B). Consistent with previous findings (30), A1AR message is too low to be detected by standard Northern analysis in a control heart.
Figure 1.

Demonstration of the A1AR transgene by Southern and Northern blotting. (A) Southern blot demonstrating presence of transgene in lines 1 and 5. (B) Northern analysis demonstrating presence of transgene message in lines 1 and 5. See under Transgene Detection for details.
Receptor Binding.
Ligand binding data revealed that transgenic hearts express approximately 1,000-fold higher A1AR than control hearts (Table 1). A1AR ligand binding was specific and saturable in transgenic tissue (Fig. 2), and the calculated Kd for CPX was comparable in control and transgenic hearts (Table 1). Percent specific binding at the Kd (1 nm) was 47 ± 9% in control and 98 ± 1% in transgenic hearts.
Table 1.
Ligand binding to membrane preparations from control and transgenic hearts
| Line 1 hearts | Line 5 hearts | Control hearts | |
|---|---|---|---|
| Antagonist and agonist binding properties | |||
| [3H]CPX Bmax, fmol⋅mg−1 | 6,574 ± 965* | 10,691 ± 1,002*† | 8 ± 5 |
| (n = 15) | (n = 8) | (n = 11) | |
| [3H]CPX Kd, nM | 0.92 ± 0.09 | 1.12 ± 0.09 | 0.73 ± 0.17 |
| (n = 15) | (n = 8) | (n = 11) | |
| 125I-ABA Bmax, fmol⋅mg−1 | 475 ± 25 | 244 ± 8† | — |
| (n=6) | (n=6) | ||
| % coupling‡ | 7.7 ± 1.0 | 2.4 ± 0.3† | — |
| Ligand Ki values (n = 6 in all cases) | |||
| Theo, nM | 17,000 ± 1,800 | 10,000 ± 2,200 | — |
| CGS, nM | 400 ± 200 | 200 ± 100 | — |
| CPX, nM | 0.4 ± 0.3 | 2.0 ± 0.6 | — |
| CPA, nM | 0.3 ± 0.2 | 0.3 ± 0.1 | — |
Total and coupled receptor number was assessed from the Bmax for antagonist (CPX) and agonist (ABA), respectively. Agonist and antagonist Ki values were determined as described in Materials and Methods. Theo, theophylline; CGS, CGS21680; CPX, 8-cyclopentyl-1,3-dipropylxanthine; CPA, N6-cyclopentyladenosine. [3H]CPX and 125I-ABA were used with competing antagonists and agonists, respectively. All binding studies were performed in triplicate.
P < 0.05 transgenic vs. control.
P < 0.05 line 5 vs. line 1.
Percent coupling is defined as Bmax agonist/Bmax antagonist.
Figure 2.

Characterization of [3H]CPX binding to membranes prepared from the hearts of transgenic mice overexpressing A1AR. (Inset) Scatchard plots of the same data. Values are means ± SEM of triplicate determination of a single experiment.
Receptor coupling was estimated by comparison of the number of high affinity agonist (125I-ABA) binding sites with total binding sites for the antagonist ([3H]CPX). Line 1 displayed a higher number of coupled receptors than line 5 despite a lower total receptor number (Table 1), and thus possessed a higher percentage of functionally coupled receptors. Calculated Ki values for different ligands (agonists and antagonists) are shown in Table 1. The calculated Ki values for theophylline, CGS-21680, CPX, and CPA are similar to published values (31–33). There were no significant differences in Ki values between the two transgenic lines.
Baseline Function in Perfused Hearts.
Baseline functional parameters for hearts from control and transgenic animals are shown in Table 2. There were no statistical differences between baseline functional parameters in hearts from lines 1 and 5. Thus, throughout the analysis, data for lines 1 and 5 are combined.
Table 2.
Baseline functional parameters in control and transgenic hearts
| Hearts | Developed tension, g | Heart rate, beats⋅min−1 | +dT/dt, g⋅s−1 | Coronary resistance, mmHg⋅ml−1⋅min−1⋅g−1 |
|---|---|---|---|---|
| Intrinsic heart rate | ||||
| Control (n = 9) | 1.59 ± 0.18 | 318 ± 14 | 30.0 ± 1.4 | 5.92 ± 0.65 |
| Transgenic (n = 10) | 1.18 ± 0.10* | 248 ± 10* | 28.6 ± 1.6 | 6.41 ± 0.43 |
| Electrically paced at 6 Hz | ||||
| Control (n = 9) | 1.61 ± 0.11 | 360 | 32.1 ± 1.5 | 6.32 ± 1.13 |
| Transgenic (n = 11) | 1.56 ± 0.10 | 360 | 32.8 ± 1.7 | 7.59 ± 1.45 |
Functional parameters were measured after 30 min of normal aerobic perfusion at a perfusion pressure of 80 mmHg. Diastolic tension was adjusted to 1.0 g in both groups. All values are means ± SEM. ∗, P < 0.05 transgenic vs. control hearts.
