

Abstract
Transcriptional dysregulation in Huntington’s disease (HD) is a well documented and broadly studied phenomenon. Its basis appears to be in huntingtin’s aberrant protein-protein interactions with a variety of transcription factors. The development of therapeutics targeting altered transcription, however, faces serious challenges. No single transcriptional regulator has emerged as a primary actor in HD. The levels of literally hundreds of RNA transcripts are altered in affected cells and it is uncertain which are most relevant. The protein-protein interactions of mutant huntingtin with transcriptional factors do not constitute conventional and easy targets for drug molecules. Nevertheless, potential therapeutic advances, targeting transcriptional deregulation in HD, have been made in recent years. In this chapter we review current progress in this area of therapeutic development. We also discuss possible drug discovery strategies targeting altered transcriptional pathways.
Keywords: Huntington’s disease, transcriptional dysregulation, mutant huntingtin, transcription factor(s), small molecule, neuroprotection
1. Preface
Huntington’s disease (HD) is an autosomal dominant, neurodegenerative disorder caused by the expansion of a CAG-triplet repeat within the coding sequences of the HD gene. This leads to the translation of an abnormal protein containing an elongated polyglutamine tract that causes a slowly progressive and fatal neurological phenotype. Dysfunction and degeneration of increasing numbers of cerebral cortical and striatal neurons especially underlie the symptoms of HD and the progressive functional decline that occurs. The precise mechanism(s) of neurodegeneration are unknown, though many aspects of neuronal homeostasis are perturbed. Although a variety of neuroprotective approaches have been demonstrated in cellular and in vivo models, none have yet been translated into treatments demonstrated to slow the progression of HD.
2. Transcriptional dysregulation in Huntington’s disease
In early studies, altered levels of dopamine receptor mRNAs have been found in HD brain tissue (Augood et al., 1997; Augood et al., 1996). In a follow-up investigation using HD transgenic mice, transcriptional dysregulation as a contributor to the pathogenesis of HD was proposed by Cha and co-workers (Cha et al., 1999) who importantly linked disease-specific decreases in RNA transcript levels to reductions of D1 and D2 dopamine receptors, specific glutamate receptors, and to the A2a adenosine receptor in HD patients and rodent models. Notably, specific decreases in RNA levels of dopamine and glutamate receptors were observed in pre-symptomatic transgenic animals suggesting that transcriptional alterations may not be a secondary consequence of neurodegeneration. The results of this study suggested that specific changes in transcription underlie neurodegeneration, rather than unspecific degradation of all RNAs in sick neurons. Transcriptional dysregulation in HD was then further investigated by Luthi-Carter and co-workers (Luthi-Carter et al., 2000). The most comprehensive analysis of transcription changes in HD has been reported in a series of published articles (Chan et al., 2002; Luthi-Carter et al., 2002a; Luthi-Carter et al., 2002b; Sipione et al., 2002). Authors compared the transcriptional profiles of different HD rodent models. Quite specific changes in RNA levels were detected in each HD model, however, overlap of common altered transcripts was incomplete. Nevertheless, this thorough work unambiguously established phenomenon of transcriptional dysregulation in HD models by detailing specific affected and unaffected genes. In follow-up studies, age and progression-dependent changes in transcript levels during disease progression were characterized. Transcriptional alterations are detectable in the blood of pre-symptomatic subjects and appreciably increased in symptomatic subjects (Borovecki et al., 2005) suggesting that transcriptional alterations could also serve as useful biomarkers reflecting HD progression and response to therapies.
The relationship between transcriptional dysregulation and neurodegeneration remains uncertain. While changes in transcript levels of specific RNAs translated into deficient protein function could certainly contribute to pathology, it is unclear whether this is an important contribution. From that perspective, it becomes critical to identify the roles of gene products, translated from deteriorating HD transcription, responsible for key pathways leading to neurodegeneration. It is counter intuitive to think that all gene transcriptional changes, translated into protein levels, contribute equally to the neurodegeneration process.
It is also conceivable that transcriptional dysregulation is a relatively benign phenomenon, in comparison to more potent causes of neurodegeneration, yet accurately underlying pathological changes in cells. Understanding the molecular basis for the origin of transcriptional dysregulation, whether there are key transcriptional pathways that are most important, and the resulting contributions to pathology are important for developing a successful drug discovery strategy. Similarly, compounds probing transcription will likely be needed to really understand how important it is to the pathogenesis of HD.
3. Probing transcriptional disease space for drug targeting
3.1. Mutant Huntington’s disease protein
Novel gain of function(s) for the mutant HD protein has been accepted as best explaining most experimental data related to the pathogenesis of HD. While transgenic knock-out mice were not viable, full-length mutant huntingtin animals are born and develop normally, indicating that huntingtin inactivation does not explain HD, though it could contribute to neurodegeneration (White et al., 1997; Zuccato et al., 2001). Neurological phenotypes comparable to that seen in human HD have been observed in mouse transgenic models expressing only fragments of mutant huntingtin protein, also suggesting the conferral of a dominant pathogenic function (Davies et al., 1997; Laforet et al., 2001). However, mutant huntingtin (Htt) appears to trigger multiple pathways of neurodegeneration, including dysregulation of transcription and it is uncertain which, if any, might have a primary position in a cascade or whether there simply are multiple parallel processes at work. Removal of mutant huntingtin by RNAi, perhaps even targeting allele specific SNPs, can be envisioned as an ultimate cure (Harper et al., 2005; Wang et al., 2006). The beauty of such an approach is the irrelevance of subsequent molecular mechanisms of neurodegeneration, since the penultimate toxin will be eliminated. Unfortunately such an approach remains distant at the moment due to lack of safe and efficient gene delivery methods that can reach most of the brain. For the purpose of this review we will limit our discussions to practical approaches to intervene with transcription with small molecule ligands.
3.2. Mutant huntingtin interactions with transcription factors
Understanding the molecular basis for transcriptional dysregulation is important for selecting an appropriate drug discovery strategy. The central cause of the disease pathogenesis, mutant huntingtin, radiates negative signals through the cellular biochemical network by direct interactions with various proteins, including transcription factors.
Because mutant huntingtin readily aggregates with itself and other proteins forming insoluble inclusions, a question that arises is whether transcriptional dysregulation occurs because huntingtin sequesters transcriptional modulators that it interacts with or whether soluble interactions are more determinant (DiFiglia et al., 1997; Scherzinger et al., 1997). Initially it was shown that mutant huntingtin polypeptides, containing extended polyglutamines, co-aggregated with basal transcriptional factors such as TATA-binding protein and CBP (Kazantsev et al., 1999; Nucifora et al., 2001; Steffan et al., 2000). Co-aggregation was based on the affinity of interactions between extended polyglutamines and wild-type polyglutamines within native sequences of TBP and CBP. It was suggested that transcription factors, precipitated in the form of insoluble aggregates, were depleted from the cellular pool, subsequently causing transcriptional dysregulation.
Results from more recent studies have altered this notion about the molecular basis of transcriptional dysregulation from the originally proposed sequestration model (Sadri-Vakili et al., 2006). It has been shown that soluble mutant huntingtin interacts with a variety of basal transcription factors such as CBP, interfering with the work of the transcription machinery (Steffan et al., 2000). The interactions with mutant huntingtin, occurring in soluble phase, affected transcriptional responses and cell viability in HD models (Cong et al., 2005; Jiang et al., 2003; Jiang et al., 2006). Furthermore, additional work has shown that polyglutamine inclusions fail to appreciably deplete transcription factor levels (Yu et al., 2002). Therefore, aggregate-mediated sequestration appears unlikely to be a major factor contributing to transcriptional dysregulation.
