

Abstract
Enzyme I (EI) is the first protein in the phosphoryl transfer sequence from phosphoenolpyruvate (PEP) to sugar in carbohydrate uptake via the bacterial PEP:glycose phosphotransferase system. The EI monomer/dimer transition may regulate the phosphotransferase system because only the EI dimer is autophosphorylated by PEP. We previously showed that the EI monomer comprises two major domains: (i) a compact, protease-resistant N-terminal domain (EI-N), containing the active site His, and (ii) a flexible, protease-sensitive C-terminal domain (EI-C), which is required for EI dimerization. EI-N interacts with the second protein, HPr, and phospho-HPr, but EI-N neither dimerizes nor is phosphorylated by PEP. We report here the molecular cloning and some properties of EI-C. EI-C is rapidly proteolyzed in vivo. Therefore, two different overexpression vectors encoding fusion proteins were constructed. Fusion Xa contains MalE (the maltose-binding protein), the four-amino acid sequence required by protease factor Xa, followed by EI-C. Fusion G contains His-Tyr between MalE and EI-C and is cleaved by the protease genenase. Homogenous EI-C was isolated from fusion G. [32P]PEP phosphorylated EI-N when supplemented with EI-C, fusion Xa, or fusion G. EI-C may act catalytically. Complementation was also demonstrated in vivo. An Escherichia coli ptsI deletion grew on mannitol as the sole source of carbon after it was transformed with two compatible vectors; one vector encoded EI-N and the other encoded fusion Xa or fusion G. The molecular details underlying important properties of EI can now be studied.
The phosphoenolpyruvate (PEP):glycose phosphotransferase system (PTS) has an essential role in several physiological processes in the bacterial cell. These include sugar transport, chemotaxis to its sugar substrates, regulation of expression of certain operons, etc. (for reviews, see refs. 1–3). A given function is mediated by a particular ensemble of PTS proteins. For example, uptake/phosphorylation of different sugars requires from three to six PTS proteins. However, the common, pivotal component of each system is enzyme I (EI), the first protein in the phosphoryl transfer sequence.
Some properties of the Escherichia coli and Salmonella typhimurium EI proteins (which are virtually identical) have been reviewed (4) and are briefly summarized as follows. (i) EI undergoes a highly temperature-sensitive monomer/dimer transition; the E. coli monomer molecular mass is 63.5 kDa. The dimer accepts the phosphoryl group from PEP, but the monomer does not. The rates of association/dissociation are surprisingly slow, much slower than the catalytic rate of the enzyme (5). (ii) The monomer contains two domains, a thermally stable, protease-resistant N-terminal domain, EI-N, and a flexible, highly protease-sensitive C-terminal domain, EI-C (6). (iii) EI-N contains the active site His residue and was originally isolated after protease digestion of intact EI (6). It was subsequently cloned from the intact gene ptsI (7, 8). Both the solution and crystal structures of cloned EI-N have been established (9, 10). (iv) The C-terminal domain contains all four Cys residues and both Trp residues of intact EI. Because EI-N does not dimerize, it was originally suggested that the C-terminal domain is responsible for dimer formation (6). The flexibility of EI-C has been well documented in thermal unfolding experiments (6) and by the reactivity of Cys-SH residues, which vary in the presence of EI ligands (11, 12). It has been suggested that the EI-C domain in intact EI confers species specificity for HPr in the N-terminal domain (8), although EI-N and EI bind E. coli HPr with about equal affinity (7).
In the phosphoryl transferase sequence, the first two reactions are as follows:
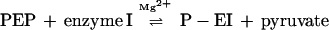 |
1 |
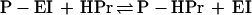 |
2 |
EI-N isolated after proteolysis of EI or cloned from ptsI actively substitutes for intact EI in reaction 2 but not in reaction 1. We show here that EI-C, cloned from ptsI, complements EI-N, i.e., EI-N accepts the phosphoryl group from PEP in the presence of EI-C. Surprisingly, this reaction also occurs both in vitro and in vivo with fusion proteins containing EI-C linked to the periplasmic maltose-binding protein, MalE.