There was no difference in coronary resistance, a slight, but significant, reduction in contractile function (indexed by developed tension), and a significant reduction in heart rate in hearts from transgene-positive animals. When rate was normalized between groups by pacing, no differences were detected in contractile function, indicating that the apparent difference in contractility is due to rate-dependent change in contractile function (e.g., a manifestation of the positive staircase phenomenon).
Functional Effects of Ischemia-Reperfusion in Control and Transgenic Hearts.
Ischemic injury was assessed by TIC in control hearts and transgenic hearts. As shown in Fig. 3, TIC was ≈10 min in control hearts. This was reduced to 7 min by treatment with 8-SPT but was unaltered by treatment with the A1 agonist CPA. Alternatively, TIC was significantly prolonged to 14 min in transgenic hearts. The improvement in transgenic hearts was reduced by competitive antagonism with 8-SPT (Fig. 3).
Figure 3.

TIC in globally ischemic hearts from control and transgenic animals. Control and transgenic (Trans) hearts were either untreated (n = 11 and 13, respectively) or pretreated with 50 μM 8-(p-sulfophenyl)theophylline (Con+8-SPT and Trans+8-SPT, n = 15 and 6, respectively). Additionally, control hearts were treated with N6-cyclopentyladenosine (Con+CPA, n = 6). Values shown are means ± SEM. ∗, P < 0.05 different from control hearts; †, P < 0.05 less than Trans hearts.
Functional parameters for control and transgenic hearts after 20 min of global normothermic ischemia are shown in Fig. 4. Global normothermic ischemia rapidly abolished contractile function and caused a gradual rise in diastolic tension. Diastolic tension rose to a maximum of 1.93 ± 0.12 g in control hearts and 1.85 ± 0.10 g in transgenic hearts. In the first 2 min of reperfusion there was a rapid recovery of contractile function (Fig. 2B). With early reperfusion (2-min recovery) developed tension recovered to 60% of the pre-ischemic value in transgenic hearts but to only 15% in control hearts (Fig. 4B; P < 0.05). This may reflect enhanced myocardial viability after the ischemic insult in transgenic hearts. At the end of reperfusion (30-min recovery), developed tension recovered to only 30% of the pre-ischemic value in control hearts whereas it recovered to 45% in transgenic hearts (Fig. 4B; P < 0.05). With early reperfusion (2-min recovery) diastolic tension was 50% above baseline in control hearts and remained elevated at the end of reperfusion (30-min recovery) at 25% above the pre-ischemic value in control hearts (Fig. 4A). In contrast, diastolic tension at 2-min recovery was initially 25% above pre-ischemic levels in transgenic hearts and recovered to pre-ischemic levels after 30-min reperfusion (Fig. 4A). Coronary flow did not differ between the two groups, displaying an initial hyperemia at onset of reperfusion followed by gradual decline to a final flow of ≈120% of baseline in both control and transgenic hearts (Fig. 4C).
Figure 4.

Postischemic recoveries of (A) diastolic tension, (B) developed tension, and (C) coronary flow after 30-min global ischemia. Reperfusion was initiated after ischemia and data are shown at 2-min and 30-min recovery in control (n = 11) and transgenic hearts (n = 13). Values shown are means ± SEM. ∗, P < 0.05 transgenic vs. control hearts.
