

Abstract
Conjugate vaccines protect vaccinated individuals against both disease from and nasopharyngeal carriage of Streptococcus pneumoniae and Haemophilus influenzae. Protection is specific to the capsular serotype(s) included in the vaccine. This specificity has raised concern that vaccination against particular (“targeted”) serotypes may cause an increase in carriage of (and diseases attributable to) nontargeted serotypes. I analyzed a mathematical model designed to predict the factors affecting, and the expected extent of, such replacement in the host population. The conditions for competitive exclusion and coexistence of serotypes under mass vaccination are derived, and the equilibrium carriage of target and nontarget serotypes is determined under various ecological and epidemiological conditions. The eradication threshold for a target serotype in the presence of competing, nontarget serotypes is always lower for serotype-specific than for bivalent vaccines. In a two-serotype model, the increase in the prevalence of any single nontargeted serotype due to vaccination will not exceed the total reduction in prevalence of a targeted serotype. However, if three or more serotypes interact epidemiologically, vaccination against one type may increase carriage of a second more than it decreases carriage of the first. Carriage of a second serotype against which the vaccine offers only partial protection may initially increase and then decrease as a function of vaccine coverage. I discuss the extent to which these theoretical results can account for existing data on serotype replacement after vaccination against H. influenzae and their implications for vaccine policy.
Polysaccharide–protein conjugate vaccines against Haemophilus influenzae and Streptococcus pneumoniae protect individuals against nasopharyngeal carriage of the targeted organisms whereas older, polysaccharide vaccines protect against invasive disease but apparently do not affect carriage (1). Protection against carriage has been demonstrated for H. influenzae type b (Hib) conjugate vaccines (2, 3) and (in more preliminary studies) for multivalent pneumococcal vaccines (4–6). The protection against carriage is specific to the capsular serotypes included in these vaccines.
H. influenzae and S. pneumoniae are characterized by a carrier state; most individuals who are nasopharyngeal carriers of these organisms do not become ill (7, 8). Because nasopharyngeal carriers are the main source of organisms transmitted to other individuals, a vaccine that protects against carriage of targeted serotypes will reduce the exposure of unvaccinated individuals to colonized hosts (9). Such indirect protection, known as herd immunity, has already been demonstrated in studies of conjugate Hib vaccines (10, 11). In addition, population-wide reduction in the carriage of particular serotypes of H. influenzae or S. pneumoniae may open ecological niches that will be filled by other serotypes (6, 12–15). Such “serotype replacement” would be likely to occur if the prevalence of bacterial serotypes and species in nasopharyngeal carriage is regulated by competition from other serotypes or species. Finally, if different serotypes compete to colonize hosts, then vaccination against multiple serotypes, as in the multivalent pneumococcal conjugate vaccines now being tested, may have less than the anticipated herd immunity effect against component serotypes. Multivalent vaccines might partially compromise their effectiveness in reducing population levels of carriage of each individual serotype by reducing the levels of carriage of competing serotypes.
This paper analyzes a mathematical model for the epidemiology of colonizing bacteria with multiple serotypes. It considers two major questions: (i) When a serotype-specific vaccine is introduced against one serotype, how much will the prevalence of a competing serotype increase? and (ii) How does the population-level effectiveness (herd immunity and eradication threshold) of a multivalent vaccine depend on the competition between targeted serotypes?
Before presenting the model, I briefly review the empirical evidence bearing on these questions in H. influenzae and S. pneumoniae.
Empirical Evidence.
H. influenzae. Concerns about serotype replacement have been raised several times with regard to the conjugate vaccines against Hib (12–15). It has been reported that some strains of Hib produce a bacteriocin active against non-b serotypes of H. influenzae (16); bacteriocins may be a means of excluding competing bacteria. Use of conjugate Hib vaccine in developed countries has reduced carriage of Hib to almost zero among vaccinated children from prevaccination levels ranging around 5–10% (2, 3, 10, 11, 17, 18). Statistically discernible increases in carriage of other H. influenzae serotypes have not been detected (3, 10). There is some evidence that the incidence of invasive disease from H. influenzae serotype f has increased since the introduction of widespread Hib vaccination (19), but the causes of this increase have not been identified.
S. pneumoniae.
A recent report from The Gambia showed that a pneumococcal conjugate vaccine reduced carriage of serotypes included in the vaccine but increased carriage of other pneumococcal serotypes (6). A multivalent pneumococcal conjugate vaccine tested in Israel reduced carriage of vaccine serotypes, but no increase was observed in nonvaccine serotypes (5). Several lines of indirect, epidemiological evidence suggest that a population of bacteria already established in the nasopharynx may lower the probability of colonization by other organisms. Prior antimicrobial use is a risk factor for carriage of drug-resistant pneumococci (20, 21). One study specifically showed that antimicrobial treatment was a risk factor for acquisition of a particular, drug-resistant strain, with a defined serotype (23F) and drug resistance profile (21). This offers particularly strong evidence for the role of competition because it excludes the possibility of spontaneous emergence of resistance and suggests, instead, that treatment increases the likelihood of colonization by a resistant serotype. However, such studies do not demonstrate that it is clearance of competing pneumococci, rather than clearance of other bacteria, that increases the risk of colonization. Several studies have shown antagonism between different species of streptococci colonizing the nasopharynx (22). Studies of nasopharyngeal pneumococcal isolates from children in Papua New Guinea (23–25) indicate that colonization with multiple serotypes is less frequent than would be expected if each serotype circulated independently of the others. With one exception (23), however, these studies sampled a small number of colonies and therefore probably underestimated the number of serotypes carried. These results are consistent with the hypothesis of interference but do not rule out such plausible alternatives as geographic heterogeneity in transmission or serotype-specific differences in host susceptibility to carriage.
