

Abstract
Modification of the C-1 ketone of salvinorin A (2a) produces analogues with opioid antagonist properties. Of particular significance is the finding that 1-deoxo-1,10-dehydrosalvinorin A (11a) is a moderately potent antagonist at all three opioid receptor subtypes, and that herkinorin (2b), a μ agonist, is converted to a weak antagonist by removal of the C-1 ketone (3b and 11b). These observations suggest that the ketone of 2b is a key structural feature responsible for μ agonist activity.
The opium poppy, Papaver somniferum, has been used for centuries for the relief of pain and to induce sleep. Among the most important constituents in opium are the alkaloids morphine (1a) and codeine (1b) (Figure 1). Many of the opiate agonists and antagonists derived from these alkaloids are essential for the effective practice of modern medicine. However, new agents are needed with fewer side effects and developing these agents can allow the exploration of the mechanisms of action that induce tolerance and dependence.1, 2
Figure 1.
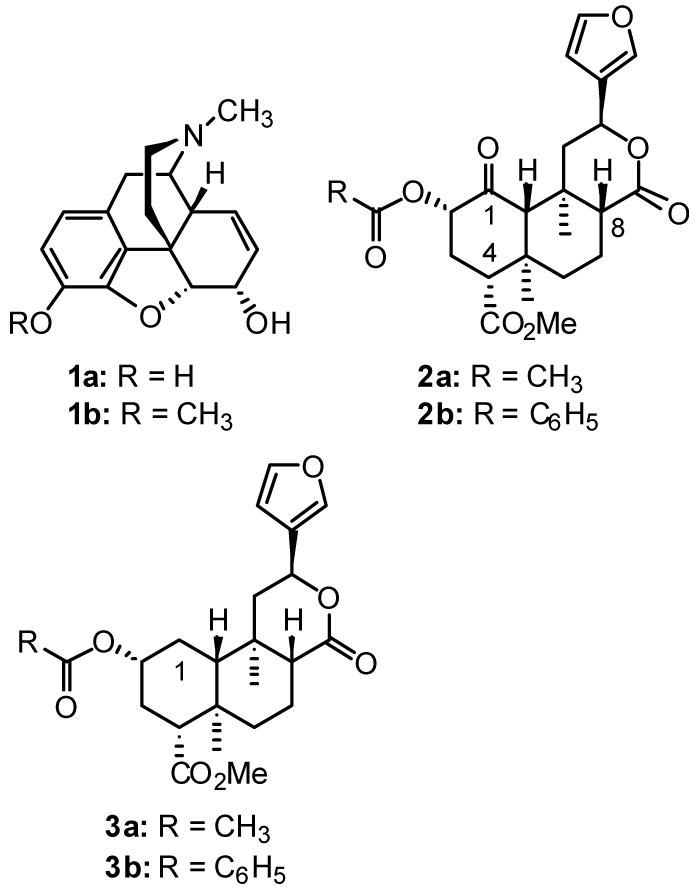
Structures of morphine (1a), codeine (1b), salvinorin A (2a), herkinorin (2b), 1-deoxosalvinorin A (3a), and 1-deoxoherkinorin (3b).
Morphine and related opiates exert their major pharmacological effects by interacting with opioid receptors (μ, δ, and κ).3 The search for potent agonists and antagonists selective for each of these opioid receptors has engaged the interest of medicinal chemists for many years because of their potential as therapeutic agents and pharmacological tools.3
As a consequence of target drug design and synthetic efforts, we have achieved a better understanding of opioid receptors. Moreover, these efforts have opened new avenues for chemical investigation. Salvinorin A (2a), a neoclerodane diterpene, is a potent and selective κ opioid receptor (κOR) agonist,4 and represents a novel scaffold for the development of opioid ligands with potentially reduced side effects.5 Several recent reports have begun to characterize the structure-activity relationships of 2a at κ opioid receptors.6-8 Our own efforts have identified analogues of 2a, such as 2b, with affinity for κORs as well as μORs and δORs.9-14
As part of a program to develop novel agents to treat drug dependence, we have prepared several C-1 modified analogues of 2a. These compounds were prepared to better elucidate the pharmacophore of 2a at opioid receptors. In particular, we were interested in better understanding the role of the C-1 ketone. In a previous report, 3a was prepared from 2a in 4 steps .6, 11 To facilitate the preparation of additional analogues, a new synthetic route to 3a and related analogues was undertaken.