DISCUSSION
The primary goal of this study was to study the affect of overexpressing A1AR in transgenic mouse heart on cardiovascular responses to ischemia-reperfusion injury. There is considerable disagreement about the impact of adenosinergic therapy on functional and metabolic responses to ischemia reperfusion, and conflicting results have been obtained regarding the ability of adenosinergic therapy to limit infarct size (e.g., refs. 5, 14, 34). A possibility not addressed in these studies is that endogenous mechanisms may normally produce maximal or near-maximal cardioprotection. Indeed, given the evidence that competitive antagonism of adenosine receptors can worsen the response to ischemia reperfusion (2, 3, 5, 6), and evidence of elevations in interstitial adenosine to high levels (>1 μM) during ischemia (10, 15), it is possible that the endogenous adenosine response is near maximal in ischemic tissue. This being the case, it may be ineffective to attempt to harness adenosinergic cardioprotection via classical pharmacological strategies (e.g., infusion of agonists, allosteric enhancers, or inhibitors of uptake and breakdown) during severe ischemic episodes. The advent of genetic engineering and the ability to transgenically engineer tissues with modified receptor number and/or function provides an opportunity to circumvent this problem. If a response is potentially cardioprotective but normally near maximal it may be possible to enhance protection by increasing the number of functional receptors. Here we describe a model in which we have successfully overexpressed the cardiac A1AR, and present evidence that this provides enhanced tolerance to ischemia and improves myocardial recovery of function on reperfusion.
Characteristics of Myocardial A1ARs on Control and Transgenic Mice.
The affinity of CPX binding is similar in control and transgenic hearts. Agonist and antagonist Ki values for transgenic hearts are similar to values for rat A1AR (31, 32). Hence, overexpressed A1ARs in transgenic lines appear to bind ligands normally. Interestingly, receptor coupling was significantly higher in line 1 than line 5 despite lower total receptor number although both lines had low-percent coupling. Leung et al (35) reported 65% ± 9% coupling of A1 adenosine receptors in bovine heart. The low-percent coupling in the transgenic hearts may reflect an upper limit for G-protein coupling in the myocyte.
Functional Effects of Overexpression of Myocardial A1ARs.
One effect caused by overexpressing A1ARs was a reduction in heart rate. Under these conditions the heart is not subject to neurohumoral influences, and the heart rate difference reflects a change in intrinsic rate (i.e. intrinsic firing of sinoatrial nodal cells). Because endogenous adenosine reduces heart rate in isolated hearts from rats, rabbits, and guinea pigs under various conditions (36, 37), and this is an A1AR response, it is not surprising that overexpression of A1ARs leads to reduced heart rate. In the absence of a change in A1AR affinity (Table 1), this indicates that endogenous adenosine levels are sufficient to activate overexpressed A1ARs. This result is consistent with modest changes in resting heart rate with adenosine antagonism in other species (36, 38). Contractile function and coronary resistance were not altered by A1AR overexpression when heart rate was normalized between groups (Table 2). These data suggest that the A1AR does not play a significant role in modifying baseline contractile function. Comparable coronary resistance in control and transgenic hearts suggests a lack of effect of transgenic manipulation on the function of A2ARs that mediate coronary dilation.
Ischemic and Post-Ischemic Function in Transgenic Versus Control Hearts.
After abolition of contractile activity during ischemia the myocardium undergoes contracture (increased diastolic or resting ventricular tension). The mechanism of contracture is not fully understood but may involve rigor bond formation as a result of impaired glycolytic ATP production (39). TIC is an indicator of the severity of the ischemic insult. In control hearts we observed a reduction in TIC in response to 8-SPT, implicating prolongation of TIC by endogenous adenosine. This is consistent with the observations of Lasley and colleagues in rat heart (2). We were unable to increase TIC by pretreatment of control hearts with the A1 agonist CPA at a dose (20 nM) that was found to reduce heart rate by more than 50% (data not shown). Taken together, these observations indicate that endogenous adenosine prolongs TIC in mouse heart and that the endogenous response is maximal, being resistant to pharmacologic augmentation. Because we predict very high levels of extracellular adenosine under these conditions (10, 15), all endogenous receptors may have been saturated, rendering CPA ineffective in prolonging TIC in ischemic mouse heart. It is not clear why our results conflict with the findings of Lasley et al. (2), who reported a decrease in TIC in ischemic rat hearts pretreated with the A1 agonist phenylisopropyladenosine (1 μM) or adenosine (100 μM). In the mouse it appears that receptor number can limit the degree of cardioprotection afforded by endogenous adenosine in the ischemic mouse heart. Provision of additional receptors provides further protection when pharmacological manipulation is ineffective. This is a potentially important point in terms of development of cardioprotective therapies for ischemic myocardium; lack of response to an exogenous receptor agonist may reflect maximal endogenous activity. Under these conditions targeting receptor expression may be a more appropriate strategy. This observation may be relevant to studies in which there are conflicting data regarding the ability of exogenous adenosinergic therapy to reduce injury from ischemia reperfusion (e.g., ref. 14).