Model.
I analyzed a compartmental model (26) for the dynamics of two serotypes of colonizing bacteria; the model’s structure is shown in Fig. 1. The model assumes that susceptible hosts enter the transmitting population at a constant rate, L, and that hosts leave at a per capita rate u. In the absence of vaccination, susceptible hosts (X) may be colonized with either bacterial serotype 1 or 2, moving them into the carrier compartments Y1 and Y2. Colonized hosts may be additionally colonized with the other serotype, moving them into the double carrier compartment Y12. These transitions occur according to mass action processes, with susceptible hosts becoming colonized with serotypes 1 and 2 at rates β1X(Y1 + Y12) and β2X(Y2 + Y12), respectively. Hosts already carrying a particular serotype are less likely to acquire the other serotype; this creates competition between the serotypes. Thus, hosts already colonized with serotype 1 are colonized with serotype 2 at a rate c2β2Y1(Y2 + Y12), and hosts colonized with serotype 2 are colonized with serotype 1 at a rate c1β1Y2(Y1 + Y12). Here 0 ≤ c1, c2 ≤ 1, and the cs are the relative risk of acquiring a serotype if already colonized with the other, compared with a noncarrier; ci = 0 corresponds to complete exclusion of dual colonization, and ci = 1 corresponds to complete independence in the behavior of the two serotypes. Colonized hosts lose each serotype independently and become susceptible to colonization again at a rate γ. Thus far, the model is very similar to those of Dietz and Gupta et al. (27, 28).
Figure 1.

Structure of the model, in which hosts can be carriers of either or both of two serotypes of colonizing bacteria. Vaccination protects hosts completely against carriage of serotype 1 (the “target” serotype) and partially, fully, or not at all against carriage of a second serotype. Hosts leave the population at a rate u per capita, shown by the unlabeled arrows.
To incorporate vaccination, the model further assumes that a fraction f of all hosts initially enter a vaccinated, uncolonized class (V) instead of the completely susceptible class; thus, the rates of entry into the susceptible and vaccinated class are (1 − f)L and fL, respectively. f is termed the “vaccine coverage.” Vaccinated hosts are completely protected against colonization with serotype 1 and protected completely, partially, or not at all against serotype 2. Vaccination only occurs for new entrants into the population; therefore, we neglect the possibility that individuals can be colonized before vaccination. The class of vaccinated carriers of serotype 2 is denoted by W; vaccinated individuals acquire serotype 2 at a rate kβ2V(Y2 + Y12 + W); vaccinated individuals’ reduction in the rate of acquiring serotype 2 is termed the “vaccine efficacy against serotype 2” and is equal to 1 − k. Vaccinated hosts carrying serotype 2 are considered to be equally infectious as unvaccinated ones; under this assumption, in the presence of vaccination, the rates at which serotype 2 colonizes susceptible hosts and carriers of serotype 1, respectively, are β2X(Y2 + Y12 + W) and c2β2Y1(Y2 + Y12 + W). To simplify calculations, define E1 ≡ Y1 + Y12 and E2 ≡ Y2 + Y12 + W. In addition, without loss of generality, set L = u = 1; this corresponds to an appropriate choice of temporal and spatial scale. Then the model is:
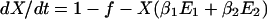 |
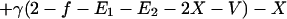 |
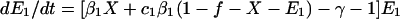 |
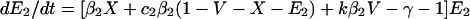 |
1 |
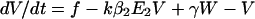 |
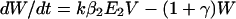 |
It should be noted that the model makes no provision for natural immunity to carriage arising from previous exposure. See Discussion for consideration of this assumption.
RESULTS
Case 1.
No vaccine. As in all models of this type, the condition for a serotype to persist in the population, in the absence of competing serotypes, is that its basic reproductive number R0—the number of secondary hosts who acquire the organism directly from one infectious host placed in an unexposed population at equilibrium—must exceed 1 (26). The basic reproductive number for serotype i is given in this model by R0i = βi/(1 + γ). Two serotypes can coexist if each has a basic reproductive number greater than 1 and each can invade the population when the other is present at its equilibrium (27, 28):
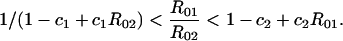 |
2 |
Case 2.