MATERIALS AND METHODS
Materials, Bacterial Strains, and Growth of Cells.
Reagents for bacterial media were obtained from Difco, J.T. Baker, and BBL. Molecular biology reagents were obtained from New England Biolabs, United States Biochemical, Stratagene, and Novagen. Radioisotopes were purchased from Amersham. Other buffers and reagents were of the highest purity available. EI and EI-N proteins were purified as described (7, 13).
E. coli strain ER2267 was obtained from New England Biolabs, and E. coli BL21 (DE3) was from Novagen. A deletion of ptsI was prepared by growing P1 phage on strain TP2815 (14) and transducing strain BL21 (DE3) to BL21 (DE3)ΔptsI. Bacteria were grown at 37°C in Luria broth or on Luria broth agar plates supplemented with ampicillin (50 μg/ml) and/or chloramphenicol (25 μg/ml), where appropriate, for selecting recombinant E. coli cells.
Construction of Fused Genes.
Standard methods were used for isolating DNA and DNA fragments, restriction mapping, construction of fused genes, etc. (15, 16). Several attempts were made to overexpress EI-C directly, by using essentially the same methods used for EI-N (7). These attempts were unsuccessful because EI-C, in sharp contrast to EI-N, is very sensitive to proteolysis. The problem was finally resolved by using a commercial gene fusion/protein purification system, obtained from New England Biolabs (kind gifts from Don Comb and Paul Riggs). Two such systems were tested. Both involved fusion of malE to suitably truncated ptsI, separated (in-frame) by a short stretch of DNA that encoded a tetrapeptide (Ile-Glu-Gly-Arg) or the dipeptide His-Tyr. The tetrapeptide was cleaved by the protease factor Xa, and the dipeptide was cleaved by a genetically engineered subtilisin, designated “genenase.”
The methods used for constructing the appropriate plasmids, pMal∷EI-C/Xa and pMal∷EI-C/G, and the vectors used for overexpressing the fusion proteins, fusion Xa and fusion G, respectively, are summarized in Fig. 1.
Figure 1.

Construction of plasmids for overexpression of fusion proteins. Two vectors (from New England Biolabs) were used for constructing the fusions. plH1148 contains the malE gene terminated by the coding sequences for His-Tyr (the amino acid sequence required for the genenase protease). The malE gene in this vector is deleted for the signal sequence so that the fusion protein is expressed in the cytoplasm. pMAL-p2 contains malE terminated by the coding sequence for Ile-Glu-Gly-Arg (required for the protease factor Xa) and contains the signal sequence so that the fusion protein is secreted into the periplasm. Two overexpression vectors were used for the constructs: pSRT7, which contains ptsI, the gene encoding EI, under control of the inducible T7 polymerase (pT7 promoter), and pET21a, which also contains pT7. pET21a was converted to pET∷MAL by inserting malE, which had been severed from pIH1148 with NdeI and HindIII. A DNA fragment encoding EI-C was cut from pSRT7 with BglII and HindIII, and the desired gene fusions were constructed by inserting the fragment downstream of the malE genes in pMAL-p2 and pET∷MAL, giving the overexpression vectors pMAL∷EI-C/Xa and pMAL∷EI-C/G, respectively.
Expression and Purification of Fusion Proteins.
pMal∷EI-C/Xa and pMal∷EI-C/G were transformed into E. coli BL21(DE30)ΔptsI (14). For purification of the fusion proteins, 4 ml of inoculum (overnight culture in Luria broth containing 50 μg/ml ampicillin) was transferred to 600 ml Difco Luria broth/ampicillin in a 6-liter flask; five such flasks were shaken at 37°C until the OD550 was 0.8. Isopropyl β-d-thiogalactoside was then added to 1 mM, and the cultures shaken for 6 h. The cells were harvested, washed, and ruptured by three passages through a French press.