Salvinorin A (2a), isolated from Salvia divinorum,15 was reduced with an aqueous solution of NaBH4 in THF to afford 1α-hydroxysalvinorin A (4a)16 in 77% yield (Scheme 1). There was no evidence of C-8 epimerization, and the major byproducts from this reaction were reduction at both C-17 and C-1 (9%). Similar conditions using 2b afforded 1α-hydroxyherkinorin (4b) in 45% yield. Treatment of 4a with methanesulfonic anhydride and DMAP in CH3CN afforded 1α-mesyloxysalvinorin A (5) in 99% crude yield. Basic hydrolysis of mesylate 5 in MeOH/CH2Cl2 at -10 °C gave crystalline 1α-mesyloxysalvinorin B (6) in 70% overall yield from 4a. Attempts to directly reduce 5 to 3a or to displace the mesyloxy group in 5 with a halide followed by reduction were unsuccessful. Similarly, efforts to convert 4a into 3a using Barton deoxygenation were unsuccessful. To circumvent this problem, 6 was reacted with DMAP in DMSO at 170 °C to afford a mixture of 1-deoxo-1,10-dehydrosalvinorin B (7) in 76% yield and 2-keto-1-deoxosalvinorin A (8) (22%). Oxidation of the allylic alcohol with MnO2 in toluene gave α,β-unsaturated ketone 9 in 78% yield. Reduction of 8 with NaBH4 in acetonitrile gave 1-deoxosalvinorin B (10)6 in 41% yield. Although reduction of 8 proceeded in high yield, it was complicated by a small amount of a byproduct, which ran just ahead of 10 on TLC. Two recrystallizations from ethyl acetate/n-hexanes removed this material, which is believed to be the 2β-epimer based on the finding that oxidation of the mixture produced pure 8. Alternatively, 10 could be prepared from 6 in 47% overall yield using a sequence of elimination, followed by Jones oxidation of the crude mixture of 7 and 8 followed by reduction of the crude oxidation product using a new selective reducing agent derived from NaBH4 and DMAP. Treatment of 10 with acetic anhydride or benzoyl chloride and a catalytic amount of DMAP afforded 1-deoxosalvinorin A (3a)6 and 1-deoxoherkinorin (3b), respectively. The reaction of 4a with methanesulfonic anhydride followed by heating with trimethylphenylammonium chloride in CH3CN produced 11a in 76% yield. Finally, treatment of 7 (obtained from hydrolysis of 11a) with benzoyl chloride using similar conditions for 3b, gave 1-deoxo-1,10-dehydroherkinorin 11b. Efforts to reduce 11a directly to 3a resulted in hydrogenolysis of the allylic acetate as the major product.
Scheme 1.

Reagents and conditions (a) NaBH4, THF/ H2O; (b) (CH3SO2)2O, DMAP, CH3CN; (c) NaOH, MeOH, CH2Cl2; (d) DMAP, DMSO; (e) MnO2, Toluene; (f) NaBH4, CH3CN; (g) Acetic anhydride or benzoyl chloride, DMAP, CH2Cl2; (h) Trimethylphenylammonium chloride, CH3CN; (i) Na2CO3, MeOH
Compounds 3a, 3b, 4a, 4b, 5, 9, 11a, and 11b were evaluated for opioid receptor activity as described previously (Tables 1 and 2).10 Removal of the 1-ketone group in 2a (3a) decreased agonist activity 6-fold at κORs (EC50 = 280 nM vs. EC50 = 45 nM). This result is in general agreement with previous work which showed 3-fold loss in activity.6 However, in our hands 3a was found to have lower efficiacy than previously reported (Emax = 50% relative to U69,593 vs. Emax = 122% relative to 2a). It should be noted that 2a has higher efficacy than U69,593.10 In addition, we found 3a to also have significant antagonist activity at μORs (Ke = 170 nM) and δORs (Ke = 100 nM). One potential reason for this discrepancy is the different functional assays used to characterize the compounds: [35S]GTP-γ-S binding17 versus the mobilization of internal calcium.18, 19 To further probe this possibility, we evaluated 3a in our own calcium mobilization assay.10 In this assay, 3a was also 3-fold less potent (EC50 = 3.34 nM vs. EC50 = 1.24 nM) than 2a, but it remained less efficacious (Emax = 93% vs. Emax = 118%). However, the efficacy of 3a increased relative to that measured in the [35S]GTPγS binding assay such that it was nearly a full agonist relative to U69,593 (Emax = 100%). This increase in efficacy was not accompanied by a change in relative potency compared to our standard compound, U69,593 (EC50 = 2.25 nM), in keeping with our [35S]GTPγS assay data.