Serotype-specific vaccine. When k = 1, the model describes competition between a target serotype, against which vaccination is completely protective, and a nontarget serotype against which vaccination offers no protection. This model is a reasonable approximation to the effects of the Hib conjugate vaccine (11). In this case, the conditions for coexistence with coverage f are:
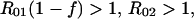 |
and
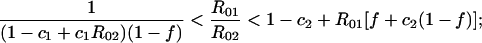 |
3 |
as before, these are the conditions under which each serotype can invade the equilibrium of the other alone.
The target serotype is eliminated when the vaccine coverage exceeds a critical fraction fc given by:
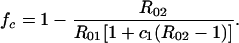 |
4 |
(See Appendix for the derivation.)
This is less than the coverage required in the absence of a competing serotype (in which case fc = 1 − 1/R01); the presence of the nontarget serotype works together with vaccination to reduce the transmission of the target serotype.
The critical coverage to eradicate serotype 1 increases with serotype 1’s basic reproductive rate and decreases with the basic reproductive number of serotype 2 and the degree to which carriage of serotype 2 inhibits carriage of serotype 1. The critical coverage does not depend on the inhibition of serotype 2 by serotype 1. Fig. 2 shows how this critical coverage depends on R01, assuming strong competition between serotypes (c1 = c2 = 0.2). The bottom most curve in each graph corresponds to a completely serotype-specific vaccine (other solid curves correspond to partially or completely bivalent vaccines, see below). The dotted curve shows the critical coverage to eliminate serotype 1 in the absence of competition. Particularly for target serotypes with low basic reproductive numbers, the contribution of competing serotypes can be considerable (e.g., reducing the critical coverage by over 50% if the two serotypes have equal basic reproductive numbers and compete strongly).
Figure 2.

Critical vaccination fraction fc required to eliminate serotype 1 as a function of serotype 1’s basic reproductive number R01 (x-axis) and degree of protection against serotype 2 (different curves). The dashed curve shows the critical fraction if no competing serotype was present, and the solid curves show the critical coverage for a vaccine with 100% (top line, k = 1) or partial (middle lines, k = 0.7, 0.2) efficacy against serotype 2 and a vaccine specific to serotype 1 (bottom line, k = 0). Parameters: R02 = 3, c1 = 0.2.
Use of a serotype-specific vaccine will always increase the carriage of a nontarget serotype that is inhibited by the target serotype, as shown in Fig. 3. The magnitude of this increase depends on various parameters, including the basic reproductive numbers of both serotypes, the vaccine coverage, and the degree of competition between serotypes (c1 and c2). In Fig. 3a, a serotype that coexists with the target serotype in the absence of vaccination increases in prevalence as the fraction of the population vaccinated increases, until the target serotype is eliminated. Fig. 3b shows that vaccination with a serotype-specific vaccine also can cause the appearance of a new serotype that was outcompeted by the target serotype in the absence of vaccination. This occurs if inequality 3 is satisfied but inequality 2 is not.
Figure 3.

Fractions of the population carrying serotype 1, serotype 2, both (1 and 2), or neither, at equilibrium, under different coverage levels with a vaccine specific to serotype 1 (a–c) or to serotypes 1 and 2 (d–f). Dual carriers are included in the rates for serotypes 1 and 2. (a) With relatively weak competition (c1 = c2 = 0.7) and a strain 1-specific vaccine, carriage of serotype 1 declines as vaccination levels increase while carriage of serotype 2 increases, but by less than the decline in serotype 1. (b) With the same parameters, but stronger competition (c1 = c2 = 0.1), serotype 2 is excluded in the absence of vaccination but appears as vaccination rates increase, reducing serotype 1 carriage. Serotype 2 carriage levels increase until the fraction vaccinated is large enough to eliminate serotype 1; above that level, increased coverage has no effect on serotype 2. Parameters for a and b: R01 = 2.2, R02 = 1.6, k = 1. (c) In a three-serotype model with vaccination against only serotype 1, a competing serotype may increase in prevalence more than the target serotype declines. Parameters: R01 = 4.0, R02 = 2.0; R03 = 3.2, cij (relative risk of acquiring serotype i for a carrier of serotype j compared with a host carrying no serotype) = 0.1 for all i, j except c21 = 0.9. Triple colonizations occur according to a multiplicative model; all occur at a relative rate 0.01 except that hosts carrying serotypes 1 and 3 acquire serotype 2 at relative rate 0.09. (d) A vaccine offering 80% protection against serotype 2 (k = 0.2) and perfect protection against serotype 1 has results similar to a serotype 1-specific vaccine if there is weak competition between serotypes (compare a). (e) When competition is stronger (c1 = c2 = 0.1), bivalent vaccines preclude replacement of serotype 1 by serotype 2 but increase the critical vaccination threshold (compare b, same parameters but a serotype-specific vaccine). Parameters for d and e: R01 = 2.2, R02 = 1.6. (f) Carriage of serotype 2 can increase until serotype 1 is eliminated, then decrease as a function of increasing vaccination coverage. Parameters: c1 = c2 = 0.1; k = 0.8; R01 = 2.2, R02 = 1.8.