Fusion proteins Xa and G were purified by a three-step procedure. The crude extracts were treated with streptomycin sulfate to precipitate nucleic acids and applied to 15-ml bed volume amylose affinity columns; after the samples were adsorbed and washed, the fusion proteins were eluted with 10 mM maltose in 20 mM Tris⋅HCl buffer (pH 7.4), 200 mM KCl, 1 mM DTT, 1 mM EDTA, 1% PMSF, and 20 mM ɛ-amino-caproic acid. The second purification step consisted of chromatography on a Sephacryl S300 column at room temperature in 10 mM potassium phosphate buffer (pH 6.5), 50 mM KCl, 1 mM EDTA, 5 mM MgCl2, 0.2 mM DTT, 1% PMSF, and 20 mM ɛ-amino-caproic acid. The apparently homogeneous preparation was applied to a 5-ml bed volume Mono Q HR5/5 column (Pharmacia) as the last step of purification by a fast protein liquid chromatography system. A 30-ml, 50–800 mM KCl gradient (1 ml/min) was used in a solution containing 10 mM Tris⋅HCl (pH 7.4), 1 mM DTT, 1 mM EDTA, 5 mM MgCl2, 1% PMSF, and 20 mM ɛ-amino-caproic acid.
Isolation of EI-C.
EI-C does not contain the amino acid sequence supposedly required for cleavage by factor Xa. Nevertheless, MalE and fragments of EI-C were obtained when the protease was incubated with fusion protein Xa. Despite extensive study, no suitable conditions could be found for isolating significant quantities of EI-C from fusion Xa.
EI-C was derived from fusion protein G. A typical preparation was as follows: 1 mg of fusion G was digested at 25°C with 30 μg of the sequence-specific protease genenase in 1 ml of 20 mM Tris⋅HCl, 2.5 M KCl, 1 mM EDTA, 1 mM DTT, 1% PMSF, and 0.01% Triton X-100 (pH 8.0). The kinetics of the reaction were followed by SDS/PAGE, and the cleavage was approximately complete at 7 h. To stop the reaction, the mixture was cooled to 0°C, concentrated by ultrafiltration (Centriprep 10, Amicon), and diluted with 20 mM Tris⋅HCl, 50 mM KCl, 1 mM DTT, 1 mM EDTA, 5 mM MgCl2, and 1% PMSF (pH 7.4). The buffer exchange was repeated three times. The reaction mixture was then applied to a 3-ml bed volume amylose column and the flow-through containing EI-C and fragments of EI-C was collected. To remove the latter, the mixture was chromatographed on a Mono Q column by using the fast protein liquid chromatography system described above. The final purification step consisted of fast protein liquid chromatography–gel filtration chromatography by using a 23-ml bed volume Superose 6 column Superose 6 (Pharmacia). EI-C was obtained in an approximately 30% yield, appeared homogeneous on SDS/PAGE, and migrated at a rate corresponding to 35 kDa (approximately the value estimated from the predicted amino acid sequence, 35.4 kDa).
In Vivo Complementation Experiments.
As described below, EI-N is complemented in vitro by EI-C, by fusion Xa, or fusion G. Attempts were therefore made to determine whether the two domains could complement each other in vivo. For this purpose, it was necessary to construct a plasmid encoding EI-N compatible with pMal∷EI-C/Xa and pMal∷EI-C/G. The construct, pSXEI-N, was generated as shown in Fig. 2. The plasmid contains intact ptsI-N under control of the T7 promoter, a Cmr cartridge, and the pSC101 origin of replication, which is compatible with a number of other ori.
Figure 2.