During reperfusion after 20 min of global ischemia there were two principal differences in contractile recovery between control and transgenic hearts. Initial contractile recovery (e.g. at 2-min reperfusion) was markedly higher in transgenic versus control hearts. This pronounced “hyper-contractile” state may reflect the viability of ischemic myocardial cells immediately after ischemic insult and before the onset of reperfusion injury (18, 19). This would be consistent with contracture data; overexpression of A1ARs improves cellular function/viability during the ischemic insult itself. The effect of A1AR overexpression in reperfused tissue was predominantly the result of reduced diastolic tension, which recovered to pre-ischemic levels in transgenic hearts but remained significantly elevated in control hearts (25% above baseline), consistent with observations in post-ischemic myocardium from other species (10, 40). As a result, developed tension was greater in transgenic versus control hearts. Because coronary flow recovered to similar values in both groups, the differences in contractile recovery do not appear to result from differences in “no-reflow.” We conclude that activation of A1ARs by endogenous adenosine improves contractile recovery predominantly via a reduction in post-ischemic diastolic dysfunction. Post-ischemic elevation in diastolic tension may reflect altered Ca2+ handling with enhanced diastolic Ca2+ (40). Endogenous adenosine acting via A1ARs may modify Ca2+ handling at the level of the sarcolemma and/or at the sarcoplasmic reticulum. Indeed, A1 receptors are functionally coupled to K+-ATP channels, which can limit Ca2+ entry when activated (41). K+-ATP channel openers have been shown to be cardioprotective (42), and effects of adenosine can be attenuated by K+-ATP channel blockers (43). Alternatively, adenosine may modify post-ischemic energy metabolism such that the SR-ATPase can more effectively sequester Ca2+. We have demonstrated receptor-mediated improvement of post-ischemic bioenergetic state by endogenous adenosine (3, 10). It is also possible that A1AR activation may directly modify sarcoplasmic reticulum channel function.
Increased expression of cardiac A1 adenosine receptors appears to be beneficial during ischemia. In a therapeutic setting, it may be desirable to have up-regulated A1 receptors in some instances, e.g., before cardiac surgery or before transplantation in the donor organ. However, chronically increased receptors is probably not desirable, because overexpression of these inhibitory receptors may be counterproductive in states that require maximal cardiac output such as shock. Hence, the possible therapeutic use of overexpression of A1 adenosine receptors in the heart may be most useful if the overexpression is limited in time. Once appropriate gene therapy vectors are available it may be possible to design a genetic manipulation to overexpress receptors on a short-term basis. With these, one could temporarily up-regulate receptors before surgery but have them return to normal within several weeks.
Conclusions.
This study indicates that A1AR activation by endogenous adenosine is beneficial in ischemic-reperfused myocardium, and that A1AR overexpression provides increased ischemic tolerance in the mouse heart. During ischemia the endogenous adenosine response appears to be near maximal, precluding additional benefit from exogenously applied adenosine agonists. However, transgenic overexpression of A1ARs provides additional cardioprotection. A1AR-mediated cardioprotection during ischemia is therefore limited by receptor number and/or coupling. During reperfusion, A1AR overexpression significantly improves contractile recovery, predominantly as a result of reduced diastolic dysfunction. The data demonstrate for the first time, to our knowledge, that genetic manipulation of the A1ARs may prove an effective method of improving outcome from ischemia reperfusion when conventional pharmacological approaches may be less effective.
Acknowledgments
We gratefully acknowledge the gift of the A1AR cDNA from Dr. Steven Reppert and the α-MHC construct from Dr. Jeff Robbins. This work was supported in part by the University of Virginia Academic Enhancement Program Grant on Gene Transfer and Gene Therapy of the Cardiovascular System, the University of Virginia Childrens Medical Center, and a grant from the National Heart Foundation of Australia.