A key prediction of this two-serotype model is that vaccination will increase carriage of the nontarget serotype less than it decreases carriage of the target serotype (this assertion is justified in the Appendix). Stated another way, vaccination will always decrease the total number of carriers. These phenomena are illustrated in Fig. 3 a and b.
If more than two serotypes are present, however, this limitation no longer applies. In a system comprised of three (or more) serotypes, vaccination against one serotype may produce an increase in the carriage of one or more other serotypes that is greater than the reduction in carriage of the target serotype. This may occur, for example, if before vaccination, the target serotype, in combination with another, coexisting serotype (“serotype 2”), competitively excludes a third serotype (“serotype 3”). If serotype 1 is eliminated by vaccination, and serotype 2 alone cannot outcompete serotype 3, serotype 3 may achieve a prevalence higher than the prevaccination prevalence of serotype 1. This possibility is illustrated in Fig. 3c.
Case 3.
Bivalent vaccine. Finally, we consider the case in which a vaccine offers partial (0 < k < 1) or full (k = 0) protection against a second serotype. Such a model applies to the multivalent pneumococcal conjugate vaccines currently undergoing field trials. As above, we assume that the vaccine gives 100% protection against carriage of the target serotype.
The condition for eliminating serotype 1 is given, as before, by f > fc, but the expression for fc (given in the Appendix) is more complicated. Fig. 2 shows how this threshold depends on the basic reproductive number of the target serotype (x-axis) and on the efficacy of the vaccine against the second serotype; the upper curves correspond to increasing efficacy against serotype 2. If the serotypes compete strongly, the threshold for eliminating serotype 1 is considerably higher with a bivalent vaccine than with a serotype-specific one.
Fig. 3 d–f shows the effects of vaccination with a bivalent vaccine on the equilibrium prevalence of two serotypes. When competition between serotypes is relatively weak, as in Fig. 3d (c1 = c2 = 0.7), the effect of a bivalent vaccine on serotype 1 is similar to the effect of a serotype-specific one (compare Fig. 3a), but the bivalent vaccine also eliminates serotype 2. Fig. 3e shows that the difference between bivalent and serotype-specific vaccines is much greater when the serotypes compete strongly (c1 = c2 = 0.1). The parameters for Fig. 3e match those of Fig. 3b, except that the vaccine is highly effective against serotype 2 (k = 0.1). This prevents the appearance of serotype 2 (compare Fig. 3b) but increases the critical coverage to eliminate serotype 1 considerably, from ≈31% of the population in Fig. 3b to ≈55% in Fig. 3e. Vaccination with a bivalent vaccine may increase carriage of a type against which it has relatively low efficacy, and the prevalence of that serotype may change nonmonotonically with vaccine coverage. In Fig. 3f, the prevalence of serotype 2 initially increases with vaccine coverage, then declines. The increase in prevalence occurs when the benefit to serotype 2 from reducing carriage of serotype 1 outweighs the cost of partial vaccine-induced immunity to serotype 2; once serotype 1 is eliminated, however, further increases in vaccination rates reduce carriage of serotype 2.
In some cases, the goal of a bivalent vaccine is to reduce or eliminate carriage of both serotypes. In eradicating two competing serotypes, the critical coverage is just the larger of the two critical coverages for each serotype in the absence of competition:
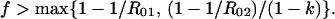 |
In the presence of a bivalent vaccine, two serotypes will coexist when:
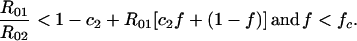 |
5 |
Two serotypes that cannot coexist in the absence of a vaccine will be unable to coexist in the presence of a vaccine that protects completely against both of them; this can be seen by noting that, for k = 0, inequality 5 implies inequality 2 but not vice-versa. With a partially protective vaccine, however, vaccination of a portion of the population can permit coexistence of serotypes that otherwise could not coexist.
DISCUSSION
This paper has analyzed a mathematical model of the transmission dynamics of two or more serotypes of bacteria colonizing a population of hosts, with special attention to the effects of vaccination against one or more serotypes. The model predicts that serotype-specific vaccines will increase the prevalence of serotypes excluded from the vaccine that compete with the vaccine serotype. This increase may include the appearance of novel serotypes that previously were unable to compete with the target serotype. In a two-serotype system, the increase in carriage of any one serotype will always be less than the decline in carriage of the vaccine serotype; thus, in such a system, the total number of individuals carrying neither serotype will always increase with vaccination. In a system with three or more serotypes, however, serotype-specific vaccination can increase the total fraction of hosts carrying a nontargeted serotype more than it reduces the fraction carrying the targeted serotype.
Competition from nontarget serotypes may be beneficial if it reduces the fraction of hosts who must be vaccinated to eliminate the target serotype. For this reason, elimination of a target serotype by a vaccine effective against many other serotypes (like the multivalent pneumococcal vaccines currently being tested) will require a higher level of coverage than if a serotype-specific vaccine were used. If a vaccine is more effective against one serotype than another, then carriage of the second serotype may increase with vaccine coverage as vaccination reduces carriage of the first, but then decline as coverage increases, due to vaccine-induced protection against the second serotype. Although the model is designed for different serotypes of the same bacterial species, it may be applied without modification to interspecific interactions (22).