Construction of plasmid for expression of EI-N for in vivo experiments. The vector pSYX39 (15) contains the pSC101 origin of replication that is compatible with a number of other origins of replication; the vector was cut with BamHI. A DNA fragment was inserted into the vector that encodes a partially truncated EI-N isolated from p9C; the fragment contained the pT7 promoter at the 5′ terminus. The truncation at the 3′ terminus was corrected by cutting p9C with XbaI and HindIII, isolating the fragment, and by inserting it into pSXEI-NΔ, which had also been cut with the two restriction enzymes. The plasmid pSXEI-N contained the gene encoding all EI-N under the control of the pT7 promoter, the Cmr gene, and the desired origin of replication.
Pairs of plasmids were introduced into E. coli BL21 (DE3) ΔptsI by transformation (16). (Plasmids were also used to transform the wild-type strain, BL21(DE3), as a control.) It should be stressed that overexpression of some of these proteins and protein fragments is lethal; therefore, we did not induce the promoters, relying instead on low levels of expression because the promoters were slightly leaky.
Mannitol utilization unequivocally requires a functional PTS, i.e., a functional EI in the present case. The transformants were therefore tested for mannitol growth on minimal agar M9 plates, supplemented with 1% mannitol as the sole carbon source, and by growth in liquid mannitol M9 medium.
Phosphorylation Studies.
Phosphorylation experiments were conducted with [32P]PEP (kindly provided by Norman Meadow, Johns Hopkins Univ.). Incubation mixtures contained the following buffer/salts (buffer A) mixture in 10 μl: 100 mM potassium phosphate buffer (pH 6.5)/1 mM EDTA/5 mM MgCl2/1 mM DTT. In addition, the mixtures contained 10 nmol [32P]PEP (specific activity, 9 × 104 cpm/nmol), and one or more of the following proteins (μg): EI, 2; EI-N, 4; EI-C, 0.4; fusion Xa, 4; fusion G, 4. Incubation times were 1 h at room temperature. To stop the reactions and to minimize hydrolysis of the phosphohistidinyl proteins during electrophoresis, 5 μl of 3-fold-concentrated SDS/PAGE sample buffer (pH 8.5) was added to each incubation mixture. The proteins were denatured by incubating for 15 min at 37°C and subjected to SDS/PAGE (17). The wet gel was not fixed but was wrapped immediately in plastic and applied to an x-ray film that was exposed for 48 h at −70°C.
The effect of concentration of EI-C on the phosphorylation reaction was studied similarly, by using 4 μg EI-N and 0.1–0.4 μg EI-C per incubation mixture.
RESULTS
Purity of Proteins.
The proteins used in these studies appeared to be homogeneous by SDS/PAGE (16, 17). Examples are shown in Fig. 3A (fusion G, lane 2; fusion Xa, lane 4; EI-C, lane 6; EI, lane 7; EI-N, lane 8).
Figure 3.

Phosphorylation of EI-N and EI. Phosphorylation experiments were conducted as described in Materials and Methods with [32P]PEP as the phosphoryl donor. Protein samples were subjected to SDS/PAGE (16, 17). (A) Coomassie stained gel. (B) Autoradiograph. Standard molecular mass markers are shown at each end of the gel. Incubation mixtures contained the following proteins: Lanes: 1, fusion G (4 μg) and EI-N (4 μg); 2, fusion G (4 μg); 3, fusion Xa (4 μg) and EI-N (4 μg); 4, fusion Xa (4 μg); 5, EI-C (0.4 μg) and EI-N (4 μg); 6, EI-C (0.4 μg); 7, EI (2 μg); 8, EI-N (4 μg).
In Vitro Phosphorylation Experiments.
The purpose of the present work was to determine whether the C-terminal domain of EI, EI-C, could complement the N-terminal domain, EI-N. Fig. 3B shows that EI autocatalytically accepts the phosphoryl group from PEP (lane 7), whereas neither EI-C nor EI-N are phosphorylated (lanes 6 and 8, respectively). However, EI-C complements EI-N as shown in Fig. 3B, lane 5, i.e., 32P is transferred from [32P]PEP to EI-N in the presence of EI-C.
