

Abstract
This report examines the structure and function of ARHGAP4, a novel RhoGAP whose structural features make it ideally suited to regulate the cytoskeletal dynamics that control cell motility and axon outgrowth. Our studies show that ARHGAP4 inhibited the migration of NIH/3T3 cells and the outgrowth of hippocampal axons. ARHGAP4 contains an N-terminal FCH domain, a central GTPase activating (GAP) domain and a C-terminal SH3 domain. Our structure/function analyses show that the FCH domain appears to be important for spatially localizing ARHGAP4 to the leading edges of migrating NIH/3T3 cells and to axon growth cones. Our analyses also show that the GAP domain and C-terminus are necessary for ARHGAP4-mediated inhibition of cell and axon motility. These observations suggest that ARHGAP4 can act as a potent inhibitor of cell and axon motility when it is localized to the leading edge of motile cells and axons.
Introduction
The regulation of cell motility typically involves the regulation of actin filament assembly at the leading edge of motile cells. RhoA, Rac1 and Cdc42 are small GTPases that regulate the signaling pathways that control actin filament assembly and disassembly, and cell motility (Dickson, 2001). The complex pathways activated by the Rho GTPases are described in several recent review articles (Luo, 2000;Huber et al., 2003;Dent and Gertler, 2003). These GTPases act as molecular on/off switches that initiate a cascade of events that directly regulate actin filament dynamics. They are switched “on” when they bind GTP. RhoGAPs enhance the hydrolysis of GTP to GDP which switches the GTPases to their “off” state, thereby inhibiting the downstream signaling that regulates actin filament dynamics and motility. This increase in GTP hydrolysis is dependent on a highly conserved arginine residue in the GAP domain that directly interacts with the active region of GTPases (Moon and Zheng, 2003;Li et al., 1997;Nassar et al., 1998;Wittmann et al., 2003;Gamblin and Smerdon, 1998).
ARHGAP4 is a complex protein that includes an N-terminal FCH (Fps/Fes/Fer/CIP4 homology) domain and a C-terminal SH3 (Src homology 3) domain (Fig. 1). SH3 domains interact with proline rich domains of many proteins, including actin binding proteins. The structure, function and specificity of the FCH domain, however, are not well understood. In total, there are approximately 100 known proteins that contain the conserved FCH domain (Greer, 2002). Present evidence suggests that FCH domain-containing proteins are involved in the regulation of cytoskeletal rearrangements, vesicular transport and endocytosis (Fuchs et al., 2001;Linder et al., 2000;Yeung et al., 1998;Qualmann and Kelly, 2000;Modregger et al., 2000;Tian et al., 2000;Tribioli et al., 1996). Some studies have suggested that the FCH domain is involved in actin binding (Aspenstrom, 1997;Fankhauser et al., 1995), but a recent study showed that recombinant FCH protein corresponding to the N-terminal 118 amino acids of CIP4 binds directly to MTs (Tian et al., 2000). The same study showed that the C-terminal SH3 domain of CIP4 binds WASP, an actin-binding protein that is known to play a role in actin regulation and in directing cell motility. The results of this study suggested that the ability of CIP4 to crosslink actin-binding proteins with MTs is important for regulation of cell migration.
Figure 1. Structural domains of the ARHGAP4 protein.