Several studies of the effects of Hib conjugate vaccines have failed to find any increase in carriage of nonvaccine serotypes of H. influenzae among vaccine recipients, despite considerable reductions in carriage of the b serotype (2, 3, 11). The mathematical model suggests that the lack of replacement may not generalize to other situations (different host populations or different organisms) in which prevalence of targeted serotypes is higher. In both Finland (3) and England (2, 10, 11), where the studies were done, prevaccination carriage of Hib was on the order of 4–7%. The model predicts that, even with 100% vaccine coverage of the population and maximum competition between serotypes, the increase in carriage of any other serotype would be at most equal to the prevaccination prevalence of Hib. Such a small increase would be difficult to detect amidst the temporal fluctuations in carriage of H. influenzae reported in the Finnish study.
Thus, the lack of replacement by competing serotypes of H. influenzae in developed countries may be a result of the low rates of carriage of Hib. Carriage of both type b and non-b H. influenzae is often considerably higher (29) in developing countries. Pneumococcal carriage is much higher than Hib carriage in developed countries (30). With higher prevalences of targeted serotypes, even if the biological interactions between serotypes are identical to those in the British and Finnish Hib studies, the number of hosts who acquire nontargeted serotypes may be considerably larger.
If replacement occurs, it will be more visible in studies of vaccines in which treatment is randomized by whole communities rather than by individuals within a community. If only the study subjects were vaccinated and these constituted a small fraction of the transmitting population, the exposure of vaccinees to each serotype circulating in the community would change little; any increase in carriage of nonvaccine serotypes by vaccinees would result only from changes in their own susceptibility to carriage. This increase would be at most ≈1/(1 − prevaccination prevalence of the target serotype). For example, if 500 people were vaccinated against Hib in a large population (so that vaccination did not change the overall carriage of Hib in the population), and carriage of Hib and another serotype (e.g., H. influenzae type f) before vaccination were 5 and 6%, respectively [as in the U.K. (10)], the expected number of vaccinated H. influenzae type f carriers would be 32 (vs. 30 in 500 controls), assuming that the maximum possible serotype replacement occurred. Such an increase would, of course, be very difficult to measure. If this reasoning is correct, then it will be much easier to see replacement (if it occurs) in populations with high vaccine coverage than in studies of a group of vaccinees in a mostly unvaccinated population.
Preliminary data on pneumococcal conjugate vaccines, although consistently showing reduction of carriage of vaccine serotypes (4–6), are split concerning the question of replacement by competing serotypes, with clear evidence of replacement in The Gambia (6) but no such evidence in an Israeli study (5). Further studies will be needed to resolve this question empirically; the model suggests that differences in the relative frequencies of different serotypes in a population and the serotype composition of the vaccine may account for different outcomes.
The model considered here describes the effects of vaccination on carriage, not invasive disease. The relationship between carriage and disease for nasopharyngeal colonizers is complex, but it is clear that, for most such bacteria, there are many more carriers than victims of invasive disease (8, 25, 31). There is considerable variation between serotypes in virulence, measured as the probability that a colonized host will develop clinical disease; for example, before the advent of vaccination, serotype b accounted for ≈90% of capsulated isolates of H. influenzae-causing disease in children (12, 19) but for a much smaller percentage of isolates from asymptomatic carriers. Similar variation has been observed between serotypes of S. pneumoniae (25). The model does not explicitly consider the complex relationship between immunity to carriage and immunity to invasive disease. If a vaccine is highly efficacious against invasive disease but less efficacious against carriage (as in the earlier generation of polysaccharide vaccines), then the importance of carriage levels will primarily be determined by residual disease in unvaccinated individuals. If vaccine-derived protection against invasive disease is lower, then carriage of the vaccine-targeted serotype, if it persists, will be manifest as invasive disease in both vaccinated and unvaccinated persons.
Given such variation, one policy in designing vaccines would be to include only the serotypes of a given species that cause the majority of invasive disease. Such a policy might be expected to produce the greatest reduction in disease, while maintaining the additional benefits of competition from serotypes not included in the vaccine. However, such vaccines may not remain effective in the long run (32). The species of colonizing bacteria considered here (as well as Neisseria meningitidis) are highly transformable, and the association between capsular serotype and other genetic characteristics, including virulence, may be short-lived (32–34), especially in the presence of vaccine-induced selective pressure. Vaccination cannot only change the prevalence of existing serotypes but also permit the appearance of serotypes that were previously absent. Such serotypes may or may not be virulent, making their impact difficult to predict (41). These considerations suggest that vaccines designed to “cover” serotypes that cause the majority of invasive disease in the absence of vaccination may require updating as the bacterial population shifts toward nontarget serotypes.