To our surprise, the same result was obtained when the fusion proteins were used in place of EI-C. Fusion G plus EI-N are shown in Fig. 3, lane 1, and fusion Xa and EI-N are shown in lane 3. As noted above for EI-C, there was no phosphorylation of the fusion proteins per se.
The experiments in Fig. 3 were conducted with an EI-N/EI-C molar ratio of approximately 10. The effects of varying the concentration of EI-C on the phosphorylation reaction are shown in Fig. 4.
Figure 4.

Effect of concentration of EI-C on phosphorylation of EI-N. The experimental conditions were the same as those used for Fig. 3 (see Materials and Methods) with the following proteins in the incubation mixtures: Lanes: 1, 2 μg EI; 2, 4 μg EI-N; 3, 4 μg EI-N (140 pmol) plus 0.4 μg EI-C (11 pmol); 4, 4 μg EI-N plus 0.2 μg EI-C; 5, 4 μg EI-N plus 0.1 μg EI-C. In the latter, the mol ratio EI-N/EI-C = 51.
In these studies, EI-N was maintained constant at 4 μg per incubation mixture, whereas EI-C was varied, 0.4 μg in lane 3, 0.2 μg in lane 4, and 0.1 μg in lane 5. Although we did not directly measure the stoichiometry or the kinetics of the EI-N/EI-C interactions, it seems possible that EI-C is a catalyst in the phosphotransfer reaction to EI-N, namely, that 1 mol EI-C can phosphorylate more than 1 mol EI-N.
In Vivo Complementation of EI-N by EI-C Fusion Proteins.
Because the fusion proteins complemented EI-N in vitro, it seemed possible that they might also be complementary in vivo. The experimental design was to use d-mannitol as the sole source of carbon in minimal medium and a deletion mutant of EI (E. coli BL21(DE3)ΔptsI). Because mannitol utilization requires the intact PTS, growth on this substrate by transformants of the deletion mutant would be definitive evidence for a functional EI. In this assay, the mutant was transformed with two compatible plasmids: pSXEI-N (Fig. 2) encoded EI-N and either pMal∷EI-C/G or pMal∷EI-C/Xa (Fig. 1) encoded the two fusion proteins G and Xa, respectively. Previous experience had shown that EI-C per se does not survive proteolysis in vivo; therefore, no attempt was made to express this fragment in the cells transformed with pSXEI-N.
Results of the growth experiments are shown in Table 1 and Fig. 5. The qualitative results of growth on mannitol/minimal media agar plates are given in Table 1. Various combinations of plasmids were tested, but growth was observed in only three cases, a control containing a plasmid that encodes intact EI and two transformants containing plasmids encoding EI-N and either fusion protein G or Xa.
Table 1.
Growth of E. coli BL21(DE3)ΔptsI transformants on mannitol
| E. coli BL21(DE3)ΔptsI transformed with plasmids* | Proteins or protein fragments encoded by plasmids | Growth† |
|---|---|---|
| pSXEI-N + pMal::EI-C/G | EI-N + fusion protein G | + |
| pSXEI-N + pMal::EI-C/Xa | EI-N + fusion protein Xa | + |
| pSXEI-N + pMal-p2 | EI-N + MalE | − |
| pSEXI-N + pET::Mal | EI-N + MalE | − |
| pSYX39 + pSRT7 | Intact EI | + |
| pSYX39 + pMal::EI-C/G | Fusion protein G | − |
| pSYX39 + pMal::EI-C/Xa | Fusion protein Xa | − |
| pSYX39 + p9C | EI-N | − |
| pSYX39 + pMal-p2 | MalE | − |
| pSYX39 + pET::Mal | MalE | − |
Figure 5.