ABBREVIATIONS
- A1AR
A1 adenosine receptors
- ABA
N6-(4-amino-3-benzyl)adenosine
- α-MHC
α-myosin heavy chain
- CPA
N6-cyclopentyladenosine
- [3H]CPX
8-cyclopentyl-1,3-[3H]dipropylxanthine
- 8-SPT
8-(p-sulfophenyl)theophylline
- TIC
time-to-ischemic contracture
References
- 1.Ely S W, Berne R M. Circulation. 1992;85:893–904. doi: 10.1161/01.cir.85.3.893. [DOI] [PubMed] [Google Scholar]
- 2.Lasley R D, Rhee J W, Van Wylen D G L, Mentzer R M., Jr J Mol Cell Cardiol. 1990;22:29–47. doi: 10.1016/0022-2828(90)90970-d. [DOI] [PubMed] [Google Scholar]
- 3.Angello D A, Headrick J P, Coddington N M, Berne R M. Am J Physiol. 1991;260:H193–H200. doi: 10.1152/ajpheart.1991.260.1.H193. [DOI] [PubMed] [Google Scholar]
- 4.Lasley R D, Mentzer R M., Jr Am J Physiol. 1992;263:H1460–H1465. doi: 10.1152/ajpheart.1992.263.5.H1460. [DOI] [PubMed] [Google Scholar]
- 5.Zhao Z Q, McGee D S, Nakanishi K, Toombs C F, Johnston W E, Ashar M S, Vinten-Johansen J. Circulation. 1993;88:709–719. doi: 10.1161/01.cir.88.2.709. [DOI] [PubMed] [Google Scholar]
- 6.Finegan B A, Lopaschuk G D, Gandhi M, Clanachan A S. Br J Pharmacol. 1996;118:355–363. doi: 10.1111/j.1476-5381.1996.tb15410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parratt J R. Trends Pharmacol Sci. 1994;15:19–25. doi: 10.1016/0165-6147(94)90129-5. [DOI] [PubMed] [Google Scholar]
- 8.Liu G S, Thornton J D, Van Winkle D M, Stanley A W H, Olsson R A, Downey J M. Circulation. 1991;84:350–356. doi: 10.1161/01.cir.84.1.350. [DOI] [PubMed] [Google Scholar]
- 9.Lawson C S, Downey J M. Cardiovasc Res. 1993;27:542–550. doi: 10.1093/cvr/27.4.542. [DOI] [PubMed] [Google Scholar]
- 10.Headrick J P. J Mol Cell Cardiol. 1996;28:1227–1240. doi: 10.1006/jmcc.1996.0113. [DOI] [PubMed] [Google Scholar]
- 11.Mizimura T, Auchampach J A, Linden J, Bruns R F, Gross G J. Circ Res. 1996;79:415–423. doi: 10.1161/01.res.79.3.415. [DOI] [PubMed] [Google Scholar]
- 12.Von Lubitz D K J E, Beenhakker M, Lin R C S, Carter M F, Paul I A, Bischofberger N, Jacobson K A. Eur J Pharmacol. 1996;302:43–48. doi: 10.1016/0014-2999(96)00101-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heurteauz C, Lauritzen I, Widmann C, Lazdunski M. Proc Natl Acad Sci USA. 1995;92:4666–4670. doi: 10.1073/pnas.92.10.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vander Heide R S, Reimer K A. Cardiovasc Res. 1996;31:711–718. doi: 10.1016/0008-6363(95)00235-9. [DOI] [PubMed] [Google Scholar]
- 15.Van Wylen D G L, Schmit T J, Lasley R D, Gingell R L, Mentzer R M., Jr Am J Physiol. 1992;262:H1934–H1938. doi: 10.1152/ajpheart.1992.262.6.H1934. [DOI] [PubMed] [Google Scholar]
- 16.Rudolphi K A, Keil M, Fastbom J, Fredholm B B. Neurosci Lett. 1989;103:275–280. doi: 10.1016/0304-3940(89)90112-2. [DOI] [PubMed] [Google Scholar]
- 17.Koch W J, Milano C A, Lefkowitz R J. Circ Res. 1996;78:511–516. doi: 10.1161/01.res.78.4.511. [DOI] [PubMed] [Google Scholar]
- 18.Marber M S, Mestril R, Chi S-H, Sayen M R, Yellon D M, Dillman W H. J Clin Invest. 1995;95:1446–1456. doi: 10.1172/JCI117815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plumier J-C L, Ross B M, Currie R W, Angelidis C E, Kazlaris H, Kollias G, Pagoulatos G N. J Clin Invest. 1995;95:1854–1860. doi: 10.1172/JCI117865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida T, Watanabe M, Engelman D T, Engelman R M, Schley J A, Maulik N, Ho Y, Oberley T D, Das D K. J Mol Cell Cardiol. 1996;28:1759–1767. doi: 10.1006/jmcc.1996.0165. [DOI] [PubMed] [Google Scholar]