3.2.1. Mutant huntingtin and basal transcription machinery
Mutant huntingtin interactions with components of the basal transcription machinery have been extensively studied. It has been shown that mutant huntingtin possibly represses basal transcription by interacting with essential subunits of RNAII complex, TBP, TFIIF, TAFII130 (Dunah et al., 2002; Zhai et al., 2005). Invasion of mutant huntingtin into basal transcriptional complexes could be particularly harmful, affecting entire transcriptional repertoires (Fig. 1A, B). In some recent works the term “transcriptional repression” has been used to emphasize the negative effects of mutant huntingtin on transcription. Clearly, many genes are also upregulated in the setting of mutant huntingtin and it can’t be assumed that these are all compensatory. While being potentially devastating, interference of ubiquitously expressed mutant huntingtin with the basal transcriptional machinery does not readily explain the relative selectivity of neurodegeneration to neurons in striatum and cortex, areas primarily affected in HD.
Figure 1.
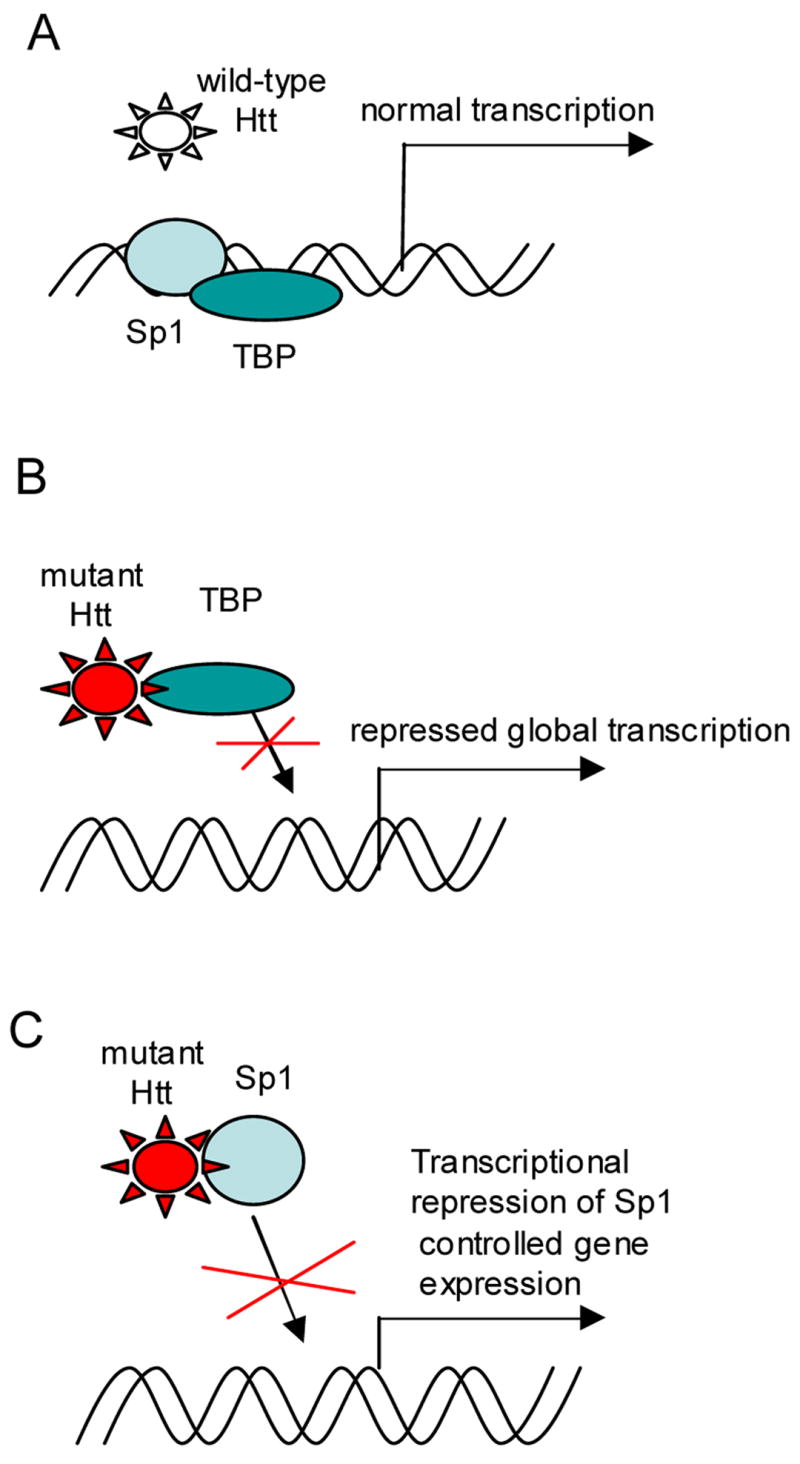
Model of transcriptional repression in Huntington’s Disease based on mutant huntingtin aberrant interactions with transcriptional factors. A. Normal transcription in wild type cells. B. Aberrant interactions of mutant huntingtin (Htt) with TATA-binding protein caused global transcriptional repression. C. Aberrant interactions of mutant huntingtin (Htt) with specificity protein 1 (Sp1) lead to transcriptional repression of Sp1 controlled gene expression.
3.2.2. Mutant huntingtin and Specificity protein 1
Aberrant interactions of mutant huntingtin with Specificity protein 1 (Sp1) have been described (Dunah et al., 2002; Li et al., 2002). The reduction of Sp1 binding to specific promoters in the presence of mutant huntingtin is consistent with Sp1 regulated genes being frequently affected in gene profiling studies (Fig.1C). The potential importance of Sp1 interactions with mutant huntingtin as a cause of transcriptional dysregulation has been examined in rodent models of HD. However, the precise effects of the relationship between these two proteins on neurodegeneration has not been clear, since reduction of Sp1 DNA binding activity or Sp1 levels appeared to be neuroprotective (Qiu et al., 2006).
3.2.3. Blocking protein interactions between mutant huntingtin and transcription factors (Sp1)
From a drug discovery point of view, disruption of huntingtin’s protein-protein interactions with small molecules is hard to achieve. To complicate matters, attempts to crystallize mutant huntingtin have failed. Thus, analyses of co-crystal structure(s) between mutant huntingtin and various transcriptional factor(s), highly desirable for rational drug design, are not yet possible. High throughput screening approaches to identify small molecules, directly disrupting the interactions of mutant huntingtin and candidate binding partners, have to rely on variations of yeast two-hybrid and pull-down assays in vitro. A high-rate of false positive hits can be expected from yeast two-hybrid based screens. To weed-out false-positives and select specific direct inhibitors of protein interactions, a stringent secondary protein-based screen has to be followed. Pull-down assays can be expected to yield compounds with poor selectivity and bioactivity. In practice, this means early and extensive medicinal chemistry campaigns on primary hits, selected from screening large compound collections, to generate selective and bioactive potent derivatives.
To by-pass these hurdles, an alternative approach was taken by D. Kranic and co-workers to find small molecules directly or indirectly reducing the burden of mutant huntingtin transcriptional repression, (unpublished results) (Fig.2.). The investigators employed a striatal neuronal cell line, derived from a double knock-in mutant huntingtin mouse, to design a cell-based drug screening assay with stable integration of a luciferase reporter, controlled by a promoter containing six SP1 binding sites. The mutant huntingtin-dependent transcriptional repression of luciferase expression in this model system was validated using mutant huntingtin RNAi. The assay was adapted for chemical or genetic high throughput screens to identify entities up-regulating luciferase expression. Eliminating false-positives, e.g., hits affecting luciferase stability, turnover, etc., is a straightforward procedure. A counter-screen is performed in the same striatal cell line expressing wild-type huntingtin to weed-out entities up-regulating reporter expression independently from mutant Htt de-repression. Testing hits further in mutant huntingtin striatal cells expressing the reporter gene under control of the same promoter, but lacking Sp1 binding sites, further eliminates nonspecific hits.