Growth of E. coli BL21(DE3) and transformants of BL21(DE3)ΔptsI on mannitol. Cells were grown on a rotary shaker at 37°C in 50-ml Erlenmeyer flasks equipped with side arm tubes for determining turbidity in the Klett colorimeter. The salts (M9) medium (9 ml) was supplemented with mannitol to a 1% concentration. Inocula consisted of 1 ml of cells grown overnight on the same medium. At the end of the growth period, plasmids were isolated from the relevant cells (see text). All cells were double transformants. ■, Control, E. coli BL21(DE3), pSYX39/pET:Mal (expresses intact EI from chromosome and MalE from plasmid). The other transformants were derived from BL21(DE3)ΔptsI: •, pSYX39/pSRT7 (intact EI); ▴, pSXEI-N/ pMal∷EI-C/G (EI-N plus fusion G); ▵, pSXEI-N/pMal∷EI-C/Xa (EI-N plus fusion Xa); ×, pSYX39/pET∷Mal (Mal E).
Quantitative data were obtained by measuring the growth rates of the same transformants in liquid culture (mannitol/M9). The results are shown in Fig. 5. Wild-type cells [E. coli BL21(DE3)] are shown as a control and grew more rapidly than the transformants of E. coli BL21(DE3)ΔptsI-harboring plasmids. Good growth was obtained with the double-transformant pSYX39/pSRT7, which encodes intact EI, and it ultimately reached about the same cell density.
The critical results were obtained with the transformant pSXEI-N/pMal∷EI-C/G, which encodes EI-N and fusion protein G, and with the corresponding double-transformant encoding EI-N and fusion protein Xa. These dual-transformants exhibited the same rates of growth as the control that expressed intact EI and, again, grew to about the same density as the wild type.
Although unlikely, it was possible that the in vivo data resulted from recombination of the DNA fragments encoding EI-N and EI-C to give intact ptsI. The transformants in Fig. 5 were therefore harvested at the end of the growth experiment, and the plasmid DNA was isolated. The expected restriction patterns were observed on electrophoresis, indicating that only the predicted proteins and protein fragments could have been expressed in the transformants (data not shown).
DISCUSSION
We previously suggested that EI may be the regulator of PTS-sugar uptake by bacterial cells (18, 19) and that the monomer/dimer transition is the key factor in this regulation because it is so slow (5). The dimer is autophosphorylated by PEP, whereas the monomer is not. The hypothesis is that other proteins and/or metabolites may affect the rate of interconversion of monomer and dimer and, therefore, the rate of sugar uptake. The molecular details of the transition and of the phosphoryl transfer reaction are therefore of great interest.
The results reported here can be summarized as follows:
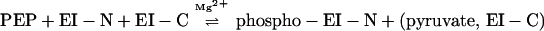 |
3 |
The two major domains of EI, EI-N and EI-C, complement each other in vitro. The fusion proteins containing MalE linked to EI-C can substitute for EI-C in this complementation assay. Furthermore, the fusion proteins complement EI-N in vivo such that transformants that express EI-N and a fusion protein grow at the same rates on mannitol as does a transformant that expresses intact EI.
Reaction 3 is written as the simplest possible form of what must be a highly complex system of four interacting species, EI-N, EI-C, PEP, and Mg2+, and it may be even more complex when the fusion proteins are substituted for EI-C. A detailed analysis of the system should show whether EI-C forms a stable complex with EI-N, which can then be phosphorylated, or whether EI-C acts catalytically such that many moles of EI-N are phosphorylated per mole of EI-C. If the latter proves to be the case, this would mean a repetitive association/dissociation interaction between the two domains. Many other questions can now be asked, such as whether EI-C (and the fusion proteins) forms a monomer/dimer transition in the absence of the N-terminal domain, the role of the PEP and Mg2+ in this transition, and whether the equivalent of the EI dimer, 2EI-N:2EI-C, must be formed for phosphoryl transfer from PEP.