There is currently little evidence about the source or effects of naturally acquired, specific immunity to colonization by these bacteria. A role for serotype-specific immunity to colonization is suggested by epidemiological evidence of an inverse correlation between immunogenicity and acquisition rates of pneumococcal serotypes (25) and by a study showing that exposure to polysaccharide crossreactive with the H. influenzae type b capsule protects against colonization with Hib (35). Immune responses against antigens that are conserved across multiple serotypes (such as the pneumococcal pneumolysin) also may offer some protection against colonization (36), but it is not clear whether such responses result from natural colonization. Sequential colonization of individuals by the same and different pneumococcal serotypes (30) suggests that the efficacy of these immune responses is limited. If substantial crossreactive immunity to colonization existed in the bacterial species considered here, it would invalidate the important assumption of the model that the presence of competing bacteria in the nasopharynx is the major determinant of competition between serotypes.
The mathematical models in this paper are related to previous models of multiple strain (or multiple serotype) pathogens. Most directly relevant is McLean’s recent study (37), which showed that vaccines could induce strain replacement in two models, one with complete crossimmunity between strains and another that allowed for the possibility of “superinfection” (38), in which one strain replaces another within a host. Unlike the present model, McLean’s model assumed that hosts became immune, rather than susceptible again, upon termination of infection, and did not allow for multiple simultaneous infection. Her assumptions, which may be more appropriate for viral infections than for the colonizing bacteria considered here, led to somewhat different conclusions. Most notably, total prevalence could increase as a result of vaccination in McLean’s two-serotype, superinfection model. Several recent papers by May and Nowak have analyzed superinfection (38, 39) and coinfection (40) (multiple simultaneous infection) for multiple-strain models and have shown that, in a superinfection model, the effect of vaccination is to remove the most virulent strains (39). The principal difference between the present model and those of McLean, Nowak, and May is that the present model, designed for bacteria that are usually carried asymptomatically for a relatively long period, assumes that hosts can carry multiple serotypes, and that competition between serotypes is mediated by reduced colonization probabilities in a host already colonized with another serotype. Previous models attribute competition to cross-reactive host immunity derived from previous infection with another strain (37) or to host death induced by other, coinfecting strains (40). The superinfection models have a similar mechanism of competition to the one considered here but assume that each host can only carry one strain; upon infection by a second strain, infection with the first strain is terminated (38, 39). Two other models have described the coexistence conditions for multistrain pathogens but have not considered vaccination (27, 28).
The model assumes that the vaccine is completely protective against carriage of the target serotype. This assumption is more nearly correct for existing Hib vaccines (11) than for pneumococcal vaccines (4). To a first approximation, less than perfect efficacy might be accounted for by multiplying the fraction vaccinated in the model by a factor equal to the vaccine efficacy. A related assumption is that multiply colonized hosts are infectious to the same degree, with each serotype, as hosts singly colonized with either serotype. In multiply colonized hosts, the proportions of different serotypes are often uneven (23); however, it is unlikely that this simplification will greatly change model dynamics. Insofar as there are serotype-specific differences in population size within a host, these should be reflected (both in the model and in reality) as differences in the transmissibility β. Finally, we have considered only equilibrium outcomes, not the approach to these equilibria. Published estimates of prevalence, acquisition rates, and loss rates for pneumococcal serotypes suggest that unvaccinated populations are very close to their expected equilibria (25).
Epidemiological studies to evaluate candidate pneumococcal and Hib conjugate vaccines in both developed and developing countries offer an opportunity to test the predictions of this model and to improve our understanding of the ecology of the nasopharyngeal bacteria. The model suggests tradeoffs between the benefits of competition in eliminating serotypes of particular concern and the risks of serotype replacement; these tradeoffs should be considered in deciding the serotype composition of candidate vaccines. In addition, the model suggests that optimism stemming from the lack of serotype replacement following widespread Hib vaccination in developed countries may not be warranted when considering other populations or other bacterial species.
Acknowledgments
I thank E. R. Moxon, P. Fine, O. Levine, G. Carlone, K. O’Brien, and the EcLF for very helpful discussions of these topics and B. R. Levin, D. Stephens, and J. Glasser for valuable criticisms of the manuscript. This work was supported by National Institutes of Health Grant GM-33782 to B.L.
ABBREVIATION
- Hib
H. influenzae type b
Appendix
Proof that the Increase in the Nontarget Serotype Will Be Less Than the Decrease in the Target Serotype in the Two-Serotype Model.
For ease of calculation, we assume that 100% of the population is vaccinated and that the vaccine gives no protection against the nontarget serotype (these assumptions are conservative because they maximize the fraction of the population infected with the nontarget serotype at the vaccinated equilibrium). We want to show that
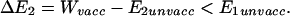 |
A1 |
The density of uninfected hosts is given by Vvacc = 1/R02. Before vaccination, the number of hosts carrying neither serotype (noncarriers) is given by Xunvacc = 1/R02 − c2Y1unvacc. Clearly, Vvacc > Xunvacc. Using the definitions in the model section above, this implies:
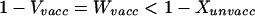 |
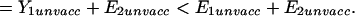 |
This is equivalent to (Eq. A1), q.e.d.
Critical Fraction to Vaccinate.