Figure 2.
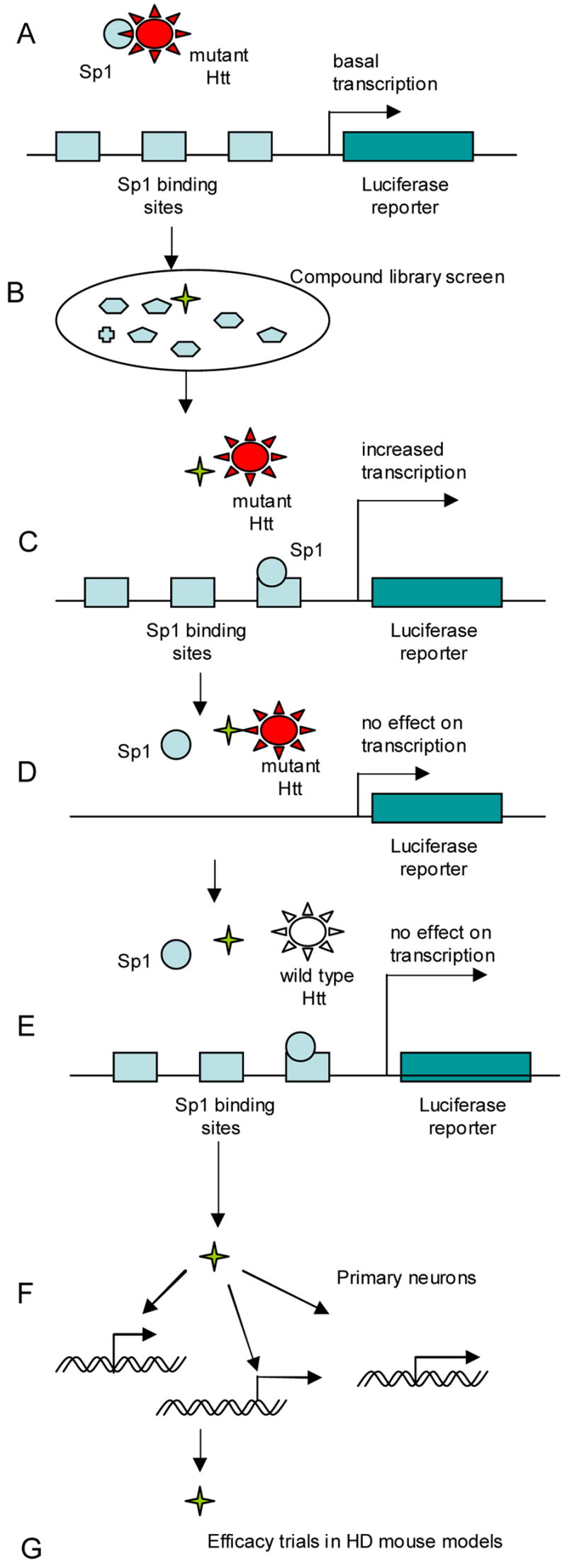
Targeting mutant huntingtin (mHtt) interaction with Sp1 transcription factor. A. Cell-based drug screening assay designed by D. Kranic et al. Cells express full-length mutant huntingtin. Promoter, controlling expression of luciferase reporter, contains 6xSp1binding sites. B. Library of small molecules (or siRNAs) for high throughput screening. C. Identified hit-compound(s) increases transcriptional expression of luciferase reporter. D, E. Validation of hit-compounds in counter-screening assays. Luciferase signal should remain unchanged in presence of compound candidate(s). D. Promoter of reporter constructs lack Sp1 sites. E. Cells express wild-type, but not mutant huntingtin. F. Compound validation in primary neuronal culture. Compound-dependent de-repression of Sp1 controlled gene transcription. G. Test efficacy of candidate compound(s) in mouse HD model(s).
It is worth pointing out that expression of the reporter, controlled by viral promoters, can be repressed by mutant Htt, providing additional complexity for development transcriptional assays and evaluation of the hits.
Further challenges remain in the identification and validation of lead compounds for restoring Sp1-dependent transcriptional dysregulation in HD neurons, and in demonstrating the efficacy of small molecules in animal trials.
3.2.4. Role of p53, NF-kB, and CBP in transcriptional dysregulation and neurodegeneration
There is additional interest in the therapeutic potentials of other specific transcription factors, such as NF-kB, p53 and CBP, which may be involved in neurodegeneration (Bae et al., 2005; Khoshnan et al., 2004; Mantamadiotis et al., 2002; Qin et al., 1998; Takano and Gusella, 2002; Yu et al., 2000). These proteins play important regulatory roles in cells and alteration of their activities in the presence of mutant huntingtin may contribute to transcriptional dysregulation in HD. Despite key roles in cell regulation played by each of these transcription factors, it seems unlikely that any one protein can be solely responsible. Nevertheless, from a drug discovery standpoint, it is feasible to assess the neuroprotective effects of genetic and chemical agonists and antagonists, modulating the respective regulatory pathways to assess their therapeutic potential. Since these proteins of interest have been widely studied over the years, key reagents for target validation are readily available. For example, analysis of progeny, generated from crosses between HD transgenic R6/2 and p53 knock-out mice, including comprehensive behavior, survival, and neuropathology studies, may clarify the role of p53 in neurodegeneration (Dumble et al., 2003).
3.3. Role of histone acetylation in Huntington’s disease
The concept of altered histone acetylation in HD was proposed and tested by Leslie Thompson and co-workers (Steffan et al., 2001). They have shown that inactivation of CBP histone transferase activity (HAT), caused by interactions with mutant huntingtin, led to decreased histone acetylation. Decrease in histone acetylation is known to be associated with transcriptional silencing, and could explain transcriptional repression in HD (Fig. 3.A, B, C). To compensate for HAT deficiency and restore histone acetylation levels, the investigators proposed to target enzymes responsible for histone deacetylation (HDAC), with small molecule inhibitors. HDAC inhibitors have been under development for some time for application in cancer therapy. HDAC inhibitors suberoylanilide hydroxamic acid (SAHA) and sodium butyrate (Fig. 3. D, E) were neuroprotective in tissue cultured cells and fly models of HD. Similar results demonstrating neuroprotective effects of HDAC inhibitors were later achieved in other HD cell-based and invertebrate models, and in HD transgenic mouse models by many researchers (Bates et al., 2006; Ferrante et al., 2003; Hockly et al., 2003; Igarashi et al., 2003). The magnitude of the benefits in mice were proportional to the best effects achieved with compounds targeting other mechanisms, but were not superior to them. In HD transgenic mice their efficacies have not been a major advance over the efficacies achieved by glutamate inhibitors, antioxidants, transglutaminase inhibitors, or energy buffers (Beal and Ferrante, 2004; Ryu et al., 2005). There are a variety of possible explanations for this. These different targets could have equivalent magnitudes. Toxicity, lack of selectivity, or difficulties with brain bioavailability may have so far prevented measurement of the true therapeutic potential of HDAC inhibition. This could be explained on the basis of partial complementation of broadly altered RNA transcripts and insufficient selectivity. This could also be a possible explanation for the cytotoxicity potent HDAC inhibitors, displayed for mammalian cells in tissue culture as well. Alternatively, in some HD cell-based models HDAC inhibitors might activate expression of mutant transgenes, controlled by viral promoters, which induces cytotoxicity and masks efficacious effects (Gomez et al, 2006). This observation is important for evaluating HDAC inhibitors in cell-based models and selecting candidates for mouse trials.