Table 1.
Comparison of Agonist Activities
| [35S]GTP-γ-S | Gα 16-h κOR Calcium Flux | |||
|---|---|---|---|---|
| Compound | κ EC50 ± SE, nM | κ Emaxa ± SE | EC50 ± SE, nM | Emaxa ± SE |
| (-)-U69,593 | 330 ± 70 | 100 ± 7 | 2.25 ± 0.82 | 100 ± 12 |
| 2a | 45 ± 10b | 108 ± 4b | 1.24 ± 0.56 | 118 ± 3 |
| 3a | 280 ± 60 | 49 ± 5 | 3.34 ± 0.11 | 93 ± 3 |
| 5 | 2700 ± 400 | 95 ± 12 | NTc | NTc |
Emax is relative to the maximum stimulation of binding observed with U69,593 run in parallel with the test compounds
Values from reference 10
Not tested, no experiments were done to determine value
Table 2.
Inhibition of Agonist Stimulated [35S]GTP-γ-S Binding by Compounds in Cloned Human Opioid Receptors
| Ke ± SD, nM | |||
|---|---|---|---|
| Cmpd | μ, DAMGO | δ, DPDPE | κ, U69,593 |
| 3a | 170 ± 20 | 100 ± 3 | NTa |
| 3b | 4700 ± 910 | 8780 ± 2130 | 580 ± 30 |
| 4a | 2300 ± 80 | 1790 ± 510 | 240 ± 5 |
| 4b | 1830 ± 930 | 2900 ± 150 | 1050 ± 600 |
| 9 | 700 ± 220 | 3500 ± 1700 | 460 ± 70 |
| 11a | 200 ± 30 | 400 ± 90 | 570 ± 140 |
| 11b | 1740 ± 230 | 2300 ± 20 | 580 ± 210 |
Not tested, no experiments were done to determine value
The Ke data represent the mean ± SD from 2 independent determinations
There are several potential explanations for the differences in 3a efficacy in our assays. The effects of agonists, and in particular partial agonists, can vary considerably depending on the number of cell surface-expressed receptors, leading to a greater proportion of receptors in an active receptor conformation recognized by the agonist and, thus, increased efficacy.20-22 The type of G-protein to whic h a GPCR is coupled can also affect efficacy.23-25 In keeping with these observations, it is not surprising that the partial agonist 3a displays assay-specific efficacies. Despite the differences in efficacy between our two assays, it is clear 3a has agonist properties and we undertook further studies to determine the role of structure in conferring this activity.
Replacement of the acetyl group in 3a with a benzoyl group (3b) resulted in an antagonist at μ, δ, and κ receptors. Benzoate 3b had highest activity at κORs (Ke = 580 nM). This change also decreased antagonist activity 28-fold at μORs (Ke = 4700 nM vs. Ke = 170 nM). and greater than 88-fold at δORs (Ke = 8780 nM vs. Ke = 100 nM). This finding is curious given our previous investigations that showed that by introducing an aromatic moiety increased activity at μORs compared to κORs.11, 13 This would suggest that the C-1 deoxo analogues may be interacting at μORs in a non-identical manner compared to C-1 keto analogues. Reduction of the 1-ketone in 2a to the corresponding α-alcohol (4a) changed the efficacy at κORs from a full agonist (2a: Emax = 108%) to an antagonist (4a: Ke = 240 nM). Alcohol 4a was 3-fold less selective over μORs (Ke = 2300 nM) and 2-fold less selective for δORs (Ke = 1800 nM). Addition of a benzene ring to 4a (4b) decreased activity 2-fold at κORs (Ke = 450 nM vs. Ke = 240 nM), whereas, the introduction of a mesylate group (5) resulted in a loss of antagonist activity at κ receptors (EC50 = 2700 nM).