Acknowledgments
We are very grateful to Dr. Norman Meadow for the gift of [32P]PEP and to Drs. Don Comb and Paul Riggs (New England Biolabs) for gifts of the proteases factor Xa and genenase. These studies were supported by grants from the National Institute of General Medical Sciences of the National Institutes of Health (GM11632 to L.B. and GM38759 to S.R.).
ABBREVIATIONS
- PEP
phosphoenolpyruvate
- PTS
phosphotransferase system
- EI
enzyme I
- PMSF
phenylmethylsulfonyl fluoride
References
- 1.Meadow N D, Fox D K, Roseman S. Annu Rev Biochem. 1990;59:497–542. doi: 10.1146/annurev.bi.59.070190.002433. [DOI] [PubMed] [Google Scholar]
- 2.Postma P W, Lengeler J W, Jacobson G R. Microbiol Rev. 1993;57:543–594. doi: 10.1128/mr.57.3.543-594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saier M H, Reizer J. Mol Microbiol. 1994;13:755–764. doi: 10.1111/j.1365-2958.1994.tb00468.x. [DOI] [PubMed] [Google Scholar]
- 4.Chauvin F, Brand L, Roseman S. Res Microbiol. 1996;147:471–479. doi: 10.1016/0923-2508(96)84001-0. [DOI] [PubMed] [Google Scholar]
- 5.Chauvin F, Brand L, Roseman S. J Biol Chem. 1994;269:20270–20274. [PubMed] [Google Scholar]
- 6.LiCalsi C, Crocenzi T S, Freire E, Roseman S. J Biol Chem. 1991;266:19519–19527. [PubMed] [Google Scholar]
- 7.Chauvin F, Fomenkov A, Johnson C R, Roseman S. Proc Natl Acad Sci USA. 1996;93:7028–7031. doi: 10.1073/pnas.93.14.7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seok Y-J, Lee B R, Zhu P-P, Peterkofsky A. Proc Natl Acad Sci USA. 1996;93:347–351. doi: 10.1073/pnas.93.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garrett D S, Seok Y-J, Liao D-I, Peterkofsky A, Gronenborn A M, Clore G M. Biochemistry. 1997;36:2517–2530. doi: 10.1021/bi962924y. [DOI] [PubMed] [Google Scholar]
- 10.Liao D-I, Silverton E, Seok Y-J, Lee B R, Peterkofsky A, Davies D R. Structure. 1996;4:861–872. doi: 10.1016/s0969-2126(96)00092-5. [DOI] [PubMed] [Google Scholar]
- 11.Han M K, Walbridge D G, Knutson J R, Brand L, Roseman S. Anal Biochem. 1987;161:479–486. doi: 10.1016/0003-2697(87)90477-5. [DOI] [PubMed] [Google Scholar]
- 12.Han M K, Roseman S, Brand L. J Biol Chem. 1990;265:1985–1995. [PubMed] [Google Scholar]
- 13.Chauvin F, Roseman S, Brand L. Biophys J. 1990;57:429a. (abstr.). [Google Scholar]
- 14.Bitoun R, de Reuse H, Touati-Schwartz D, Danchin A. FEMS Microbiol Lett. 1983;16:163–167. [Google Scholar]
- 15.Xu S, Fomenkov A. BioTechniques. 1994;17:57. [PubMed] [Google Scholar]
- 16.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current Protocols in Molecular Biology. New York: Wiley; 1995–1998. , Vols. 1–3. [Google Scholar]
- 17.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 18.Waygood E B, Weigel N, Nakazawa A, Kukuruzinska M A, Roseman S. Can Fed Biol Soc Proc. 1977;20:54. (abstr.). [Google Scholar]
- 19.Waygood E B, Meadow N D, Roseman S. Anal Biochem. 1979;95:293–304. doi: 10.1016/0003-2697(79)90219-7. [DOI] [PubMed] [Google Scholar]