Figure 3.
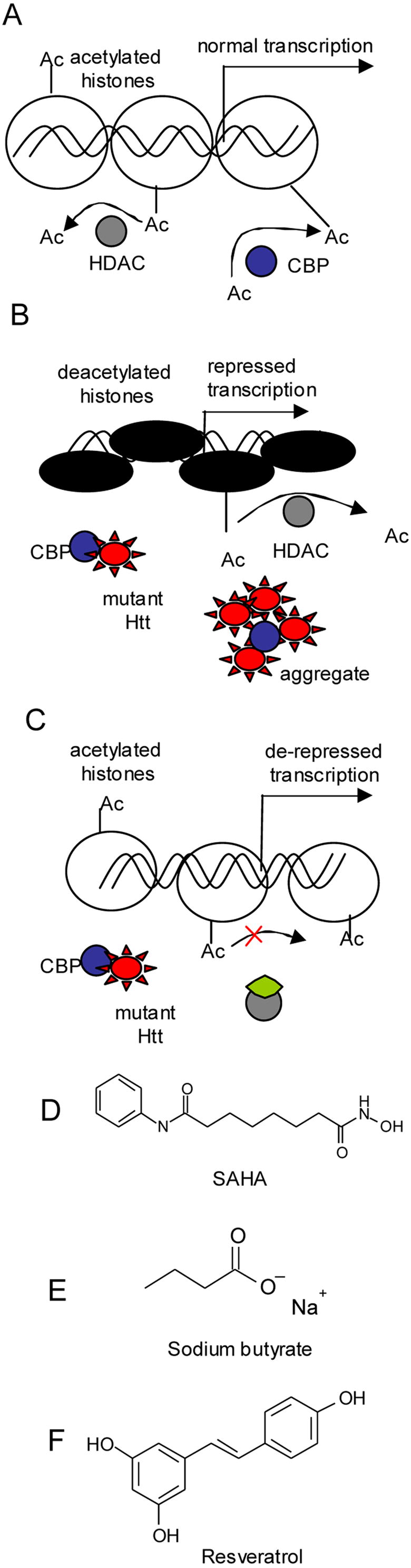
Model of transcriptional repression in Huntington’s Disease based on mutant huntingtin aberrant interactions with CBP (histone acetyl transferase(s)). A. Transcription depends on the acetylation status of histones, regulated by activities of histone acetyl transferases (HATs) and histone deacetylases (HDACs). B. Mutant Htt interactions with CBP and other HATs inhibit proper histone acetylation and cause repression of transcription. Aberrant interactions between mHtt and CBP appear to proceed in soluble phase. C. Increase histone acetylation and up-regulation of transcription is achieved by inhibition of HDAC activities with small molecules. D, E. Small molecule inhibitors, used in animal studies to validate neuroprotective effects of HDAC inhibition in HD. Inhibitors of HDAC class I and II: (D) SAHA (E) Sodium butyrate. F. Resveratrol, activator of sirtuin 1, class III HDAC enzymes shows neuroprotection in various HD models.
3.3.1. Targeting histone deacetylases
Histone deacetylase (HDAC) enzymes play important regulatory functions in cells by modulating a broad spectrum of transcriptional responses. The HDAC family consists of enzymes belonging to three evolutionarily divergant classes. Much cancer therapeutic development has been concentrated in recent years on small molecule inhibitors targeting classes I and II of HDAC enzymes (Hildmann et al., 2006). There has been a major effort made by the pharmaceutical industry to develop selective HDAC inhibitors to improve their therapeutic potentials (Wang et al., 2005). There have been two reports on the discovery of selective class II HDAC6 inhibitors, which could be used for target validation in cell-based and invertebrate HD models. Unfortunately both molecules are predicted to penetrate the blood brain barrier poorly (Mai et al. 2005; Suzuki et al, 2006). The development of HDAC inhibitors for CNS has been slow, and discovered brain-penetrable potent pan HDAC inhibitor MS-275 appeared not to localize in cortex, an area affected in HD (Simonini et al, 2006).
Sirtuins, human histone deacetylases belonging to class III of HDAC enzymes, have been a recent focus of therapeutic development for neurodegenerative diseases (Blander and Guarente, 2004). Interest in sirtuins was precipitated by experiments demonstrating the regulatory effects of sirtuins on aging and longevity in many biological models, and further by the discovery of neuroprotective effects of the sirtuin ligand, resveratrol (Fig. 3F) (Baur and Sinclair, 2006; Guarente and Kenyon, 2000; Viswanathan et al., 2005).
Sirtuins are NAD-dependent deacetylases, which uniquely distinguishes them from other HDACs. The first sirtuin (Sir2) was discovered in yeast, and its role was originally associated with mating switch silencing (Guarente, 2000; Imai et al., 2000; Kennedy et al., 1997). Later it was shown in yeast, fly and worm models that SIR2 controls the aging process (Kaeberlein et al., 1999; Tissenbaum and Guarente, 2001). Humans have seven distinct sirtuin gene products, localized in the nucleus, cytoplasm, and mitochondria, and they are thought to play important and diverse regulatory roles in specialized mammalian cells. The closest human structural homologue to the yeast Sir2 is the SirTT1 protein. Interestingly, activation, rather than inhibition of SirT1, with the small molecule resveratrol was neuroprotective in a HD worm model (Borra et al., 2005; Parker et al., 2005). Currently, resveratrol is being tested in HD mouse models.
Another interesting candidate drug target is human sirtuin 2 (SirT2). SirT2 is a cytoplasmic protein, implicated in the regulation of cell division via deacetylation of α̃tubulin, the best-known substrate for this enzyme (North et al., 2003). Substrate specificity might suggest an involvement of SirT2 in microtubule organization and in maintaining cytoskeleton integrity, perhaps in a functional complex with HDAC6 (Iwata et al., 2005). Recently published results have identified histone H4 as a SirT2 substrate, implicating a broader regulatory role for SIRT2 in the cell (Vaquero et al., 2006). To assess the therapeutic potential for these enzyme targets for HD, it will be necessary to develop potent and selective activators and inhibitors of human sirtuins.
3.4. Alternative models of transcriptional dysregulation
As we discussed, the repression of Sp1-mediated transcription by mutant Huntingtin has been demonstrated (Li et al., 2002; Dunah et al., 2002; Chen-Plotkin et al., 2006), and yet in HD transgenic mice reduction of Sp1 DNA binding or levels is neuroprotective (Qiu et al., 2006). The same dichotomy has been demonstrated for p53 (Steffan et al., 2000; Bae et al., 2005) and CREB (Nucifora et al., 2001; Wyttenbach et al., 2001; Sugars et al., 2004; Obrietan et al., 2004). These results challenge the current model of transcriptional dysregulation that implicates an inhibitory effect of mutant Htt protein-protein interactions on the performance of transcription factors. Given the similarities between Sp1, p53 and CREB, it is tempting to speculate that mutant Htt represses transcription only in proximity to DNA. Hence, a role for transcription factors may be to deliver mutant Htt to DNA. Once loaded onto DNA, mutant Htt might intercalate in the double helix, potentially form oligomers and small aggregates, which distort histones, and alter transcription locally. The selectivity of these alterations is determined by the transcription factors binding to specific DNA sequences, enriched in promoter regions.