Finally, introduction of a 1,10-alkene functionality was explored. This modification was chosen for several reasons. First, it would probe the role of sp2 hybridization at C-1 and second it would also examine the effect of stereochemistry at the C-10 position on the activity of 3a. The presence of the 1,10-alkene (11a) resulted in a switch of efficacy from partial agonist (3a: κ EC50 = 340 nM, Emax = 49% relative to U69,593) to antagonist (Ke = 570 nM). Furthermore, 11a had higher antagonist activity at μORs (Ke = 200 nM) and δORs (Ke = 400 nM). When compared to 3a, 11a had similar antagonist activity at μORs (Ke = 200 nM vs. Ke = 170 nM) and reduced activity at δORs (Ke = 400 nM vs. Ke = 100 nM). Replacement of the acetyl of 11a with benzoyl group (11b) decreased activity 9-fold at μORs (Ke = 1740 nM vs. Ke = 200 nM) and 6-fold at δORs (Ke = 2310 nM vs. Ke = 400 nM) but had little effect on κORs (Ke = 720 nM vs. Ke = 570 nM). This also suggests that the 1,10-dehydro analogues are not interacting in a similar manner to analogues which contain a C-1 keto group. To further confirm this observation, we evaluated ketone 9. If these analogues were interacting in an identical manner to their C-1 ketone analogues, removal of the acetyl group would have a deleterious effect on activity. However, 9 was found to have similar antagonist activity as 11a at κORs (Ke = 460 nM vs. Ke = 570 nM) but reduced activity at μORs (Ke = 700 nM vs. Ke = 200 nM) and δORs (Ke = 3500 nM vs. Ke = 400 nM). This finding also suggests that the 1,10-alkene analogues may be a productive scaffold for the development of selective opioid antagonists. This, however, needs validation through further synthesis and testing.
The results presented here indicate that the interaction of neoclerodane diterpenes with opioid receptors may be more complicated than first thought. Previous work in the opioid field has highlighted the importance of the message-address concept in the recognition of ligands by opioid receptors.26 This work was originally applied to the recognition elements of peptide hormones such as dynorphin,27 but later work illustrated its utility in the design of selective nonpeptide opioid antagonists.28 Based on our previous finding that the introduction of a benzene ring increases activity at the μOR,11 we synthesized 3b and 11b. It was thought that perhaps the benzene ring of 2b was functioning as an address moiety to μORs. However, this proved not to be the case. It is therefore possible that once the recognition elements of neoclerodane diterpenes with opioid receptors are better understood, more selective agents for μORs and δORs can be better designed.
In summary, modification of the C-1 ketone of 2a has been found to alter its potent κ opioid agonist activity to produce, in almost every case, compounds that are antagonists at all three opioid receptor subtypes. The major exception is 3a, which is a partial agonist at κ receptors but is, however, a moderately potent antagonist at μ and δ receptors. These studies suggest that the C-1 ketone of 2a imparts agonist activity and that analogues lacking this structural feature bind to opioid receptors in a different manner, generally producing antagonist responses. Last, one should keep in mind that partial agonists have the potential to have different efficacies in different tissues or brain regions depending on the number of target receptors expressed and their effector coupling, and caution must be used in comparison of data from different in vitro functional assays.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health, National Institute on Drug Abuse Grant DA018151 (to TEP). The authors also thank Keith Warner for technical assistance and the American Foundation for Pharmaceutical Education (KT) and the University of Iowa Graduate College (AM) for predoctoral fellowships.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Coop A, MacKerell AD. Am J Pharm Educ. 2002;66:153. [Google Scholar]
- 2.Martin TJ, Eisenach JC. J. Pharmacol. Exp. Ther. 2001;299:811. [PubMed] [Google Scholar]
- 3.Casy AF, Parfitt RT. Opioid analgesics: chemistry and receptors. Plenum Press; New York: 1986. [Google Scholar]