ARHGAP4 contains N-terminal FCH (Fps/Fes/Fer/CIP4 homology), coiled-coil (CC) and ARNEY domains. The GAP domain is centrally located and the C-terminal contains a SH3 (Src homology 3) domain.
ARHGAP4 shows structural similarity to CIP4 in that both have N-terminal FCH and C-terminal SH3 domains. Our results show that the 1-71 fragment of the FCH domain of ARHGAP4 is responsible for its localization to the leading edge of NIH/3T3 cells and axons and growth cones. However, our results suggest that this localization of ARHGAP4 is dependent on actin filaments rather than MTs. This spatial targeting to the leading edge of migrating cells appears to be critical to ARHGAP4’s regulation of motility. Although the GAP and SH3 domains do not appear to play a role in localizing ARHGAP4 to axons and growth cones, all three domains appear to be important in the regulation of ARHGAP4-mediated cell and axon motility.
Results
Endogenous ARHGAP4 is localized to the leading edge of migrating NIH/3T3 cells and to axons and growth cones
Endogenous ARHGAP4 has been shown to be expressed in NIH/3T3 fibroblasts, NRK epithelial cells and PC12 cells (Foletta et al., 2002). These data showed ARHGAP4 was localized to the golgi, along microtubules in NRK cells, and at the tips of extending neurites of NGF-treated (neuronally differentiated) PC12 cells. Our data showed that ARHGAP4 was also localized to the leading edge of migrating NIH/3T3 cell fibroblasts (Figs. 2A–C). Mossy fiber (MF) growth cones from dissociated dentate granule cell cultures were immunostained for ARHGAP4 and costained using an antibody against β-tubulin III (Figs. 2D–F). These data show that ARHGAP4 is localized to axons and growth cones, including the tips of filopodia.
Figure 2. Endogenous ARHGAP4 protein is present in the peripheral zone of NIH/3T3 cells and growth cones.

NIH/3T3 cells (A–C) and growth cones from dissociated dentate granule cell cultures (D–F) were immunostained for endogenous ARHGAP4 using a rabbit antibody (Foletta et al, 2002) and a mouse monoclonal antibody against β-tubulin III to label axons and the central region of growth cones. Fluorescence was visualized using biotinylated secondary antibodies labeled with avidin-Alexa 594 (red) and avidin-Alexa 488 (green). Images were viewed using a Leica confocal laser scanning microscope. Yellow indicates regions of overlapping immunoreactivity. Panels A–C: NIH/3T3 cells, Panels D–F: MF growth cones. The leading edge of the NIH/3T3 cell and the distal tips of filopodia of dentate granule cells are indicated by arrows in these representative images (n =3). Calibration = 5μm.
The FCH domain is important for localizing ARHGAP4 to growth cones and to the leading edge of NIH/3T3 cells
Structural and functional analyses were performed to identify the role of the different domains in targeting ARHGAP4 to the extreme peripheral tips of NIH/3T3 cells (Figs. 3A, 3B). Western analysis confirmed that our transfection methods resulted in the expression of FLAG-tagged proteins of appropriate size (Supplemental Figs. 1A). Fluorescent images of full-length protein (1-965)-FLAG and the C-terminal truncation (1-770)-FLAG are shown in Figures. 4 and 5. The wealth of knowledge about the regulation of MT and actin cytoskeletal dynamics in NIH/3T3 cells during motility makes them a valuable model for the study of RhoGAP function. For these studies, the location of a full-length ARHGAP4 FLAG-tagged fusion protein was compared to that of FLAG-tagged N-terminal and C-terminal ARHGAP4 truncation mutants. Fluorescence intensity was analyzed in a semi-quantitative fashion to calculate the ratio of fluorescence intensity at the peripheral tip of the cell compared to the intensity 20μm from the tip (Fig. 3A). These data show that the ARHGAP4 N-terminal truncation mutant (72-965)-FLAG was less specifically localized to the peripheral region of NIH/3T3 cells than the full-length protein (1-965)-FLAG, the C-terminal truncation mutant (1-770)-FLAG, or the GAP loss-of-function (GAP LOF) mutant (R562A)-FLAG (Fig. 3B) (see Supplemental Figure 4 for GAP-LOF data).
Figure 3. The ARHGAP4 N-terminus is required for normal protein localization in NIH/3T3 cells and axon terminals.
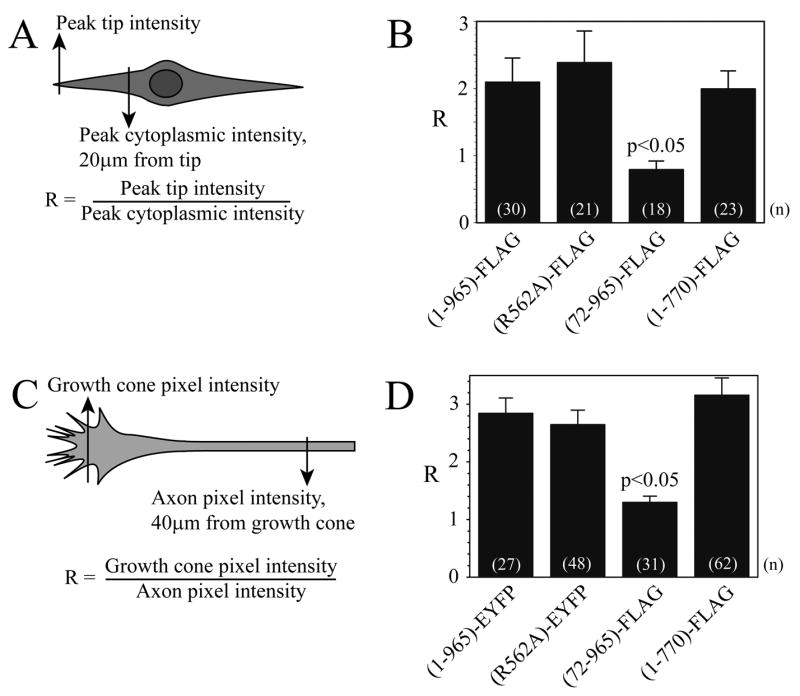
A,B) Localization of FLAG-tagged full-length ARHGAP4 protein was compared to FLAG-tagged N-terminal and C-terminal truncation mutants in NIH/3T3 cells transiently transfected using Lipofectamine 2000 (Invitrogen), as described in Methods. Anti-FLAG immunohistochemistry was used to label mutant proteins using a biotinylated secondary antibody and avidin-Alexa 488. A)Fluorescence intensity was analyzed in a semi-quantitative fashion using NIH Image software. The ratio (R) of fluorescence intensity at the tip of the leading edge to the intensity 20μm from the tip was calculated. B)Results of the analysis are shown as mean ± SEM, and ANOVA was performed as described in Methods. Supplemental Figure 1 shows Western analysis of cells transfected to express each protein using the M2 anti-FLAG antibody.
C,D) Localization of ARHGAP4 in axons and growth cones. C) The distribution of full-length ARHGAP4 and of 3 mutant proteins was quantified by dividing the pixel intensity of the respective tags at the growth cone by the pixel intensity at the axon 40μm from the growth cone, similar to the method used for NIH/3T3 cells. B)Results are shown as mean ± SEM, and ANOVA was performed using SigmaStat 3.1 software.
Figure 4. Effects of actin and microtubule destabilizing drugs on the localization of full-length ARHGAP4 (1-965-FLAG).