3.4.1. Distortion of chromatin DNA complexes by mutant huntingtin
Given that huntingtin interacts with a variety of transcription factors, it seems conceivable that nuclear, mutant huntingtin, might invade DNA-chromatin complexes, causing global aberrations in chromatin organization and remodeling (Fig. 4). It is tempting to speculate further that cells could respond to nuclear huntingtin by tightening-up chromatin to protect DNA integrity, leading to global transcriptional repression. Consistent with a model of global transcriptional repression in HD, Robert Ferrante and colleagues found increased methylation of lysine 9 in histone H3, a well-established mechanism of gene silencing (Ferrante et al., 2004). In the same study investigators demonstrated that Mythromicin A, a DNA binding antibiotic that recognizes guanosine-cytosine-rich regions has neuroprotective potency in that it is significantly greater than all other drugs tested so far in HD transgenic mice. Its efficacy is likely to be relevant to its DNA binding activity. Mutant huntingtin affinity for DNA in vitro was previously demonstrated in early work (Kegel et al., 2002; Steffan et al., 2000). Mithromycin A could block mutant huntingtin binding to DNA in a competitive manner to prevent transcriptional repression. A recent paper on the negative effect of Sp1 over-expression on the survival of HD transgenic mice indirectly supports such a model (Qiu et al., 2006). By residing in GC rich sequences, enriched in promoters, Mithromycin A could reduce the binding of protein Sp1-mutant huntingtin complexes to DNA.
Figure 4.
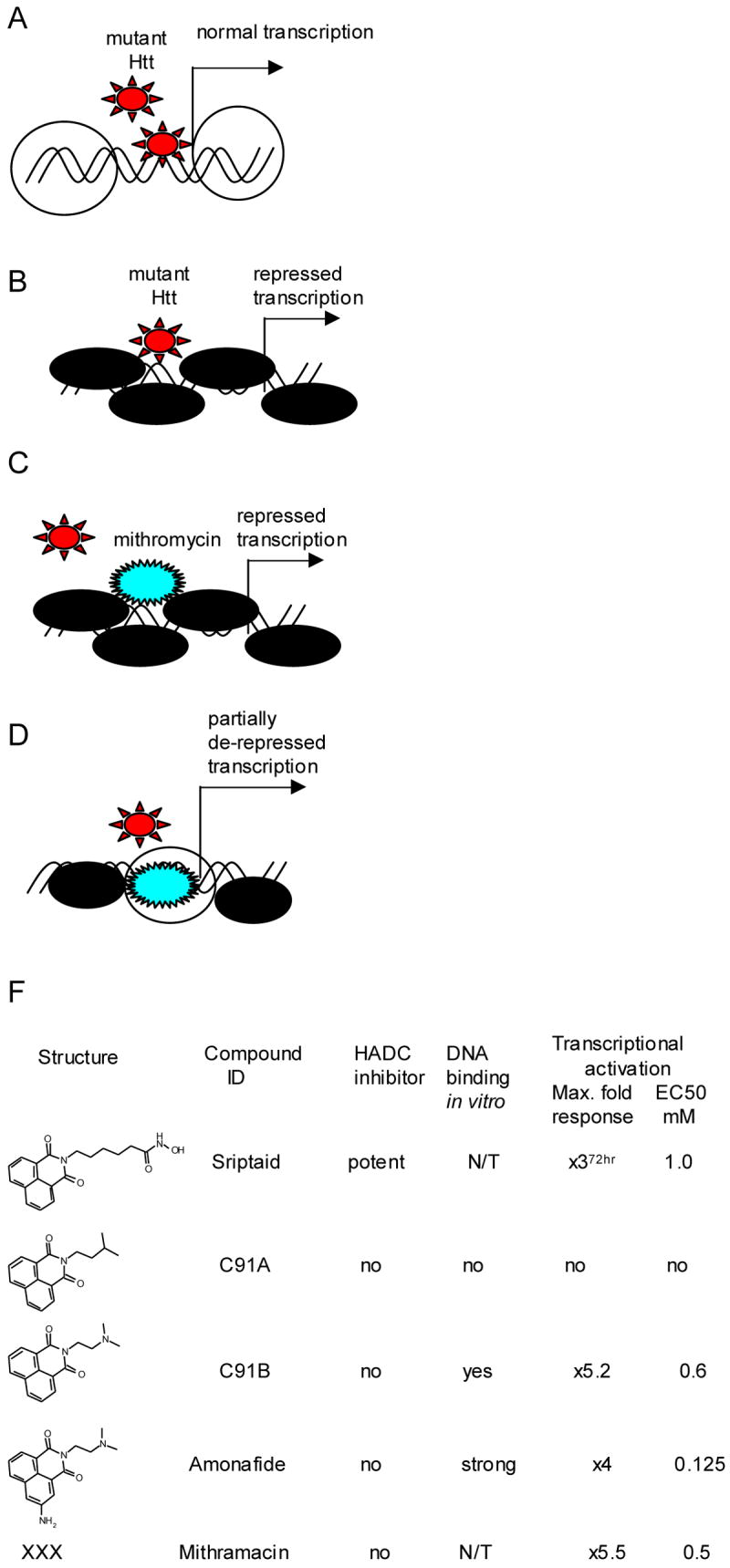
Model of transcriptional repression based on affinity mutant Htt to DNA. A. Invasion mHtt in DNA-chromatin complex. B. Increased chromatin condensation to block mHtt invasion leads to transcriptional repression. C. Antibiotic drug Mithromycin A, has strong affinity to GC rich DNA sequences. D. Binding Mithromycin A to DNA causes partial transcriptional de-repression. F. DNA intercalators possibly have similar mechanism of transcriptional activation as Mithromycin A. Compound DNA binding activity in vitro was determined by displacement of Ethidium Bromide. Compound-dependent activation of transcription was determined in a cell-based assay, designed by D. Kranic et al.
Mithromycin A is an obsolete compound no longer used in humans because of its toxicity, the availability of safer compounds for its principal use in hypercalcemia, and because it has very poor oral bioavailability. We hypothesized that other DNA binding compounds may have similar effects as Mithromycin A on transcription (Fig.4F). To elucidate this notion further we tested the effects of various DNA intercalators on transcription in a mutant huntingtin cell environment. We tested a series of structural analogs (Fig.4F), including the known DNA intercalators, Amonafide and Mitonafide, in the transcriptional assay described above. We found that DNA binding activity in vitro correlated with transcriptional activation. In contrast to the early effects of HDAC inhibitors, transcriptional responses in cells mediated by DNA binding compounds, such as Mithromycin A, were delayed by 24-48 hours. It will be interesting to explore other classes of DNA binding molecules in different HD assays: major and minor groove binders, preferential binders to GC versus AT rich sequences, etc.
While DNA intercalators could all have undesirable side effects, this model suggests potential therapeutic benefits from small molecules modifying chromatin/DNA binding and structure.
3.4.2. REST model of transcriptional repression
Reduction in levels of the neurotrophic factor BDNF in HD has been described (Zuccato et al., 2001). Study of the cause of BDNF deficiency led to the intriguing discovery by Elena Cattaneo and colleagues that one of huntingtin’s native functions was sequestration of the transcriptional repressor REST, a negative regulator of BDNF expression (Zuccato et al., 2003). Furthermore, the HD mutation caused loss of this huntingtin native function, leading to the release of REST protein, which repressed BDNF production. More broadly, these results suggested that in the HD mutant environment, free REST protein might negatively affect the transcription of many genes, contributing to transcriptional repression in HD. Hence, targeting REST could be an attractive therapeutic approach, leading to transcriptional de-repression.