- 21.Subramaniam A, Jones W K, Gulick J, Wert S, Neumann J, Robbins J. J Biol Chem. 1991;266:24613–24620. [PubMed] [Google Scholar]
- 22.Reppert S M, Weaver D R, Stehle J H, Rivkees S A. Mol Endocrinol. 1991;5:1037–1048. doi: 10.1210/mend-5-8-1037. [DOI] [PubMed] [Google Scholar]
- 23.Adolph E A, Subramaniam A, Cserjes P, Olson E N, Robbins J. J Biol Chem. 1993;268:5349–5352. [PubMed] [Google Scholar]
- 24.Hogan B, Costantini F, Lacy E. Manipulating the Mouse Embryo. Plainview NY: Cold Spring Harbor Lab. Press; 1986. [Google Scholar]
- 25.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 26.Church G M, Gilbert W. Proc Natl Acad Sci USA. 1974;81:1991–1995. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cothran D L, Lloyd T R, Taylor H, Linden J, Matherne G P. Biol Neonate. 1995;68:111–118. doi: 10.1159/000244226. [DOI] [PubMed] [Google Scholar]
- 28.Linden J, Patel A, Sadek S. Circ Res. 1985;56:279–284. doi: 10.1161/01.res.56.2.279. [DOI] [PubMed] [Google Scholar]
- 29.Linden J. J Cyclic Nucleotide Res. 1982;8:163–172. [PubMed] [Google Scholar]
- 30.Matherne G P, Byford A M, Gilrain J T, Dalkin A C. Biol Neonate. 1996;873:1–6. doi: 10.1159/000244365. [DOI] [PubMed] [Google Scholar]
- 31.Bruns R F, Fergus J H, Badger E W, Bristol J A, Santay L A, Hartman J D, Hays S J, Huang C C. Naunyn-Schmiedeberg’s Arch Pharmacol. 1987;335:59–63. doi: 10.1007/BF00165037. [DOI] [PubMed] [Google Scholar]
- 32.Bruns R F, Pugsley T A. In: Topics and Perspectives in Adenosine Research. Gerlach E, Becker B F, editors. Berlin: Springer; 1987. pp. 59–73. [Google Scholar]
- 33.Luthin D R, Olsson R A, Thompson R D, Sawmiller D R, Linden J. Mol Pharmacol. 1995;47:307–313. [PubMed] [Google Scholar]
- 34.Toombs C F, McGee S, Johnston W E, Vinten-Johansen J. Circulation. 1992;86:986–994. doi: 10.1161/01.cir.86.3.986. [DOI] [PubMed] [Google Scholar]
- 35.Leung E, Jacobson K A, Green R D. Naunyn-Schmiedeberg’s Arch Pharmacol. 1991;344:639–644. doi: 10.1007/BF00174747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Headrick J, Willis R J. Pflügers Arch. 1988;412:618–623. doi: 10.1007/BF00583763. [DOI] [PubMed] [Google Scholar]
- 37.Belardinelli L, Linden J, Berne R M. Prog Cardiovasc Dis. 1989;32:73–97. doi: 10.1016/0033-0620(89)90015-7. [DOI] [PubMed] [Google Scholar]
- 38.Headrick J P. Am J Physiol. 1996;270:H897–H906. doi: 10.1152/ajpheart.1996.270.3.H897. [DOI] [PubMed] [Google Scholar]
- 39.Kingsley P B, Sako E Y, Yang M Q, Zimmer SD, Ugurbil K, Foker J E. Am J Physiol. 1991;261:H469–H478. doi: 10.1152/ajpheart.1991.261.2.H469. [DOI] [PubMed] [Google Scholar]
- 40.Meissner A, Morgan J P. Am J Physiol. 1995;268:H100–H111. doi: 10.1152/ajpheart.1995.268.1.H100. [DOI] [PubMed] [Google Scholar]
- 41.Kirsch G E, Codina J, Birnbaumer L, Brown A M. Am J Physiol. 1990;259:H820–H826. doi: 10.1152/ajpheart.1990.259.3.H820. [DOI] [PubMed] [Google Scholar]
- 42.Grover G J. Cardiovasc Res. 1994;28:778–782. doi: 10.1093/cvr/28.6.778. [DOI] [PubMed] [Google Scholar]
- 43.Toombs C F, McGee D S, Johnston W E, Vinten-Johansen J. Cardiovasc Res. 1993;27:623–629. doi: 10.1093/cvr/27.4.623. [DOI] [PubMed] [Google Scholar]