- 4.Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Proc. Natl. Acad. Sci. U.S.A. 2002;99:11934. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, Bohn LM. Mol. Pharmacol. 2007;71:549. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munro TA, Rizzacasa MA, Roth BL, Toth BA, Yan F. J. Med. Chem. 2005;48:345. doi: 10.1021/jm049438q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beguin C, Richards MR, Li J-G, Wang Y, Xu W, Liu-Chen L-Y, Carlezon JWA, Cohen BM. Bioorg. Med. Chem. Lett. 2006;16:4679. doi: 10.1016/j.bmcl.2006.05.093. [DOI] [PubMed] [Google Scholar]
- 8.Bikbulatov RV, Yan F, Roth BL, Zjawiony JK. Bioorg. Med. Chem. Lett. 2007;17:2229. doi: 10.1016/j.bmcl.2007.01.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harding WW, Schmidt M, Tidgewell K, Kannan P, Holden KG, Dersch CM, Rothman RB, Prisinzano TE. Bioorg. Med. Chem. Lett. 2006;16:3170. doi: 10.1016/j.bmcl.2006.03.062. [DOI] [PubMed] [Google Scholar]
- 10.Harding WW, Schmidt M, Tidgewell K, Kannan P, Holden KG, Gilmour B, Navarro H, Rothman RB, Prisinzano TE. J. Nat. Prod. 2006;69:107. doi: 10.1021/np050398i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harding WW, Tidgewell K, Byrd N, Cobb H, Dersch CM, Butelman ER, Rothman RB, Prisinzano TE. J. Med. Chem. 2005;48:4765. doi: 10.1021/jm048963m. [DOI] [PubMed] [Google Scholar]
- 12.Harding WW, Tidgewell K, Schmidt M, Shah K, Dersch CM, Snyder J, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. Org Lett. 2005;7:3017. doi: 10.1021/ol0510522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tidgewell K, Harding WW, Lozama A, Cobb H, Shah K, Kannan P, Dersch CM, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. J. Nat. Prod. 2006;69:914. doi: 10.1021/np060094b. [DOI] [PubMed] [Google Scholar]
- 14.Rothman RB, M urphy DL, Xu H, Godin JA, Dersch CM, Partilla JS, Tidgewell K, Schmidt M, Prisinzano TE. J. Pharmacol. Exp. Ther. 2007;320:801. doi: 10.1124/jpet.106.113167. [DOI] [PubMed] [Google Scholar]
- 15.Tidgewell K, Harding WW, Schmidt M, Holden KG, Murry DJ, Prisinzano TE. Bioorg. Med. Chem. Lett. 2004;14:5099. doi: 10.1016/j.bmcl.2004.07.081. [DOI] [PubMed] [Google Scholar]
- 16.Valdes LJ, III, Butler WM, Hatfield GM, Paul AG, Koreeda M. J. Org. Chem. 1984;49:4716. [Google Scholar]
- 17.Traynor JR, Nahorski SR. Mol. Pharmacol. 1995;47:848. doi: 10.1016/S0026-895X(25)08634-1. [DOI] [PubMed] [Google Scholar]
- 18.Chavkin C, Sud S, Jin W, Stewart J, Zjawiony JK, Siebert DJ, Toth BA, Hufeisen SJ, Roth BL. J. Pharmacol. Exp. Ther. 2004;308:1197. doi: 10.1124/jpet.103.059394. [DOI] [PubMed] [Google Scholar]
- 19.Pauwels PJ, Colpaert FC. Neuropharmacology. 2000;39:2101. doi: 10.1016/s0028-3908(00)00040-x. [DOI] [PubMed] [Google Scholar]
- 20.Gazi L, Bobirnac I, Danzeisen M, Schupbach E, Langenegger D, Sommer B, Hoyer D, Tricklebank M, Schoeffter P. Br. J. Pharmacol. 1999;128:613. doi: 10.1038/sj.bjp.0702849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ko MC, Lee H, Harrison C, Clark MJ, Song HF, Naughton NN, Woods JH, Traynor JR. J. Pharmacol. Exp. Ther. 2003;306:179. doi: 10.1124/jpet.103.050625. [DOI] [PubMed] [Google Scholar]
- 22.Remmers AE, Clark MJ, Alt A, Medzihradsky F, Woods JH, Traynor JR. Eur. J. Pharmacol. 2000;396:67. doi: 10.1016/s0014-2999(00)00212-0. [DOI] [PubMed] [Google Scholar]
- 23.Kenakin T. Annu. Rev. Pharmacol. Toxicol. 2002;42:349. doi: 10.1146/annurev.pharmtox.42.091401.113012. [DOI] [PubMed] [Google Scholar]
- 24.Kinzer-Ursem TL, Linderman JJ. PLoS computational biology. 2007;3:e6. doi: 10.1371/journal.pcbi.0030006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saidak Z, Blake -Palmer K, Hay DL, Northup JK, Glass M. Br. J. Pharmacol. 2006;147:671. doi: 10.1038/sj.bjp.0706661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwyzer R, Ann NY. Acad. Sci. 1977;297:3. doi: 10.1111/j.1749-6632.1977.tb41843.x. [DOI] [PubMed] [Google Scholar]
- 27.Chavkin C, Goldstein A. Proc. Natl. Acad. Sci. USA. 1981;78:6543. doi: 10.1073/pnas.78.10.6543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Portoghese PS. Trends Pharmacol. Sci. 1989;10:230. doi: 10.1016/0165-6147(89)90267-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.