Since BDNF may be critical for neuronal survival, restoration of endogenous BDNF levels to normal may be a therapeutic approach for HD (Mattson et al., 2004). To test this possibility, Elena Cattaneo, Dorothea Rigamonti and co-workers developed a cell-based reporter assay to monitor REST activity (personal communication) (Fig.5). A parental ST14A rat cell line of neuronal origin was employed for assay development. Two DNA reporter constructs were stably integrated and constitutively expressed in cells. Expression of a luciferase reporter, controlled by the BDNF promoter, contains repressor-specific regulatory elements. A promoter lacking REST regulatory binding sites controlled expression of a second independent reporter. Thus, this double read-out assay, combining features of primary and counter screening, was developed to identify compounds specifically up-regulating BDNF expression. Compounds identified in high throughput screens as BDNF up-regulators could alleviate REST-dependent repression and ameliorate global transcription repression in HD cells.
Figure 5.
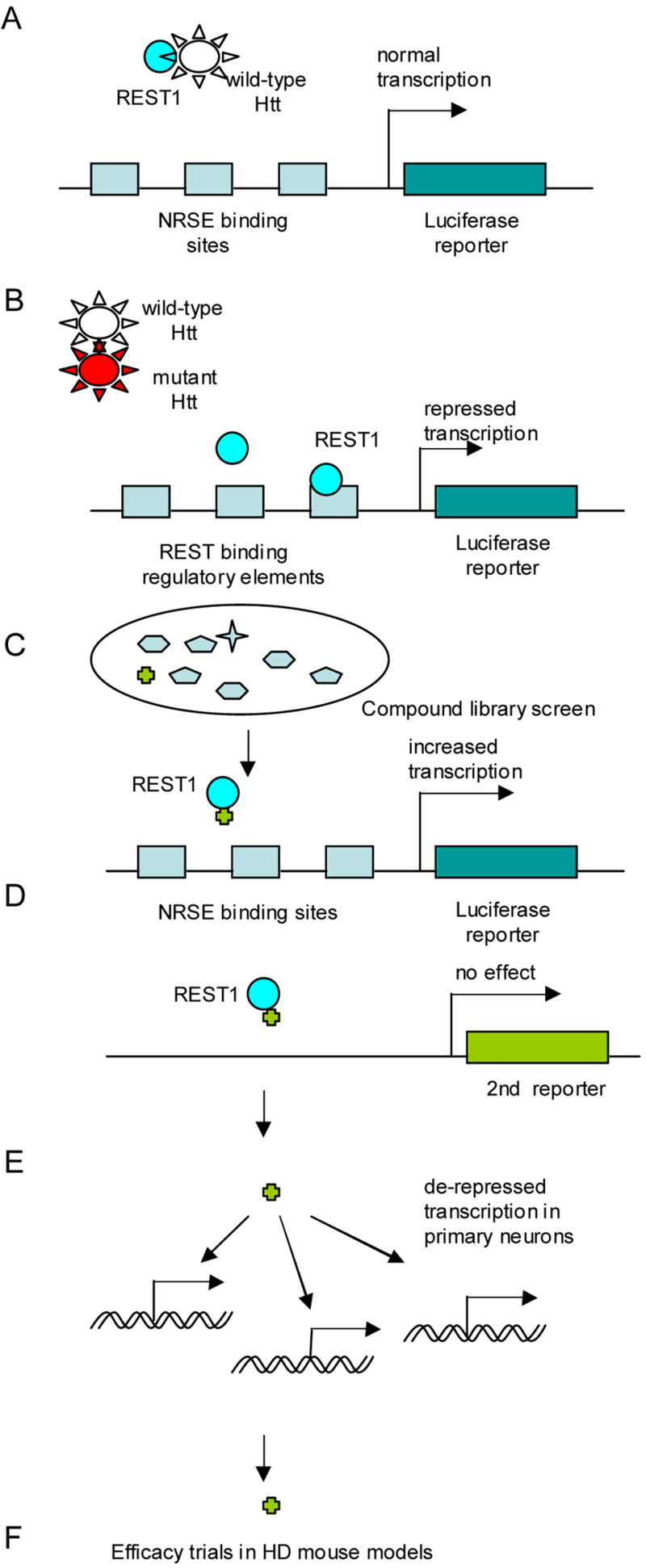
Targeting REST1 mediated repression. A. Cell-based assay for drug screening designed by D. Rigamonti et al. Promoter, controlling expression of luciferase reporter, contains REST binding regulatory elements (NSRE binding sites). B. Library of small molecules (or siRNAs) for high throughput screening. C. Identified hit-compound(s) increases expression of luciferase reporter. Candidate compound does not change read-out of second reporter, controlled by REST independent promoter. D. Compound validation in primary neuronal culture. E. Compound-mediated de-repression of REST1 dependent gene transcription. F. Test efficacy of candidate compound(s) in mouse HD model(s).
4. Towards a cure
Despite significant achievements in our understanding of the pathogenesis of HD, the drug discovery process is still in a slow phase. With few exceptions, interesting and sometimes astounding results generated by basic scientific research may be poorly translated into conventional drug discovery projects. An important bottleneck has also been the great time and effort needed to perform proof of concept studies in HD mouse models, which have limited throughput for screening and comparing possible small molecule therapies.
4.1. Diversity of potential drug targets
Multiple biochemical pathways appear to be affected in HD. Numerous abnormalities have been detected in human disease models by many read-outs. It has not been clear which are critical for neurodegeneration, which are benign disease biomarkers, and which are protective, benign, or toxic responses to injury. Some appear to have greater appeal because of associations with huntingtin, with aging, with the striatum, or with already implicated mechanisms. This richness and diversity of potential targets suggests that some will be more proximal, more specific for HD, and possibly more potent. Some may be more distant from mutant huntingtin and represent more generic neurodegeneration targets of greater potential use in other disorders but possibly not as potent in HD. Some may not be significant targets at all but related to epiphenomenon. Until the data on a given target rests on sufficient proof of concept studies in HD models, especially mouse models, it may be difficult to determine. Fortunately, the growing knowledge of HD pathogenesis provides context for such considerations and reagents able to probe potential targets are often available. The target diversity also provides more choices, which are important for HD given that not all of the potential targets are amenable to high-throughput screening, to hitting well-demarcated active sites in enzymes or receptors, or to molecular modeling of an interaction.
4.2. Drug target identification
Drug discovery progress suffers from a lack of useful and conventional drug targets. The discovery of small RNA interference may greatly facilitate the process of target identification and assessment. In the future we expect the identification of novel molecular target candidates to emerge from siRNA screens based on assays derived from embryonic stem cells. Biomarkers of transcriptional pathology in HD and more generically of responses to therapy are emerging which will also provide essential tools as primary read-outs of these screens.
A chemical-genetic approach, combining genetic and pharmacological screening, is a powerful tool for drug discovery. Identified gene products can be used as a basis for cell-free assay development and drug screens in vitro.
Biochemical profiling of lead-compounds identified in phenotypic cell-based screens appeals as another useful approach in identifying molecular target candidates. These small molecules, validated through secondary confirmatory assays to ensure their specificity, can be subjected to target profiling and also can be used as reference compounds to calibrate the hit selection threshold of the primary assays.
4.3. Identification pre-clinical candidates
Accurate modeling of disease pathophysiology in vitro is essential for target identification, assay development, compound screens, hit validation and optimization, etc. Currently employed for drug discovery, cell-based assays utilize immortalized cell cell-lines. While indefinitely proliferating cancer cells provide sufficient material for high throughput screens, dose-response, and structure activity relationship studies, their gene expression profile is apparently different from gene expression repertoire of the terminally differentiated CNS neurons, affected in HD. A breakthrough in the generation of physiologically relevant cell-based disease models, providing sufficient material for pharmacological and genetic screens, is anticipated from embryonic stem cell research. Such screening platforms, utilizing embryonic stem cells from HD mice, may also be available in the near future (Mee et al., 2006).