The critical fraction that must be vaccinated to eliminate serotype 1 is that value of f that makes the realized reproductive rate of serotype 1 equal to unity. This is given by the equation:
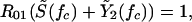 |
A2 |
where S̃(fc) and Ỹ2(fc) are the densities of unvaccinated, noncarrier hosts and unvaccinated carriers of serotype 2, respectively, as calculated at equilibrium in the absence of serotype 1 with vaccine coverage fc.
If the vaccine gives no protection against serotype 2 (k = 1), then, using (Eq. A2),
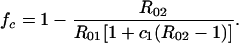 |
If the vaccine gives partial protection against serotype 2 and if serotype 1 can coexist in the presence of serotype 2 without vaccination, then (again, using Eq. A2), the critical coverage to eliminate serotype 1 is given by:
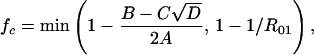 |
where: A = c12(1 − k)R01R02; B = R01(k − c1)(1 − c1) + 2R02c1(1 − k) + R01R02c1k(1 − c1); C = (1 − c1); and D = R01[R01(c1 − k)2 + 4R02c1k(1 − k) + c1kR01R02(2k − 2c1 + c1kR02)].
The quadratic form applies if serotype 2 is present when serotype 1 is eradicated; the simpler form of 1–1/R01 applies if serotype 2 is not present.
References
- 1.Douglas R M, Hansman D, Miles H B, Paton J C. Am J Dis Child. 1986;140:1183–1185. doi: 10.1001/archpedi.1986.02140250109044. [DOI] [PubMed] [Google Scholar]
- 2.Barbour M L, Booy R, Crook D W M, Griffiths H, Chapel H M, Moxon E R, Mayon-White D. Pediatr Infect Dis J. 1993;12:478–484. doi: 10.1097/00006454-199306000-00005. [DOI] [PubMed] [Google Scholar]
- 3.Takala A K, Eskola J, Leinonen M, Kayhty H, Nissinen A, Pekkanen E, Makela P H. J Infect Dis. 1991;164:982–986. doi: 10.1093/infdis/164.5.982. [DOI] [PubMed] [Google Scholar]
- 4.Dagan R, Muallem M, Melamed R, Leroy O, Yagupsky P. 36th Interscience Conference on Antimicrobial Agents and Chemotherapy. New Orleans, LA: American Society for Microbiology; 1996. , abstract G39. [Google Scholar]
- 5.Dagan R, Melamed R, Muallem M, Piglansky L, Greenberg D, Abramson O, Mendelman P M, Bohidar N, Yagupsky P. J Infect Dis. 1996;174:1271–1278. doi: 10.1093/infdis/174.6.1271. [DOI] [PubMed] [Google Scholar]
- 6.Obaro S K, Adegbola R A, Banya W A S, Greenwood B M. Lancet. 1996;348:271–272. doi: 10.1016/s0140-6736(05)65585-7. [DOI] [PubMed] [Google Scholar]
- 7.Austrian, R. (1986) J. Antimicrob. Chemother. 18, Suppl. A, 35–45. [DOI] [PubMed]
- 8.Moxon, E. R. (1986) J. Antimicrob. Chemother. 18, Suppl. A, 17–24. [DOI] [PubMed]
- 9.Aniansson, G., Alm, B., Andersson, B., Larsson, P., Nylen, O., Peterson, H., Rigner, P., Svanborg, M. & Svanborg, C. (1992) J. Infect. Dis. 165, Suppl. 1, S38–S40. [DOI] [PubMed]
- 10.Barbour M L, Mayon-White R T, Coles C, Crook D W M, Moxon E R. J Infect Dis. 1995;171:93–98. doi: 10.1093/infdis/171.1.93. [DOI] [PubMed] [Google Scholar]
- 11.Barbour M L. Emerging Infect Dis. 1996;2:176–182. doi: 10.3201/eid0203.960303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wenger, J. D., Pierce, R., Deaver, K., Franklin, R., Bosley, G., Pigott, N. & Broome, C. V. (1992) J. Infect. Dis. 165, Suppl. 1, S34–S35. [DOI] [PubMed]
- 13.Farley M M, Stephens D S, Brachman P S., Jr Ann Intern Med. 1992;116:806–812. doi: 10.7326/0003-4819-116-10-806. [DOI] [PubMed] [Google Scholar]
- 14.Nitta D M, Jackson M A, Burry V F, Olson L C. Pediatr Infect Dis J. 1995;14:157–160. [PubMed] [Google Scholar]
- 15.Greene G R. Pediatrics. 1978;62:1021–1025. [PubMed] [Google Scholar]
- 16.Venezia R A, Robertson R G. Can J Microbiol. 1975;21:1587–1594. doi: 10.1139/m75-232. [DOI] [PubMed] [Google Scholar]
- 17.Madore D V. Infect Agents Dis. 1996;5:8–20. [PubMed] [Google Scholar]
- 18.Murphy T V, Pastor P, Medley F, Osterholm M T, Granoff D M. J Pediatr. 1993;122:517–523. doi: 10.1016/s0022-3476(05)83529-2. [DOI] [PubMed] [Google Scholar]