Based on an operational definition of transcriptional dysregulation, compound-dependent complementation of altered RNA levels in striatal and cortex of HD neurons is expected. Therefore, compounds identified in drug screens or rationally selected have to demonstrate restoration of gene transcripts, constituting biomarkers of transcriptional dysregulation, in disease relevant confirmatory assays. Successful validation and selection of lead candidates for animal trials depend on the power, reliability, and physiological disease relevance of these secondary assays. The secondary assays can be based on embryonic stem cell, primary neuronal cultures, and embryonic cultured brain slices ex vivo derived from HD mice. Small molecules, restoring normal levels of altered RNA transcripts, can then be subjected to further optimization for potency and ADMET qualities, followed by efficacy trials in rodent HD models.
5. Concluding remarks
Decades of basic research have demonstrated that the pathogenesis of Huntington’s disease (HD) seems to involve the recruitment of multiple biochemical pathways, including transcription. It is likely that some of these pathways may be amenable to modulation by small molecules, which are beginning to be identified. Since there is insufficient knowledge to prioritize the different targets for HD, and therefore predict the therapeutic potentials of identified lead-compounds, the experimental approach is to assess their therapeutic values in genetic mouse models. In this way the most promising leads can be moved forward towards human drug development unhindered by any scientific biases about which mechanisms are most important. We believe that this strategy can successfully identify therapeutic leads that can lead to Investigational New Drugs (INDs) and clinical testing in the near future.
Acknowledgments
We express gratitude to our colleagues Dr. D. Krainc and Dr. D. Rigamonti for sharing with us the design and details of unpublished screening assays. This work was supported by NIH: NS35255, AT00613 and, NS045242; the Huntington’s Disease Society of America Coalition for the Cure; Hereditary Disease Foundation.
Abbreviation List
- HD
Huntington’s Disease
- Htt
Huntingtin
- IND
Investigational New Drug
- HAT
Histone Acetyl Transferase
- HDAC
Histone deacetylase
- SAHA
Suberoylanilide hydroxamic acid
- Sir2
Silent Information Regulator (Sirtuin)
- SirT1
Sirtuin 1
- SirT2
Sirtuin 2
- BDNF
Brain-Derived Neurotrophic Factor
- REST
RE1-Silencing Transcription Factor
- CBP
CREB Binding Protein
- ADMET
Absorption, Distribution, Metabolism, Excretion and Toxicity
- SNP
Single Nucleotide Polymorphism
- TBP
TATA-box Binding Protein
- Sp1
Specificity Protein 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Augood SJ, Faull RL, Emson PC. Dopamine D1 and D2 receptor gene expression in the striatum in Huntington’s disease. Ann Neurol. 1997;42:215–21. doi: 10.1002/ana.410420213. [DOI] [PubMed] [Google Scholar]
- Augood SJ, Faull RL, Love DR, Emson PC. Reduction in enkephalin and substance P messenger RNA in the striatum of early grade Huntington’s disease: a detailed cellular in situ hybridization study. Neuroscience. 1996;72:1023–36. doi: 10.1016/0306-4522(95)00595-1. [DOI] [PubMed] [Google Scholar]
- Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, Snyder SH, Sawa A. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron. 2005;47:29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Bates EA, Victor M, Jones AK, Shi Y, Hart AC. Differential contributions of Caenorhabditis elegans histone deacetylases to huntingtin polyglutamine toxicity. J Neurosci. 2006;26:2830–8. doi: 10.1523/JNEUROSCI.3344-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- Beal MF, Ferrante RJ. Experimental therapeutics in transgenic mouse models of Huntington’s disease. Nat Rev Neurosci. 2004;5:373–84. doi: 10.1038/nrn1386. [DOI] [PubMed] [Google Scholar]
- Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–35. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- Borovecki F, Lovrecic L, Zhou J, Jeong H, Then F, Rosas HD, Hersch SM, Hogarth P, Bouzou B, Jensen RV, Krainc D. Genome-wide expression profiling of human blood reveals biomarkers for Huntington’s disease. Proc Natl Acad Sci U S A. 2005;102:11023–8. doi: 10.1073/pnas.0504921102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borra MT, Smith BC, Denu JM. Mechanism of human SIRT1 activation by resveratrol. J Biol Chem. 2005;280:17187–95. doi: 10.1074/jbc.M501250200. [DOI] [PubMed] [Google Scholar]
- Cha JH, Frey AS, Alsdorf SA, Kerner JA, Kosinski CM, Mangiarini L, Penney JB, Jr, Davies SW, Bates GP, Young AB. Altered neurotransmitter receptor expression in transgenic mouse models of Huntington’s disease. Philos Trans R Soc Lond B Biol Sci. 1999;354:981–9. doi: 10.1098/rstb.1999.0449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EY, Luthi-Carter R, Strand A, Solano SM, Hanson SA, DeJohn MM, Kooperberg C, Chase KO, DiFiglia M, Young AB, Leavitt BR, Cha JH, Aronin N, Hayden MR, Olson JM. Increased huntingtin protein length reduces the number of polyglutamine-induced gene expression changes in mouse models of Huntington’s disease. Hum Mol Genet. 2002;11:1939–51. doi: 10.1093/hmg/11.17.1939. [DOI] [PubMed] [Google Scholar]
- Cong SY, Pepers BA, Evert BO, Rubinsztein DC, Roos RA, van Ommen GJ, Dorsman JC. Mutant huntingtin represses CBP, but not p300, by binding and protein degradation. Mol Cell Neurosci. 2005;30:560–71. [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–48. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–3. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Dumble ML, Donehower LA, Lu X. Generation and characterization of p53 mutant mice. Methods Mol Biol. 2003;234:29–49. doi: 10.1385/1-59259-408-5:29. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science. 2002;296:2238–43. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci. 2003;23:9418–27. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Ryu H, Kubilus JK, D’Mello S, Sugars KL, Lee J, Lu P, Smith K, Browne S, Beal MF, Kristal BS, Stavrovskaya IG, Hewett S, Rubinsztein DC, Langley B, Ratan RR. Chemotherapy for the brain: the antitumor antibiotic mithramycin prolongs survival in a mouse model of Huntington’s disease. J Neurosci. 2004;24:10335–42. doi: 10.1523/JNEUROSCI.2599-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 2000;14:1021–6. [PubMed] [Google Scholar]
- Guarente L, Kenyon C. Genetic pathways that regulate ageing in model organisms. Nature. 2000;408:255–62. doi: 10.1038/35041700. [DOI] [PubMed] [Google Scholar]
- Harper SQ, Staber PD, He X, Eliason SL, Martins IH, Mao Q, Yang L, Kotin RM, Paulson HL, Davidson BL. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc Natl Acad Sci U S A. 2005;102:5820–5. doi: 10.1073/pnas.0501507102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildmann C, Wegener D, Riester D, Hempel R, Schober A, Merana J, Giurato L, Guccione S, Nielsen TK, Ficner R, Schwienhorst A. Substrate and inhibitor specificity of class 1 and class 2 histone deacetylases. J Biotechnol. 2006;124:258–70. doi: 10.1016/j.jbiotec.2006.01.030. [DOI] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci U S A. 2003;100:2041–6. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi S, Morita H, Bennett KM, Tanaka Y, Engelender S, Peters MF, Cooper JK, Wood JD, Sawa A, Ross CA. Inducible PC12 cell model of Huntington’s disease shows toxicity and decreased histone acetylation. Neuroreport. 2003;14:565–8. doi: 10.1097/00001756-200303240-00007. [DOI] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–92. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- Jiang H, Nucifora FC, Jr, Ross CA, DeFranco DB. Cell death triggered by polyglutamine-expanded huntingtin in a neuronal cell line is associated with degradation of CREB-binding protein. Hum Mol Genet. 2003;12:1–12. doi: 10.1093/hmg/ddg002. [DOI] [PubMed] [Google Scholar]
- Jiang H, Poirier MA, Liang Y, Pei Z, Weiskittel CE, Smith WW, Defranco DB, Ross CA. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol Dis. 2006 doi: 10.1016/j.nbd.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–80. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman D. Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci U S A. 1999;96:11404–9. doi: 10.1073/pnas.96.20.11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegel KB, Meloni AR, Yi Y, Kim YJ, Doyle E, Cuiffo BG, Sapp E, Wang Y, Qin ZH, Chen JD, Nevins JR, Aronin N, DiFiglia M. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J Biol Chem. 2002;277:7466–76. doi: 10.1074/jbc.M103946200. [DOI] [PubMed] [Google Scholar]
- Kennedy BK, Gotta M, Sinclair DA, Mills K, McNabb DS, Murthy M, Pak SM, Laroche T, Gasser SM, Guarente L. Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell. 1997;89:381–91. doi: 10.1016/s0092-8674(00)80219-6. [DOI] [PubMed] [Google Scholar]
- Khoshnan A, Ko J, Watkin EE, Paige LA, Reinhart PH, Patterson PH. Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J Neurosci. 2004;24:7999–8008. doi: 10.1523/JNEUROSCI.2675-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laforet GA, Sapp E, Chase K, McIntyre C, Boyce FM, Campbell M, Cadigan BA, Warzecki L, Tagle DA, Reddy PH, Cepeda C, Calvert CR, Jokel ES, Klapstein GJ, Ariano MA, Levine MS, DiFiglia M, Aronin N. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington’s disease. J Neurosci. 2001;21:9112–23. doi: 10.1523/JNEUROSCI.21-23-09112.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H, Li XJ. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol. 2002;22:1277–87. doi: 10.1128/mcb.22.5.1277-1287.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, Woods AM, Chan EY, Kooperberg C, Krainc D, Young AB, Tapscott SJ, Olson JM. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet. 2002a;11:1911–26. doi: 10.1093/hmg/11.17.1911. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, Ross CA, Borchelt DR, Tapscott SJ, Young AB, Cha JH, Olson JM. Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum Mol Genet. 2000;9:1259–71. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Strand AD, Hanson SA, Kooperberg C, Schilling G, La Spada AR, Merry DE, Young AB, Ross CA, Borchelt DR, Olson JM. Polyglutamine and transcription: gene expression changes shared by DRPLA and Huntington’s disease mouse models reveal context-independent effects. Hum Mol Genet. 2002b;11:1927–37. doi: 10.1093/hmg/11.17.1927. [DOI] [PubMed] [Google Scholar]
- Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A, Tronche F, Kellendonk C, Gau D, Kapfhammer J, Otto C, Schmid W, Schutz G. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet. 2002;31:47–54. doi: 10.1038/ng882. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Maudsley S, Martin B. BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2004;27:589–94. doi: 10.1016/j.tins.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Mee PJ, O’Brien CM, Thomson H, van der Sar S, Lakics V, Allsopp TE. Embryonic stem cells as a source of differentiated neural cells for pharmacological screens. Methods Mol Biol. 2006;329:353–69. doi: 10.1385/1-59745-037-5:353. [DOI] [PubMed] [Google Scholar]
- North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–44. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–8. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- Parker AJ, Arango M, Abderrahmane S, Lambert E, Tourette C, Catoire H, Neri C. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Med Sci (Paris) 2005;21:556–7. doi: 10.1051/medsci/2005215556. [DOI] [PubMed] [Google Scholar]
- Qin ZH, Wang Y, Nakai M, Chase TN. Nuclear factor-kappa B contributes to excitotoxin-induced apoptosis in rat striatum. Mol Pharmacol. 1998;53:33–42. doi: 10.1124/mol.53.1.33. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Norflus F, Singh B, Swindell MK, Buzescu R, Bejarano M, Chopra R, Zucker B, Benn CL, DiRocco DP, Cha JH, Ferrante RJ, Hersch SM. Sp1 is up-regulated in cellular and transgenic models of Huntington disease, and its reduction is neuroprotective. J Biol Chem. 2006;281:16672–80. doi: 10.1074/jbc.M511648200. [DOI] [PubMed] [Google Scholar]
- Ryu H, Rosas HD, Hersch SM, Ferrante RJ. The therapeutic role of creatine in Huntington’s disease. Pharmacol Ther. 2005;108:193–207. doi: 10.1016/j.pharmthera.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Sadri-Vakili G, Menon AS, Farrell LA, Keller-McGandy CE, Cantuti-Castelvetri I, Standaert DG, Augood SJ, Yohrling GJ, Cha JH. Huntingtin inclusions do not down-regulate specific genes in the R6/2 Huntington’s disease mouse. Eur J Neurosci. 2006;23:3171–5. doi: 10.1111/j.1460-9568.2006.04871.x. [DOI] [PubMed] [Google Scholar]
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–58. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- Sipione S, Rigamonti D, Valenza M, Zuccato C, Conti L, Pritchard J, Kooperberg C, Olson JM, Cattaneo E. Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum Mol Genet. 2002;11:1953–65. doi: 10.1093/hmg/11.17.1953. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–43. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97:6763–8. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano H, Gusella JF. The predominantly HEAT-like motif structure of huntingtin and its association and coincident nuclear entry with dorsal, an NF-kB/Rel/dorsal family transcription factor. BMC Neurosci. 2002;3:15. doi: 10.1186/1471-2202-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–30. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW, Serrano L, Sternglanz R, Reinberg D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006;20:1256–61. doi: 10.1101/gad.1412706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan M, Kim SK, Berdichevsky A, Guarente L. A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans life span. Dev Cell. 2005;9:605–15. doi: 10.1016/j.devcel.2005.09.017. [DOI] [PubMed] [Google Scholar]
- Wang DF, Helquist P, Wiech NL, Wiest O. Toward selective histone deacetylase inhibitor design: homology modeling, docking studies, and molecular dynamics simulations of human class I histone deacetylases. J Med Chem. 2005;48:6936–47. doi: 10.1021/jm0505011. [DOI] [PubMed] [Google Scholar]
- Wang H, Lim PJ, Yin C, Rieckher M, Vogel BE, Monteiro MJ. Suppression of polyglutamine-induced toxicity in cell and animal models of Huntington’s disease by ubiquilin. Hum Mol Genet. 2006;15:1025–41. doi: 10.1093/hmg/ddl017. [DOI] [PubMed] [Google Scholar]
- White JK, Auerbach W, Duyao MP, Vonsattel JP, Gusella JF, Joyner AL, MacDonald ME. Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat Genet. 1997;17:404–10. doi: 10.1038/ng1297-404. [DOI] [PubMed] [Google Scholar]
- Yu Z, Zhou D, Cheng G, Mattson MP. Neuroprotective role for the p50 subunit of NF-kappaB in an experimental model of Huntington’s disease. J Mol Neurosci. 2000;15:31–44. doi: 10.1385/JMN:15:1:31. [DOI] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Nguyen HP, Li XJ. Huntingtin inclusions do not deplete polyglutamine-containing transcription factors in HD mice. Hum Mol Genet. 2002;11:905–14. doi: 10.1093/hmg/11.8.905. [DOI] [PubMed] [Google Scholar]
- Zhai W, Jeong H, Cui L, Krainc D, Tjian R. In vitro analysis of huntingtin-mediated transcriptional repression reveals multiple transcription factor targets. Cell. 2005;123:1241–53. doi: 10.1016/j.cell.2005.10.030. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293:493–8. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]