- 19.Urwin G, Krohn J A, Deaver-Robinson K, Wenger J D, Farley M M. Clin Infect Dis. 1996;22:1069–1076. doi: 10.1093/clinids/22.6.1069. [DOI] [PubMed] [Google Scholar]
- 20.Pradier C, Dunais B, Carsenti-Etesse H, Largillier R, Bernard E, Dellamonica P. 36th Interscience Conference on Antimicrobial Agents and Chemotherapy. New Orleans, LA: American Society for Microbiology; 1996. , abstract C56. [Google Scholar]
- 21.Reichler M R, Allphin A A, Breiman R F, Schreiber J R, Arnold J E, McDougal L K, Facklam R R, Boxerbaum B, May D, Walton R O, Jacobs M R. J Infect Dis. 1992;166:1346–1353. doi: 10.1093/infdis/166.6.1346. [DOI] [PubMed] [Google Scholar]
- 22.Sanders C C, Sanders W E, Jr, Harrowe D J. Infect Immunity. 1976;13:808–812. doi: 10.1128/iai.13.3.808-812.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gratten M, Montgomery J, Gerega G, Gratten H, Siwi H, Poli A, Koki G. Southeast Asian J Tropical Med Public Health. 1989;20:501–509. [PubMed] [Google Scholar]
- 24.Gratten M, Manning K, Dixon J, Morey F, Torzillo P, Hanna J, Erlich J, Asche V, Riley I. Southeast Asian J Tropical Med Public Health. 1994;25:123–131. [PubMed] [Google Scholar]
- 25.Smith T, Lehmann D, Montgomery J, Gratten M, Riley I D, Alpers M P. Epidemiol Infect. 1993;111:27–39. doi: 10.1017/s0950268800056648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson R M, May R M. Infectious Diseases of Humans: Dynamics and Control. Oxford: Oxford Univ. Press; 1991. [Google Scholar]
- 27.Dietz K. J Math Biol. 1979;8:291–300. doi: 10.1007/BF00276314. [DOI] [PubMed] [Google Scholar]
- 28.Gupta S, Swinton J, Anderson R M. Proc R Soc London Ser B. 1994;256:231–238. doi: 10.1098/rspb.1994.0075. [DOI] [PubMed] [Google Scholar]
- 29.Montgomery, J. M., Lehmann, D., Smith, T., Michael, A., Joseph, B., Lupiwa, T., Coakley, C., Spooner, V., Best, B., Riley, I. D. & Alpers, M. P. (1990) Rev. Infect. Dis. 12, Suppl. 8, S1006–S1016. [DOI] [PubMed]
- 30.Gray B M, Converse G M, III, Dillon H C., Jr J Infect Dis. 1980;142:923–933. doi: 10.1093/infdis/142.6.923. [DOI] [PubMed] [Google Scholar]
- 31.Caugant D A, Hoiby E A, Magnus P, Scheel O, Hoel T, Bjune G, Wedege E, Eng J, Froholm L O. J Clin Microbiol. 1994;32:323–330. doi: 10.1128/jcm.32.2.323-330.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swartley J S, Marfin A A, Edupuganti S, Liu L-J, Cieslak P, Perkins B, Wenger J D, Stephens D S. Proc Natl Acad Sci USA. 1997;94:271–276. doi: 10.1073/pnas.94.1.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barnes D M, Whittier S, Gilligan P H, Soares S, Tomasz A, Henderson F W. J Infect Dis. 1995;171:890–896. doi: 10.1093/infdis/171.4.890. [DOI] [PubMed] [Google Scholar]
- 34.Takala A K, Vuopio-Varkila J, Tarkka E, Leinonen M, Musser J M. J Infect Dis. 1996;173:128–135. doi: 10.1093/infdis/173.1.128. [DOI] [PubMed] [Google Scholar]
- 35.Moxon E R, Anderson P. J Infect Dis. 1979;140:471–478. doi: 10.1093/infdis/140.4.471. [DOI] [PubMed] [Google Scholar]
- 36.Alexander J E, Lock R A, Peeters C C A M, Poolman J T, Andrew P W, Mitchell T J, Hansman D, Paton J C. Infect Immunity. 1994;62:5683–5688. doi: 10.1128/iai.62.12.5683-5688.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McLean A R. Proc R Soc London Ser B. 1995;261:389–393. [Google Scholar]
- 38.Nowak M A, May R M. Proc R Soc Lond Serv B. 1994;255:81–89. [Google Scholar]
- 39.May R M, Nowak M A. J Theor Biol. 1994;170:95–114. doi: 10.1006/jtbi.1994.1171. [DOI] [PubMed] [Google Scholar]
- 40.May R M, Nowak M A. Proc R Soc London Ser B. 1995;261:209–215. [Google Scholar]
- 41.Kelly T, Dillard J P, Yother J. Infect Immunity. 1994;62:1813–1819. doi: 10.1128/iai.62.5.1813-1819.1994. , [DOI] [PMC free article] [PubMed] [Google Scholar]