Wound assay experiments were performed on NIH/3T3 cells that expressed full length FLAG-tagged ARHGAP4 (1-965-FLAG). Three hours after wounding, the cells were treated with vehicle alone (DMSO), nocodazole, or cytochalasin-D to disrupt MTs and actin filaments, respectively. Fluorescent images were merged with brightfield images (C,F,I) to show leading edge boundaries.
Figure 5. Effects of actin and microtubule destabilizing drugs on the localization of the C-terminal truncation mutant ARHGAP4 (1-770-FLAG).

Wound assay experiments were performed on NIH/3T3 cells that expressed C-terminal deletion mutant FLAG-tagged ARHGAP4 (1-770-FLAG). Three hours after wounding, the control cells were treated with vehicle alone (DMSO), nocodazole, or cytochalasin-D to disrupt MTs and actin filaments, respectively. Fluorescent images were merged with brightfield images (C,F,I) to show leading edge boundaries.
The same proteins were expressed in dissociated cultured granule cell neurons to identify regions responsible for targeting ARHGAP4 to axons and the peripheral region of growth cones and the data were quantified (Figs. 3C, 3D). As in NIH/3T3 cells, only the N-terminal truncation mutant, (72-965)-FLAG, exhibited a reduced localization in growth cones compared to full-length ARHGAP4 (fluorescent images not shown). These data suggest that the N-terminal FCH domain is necessary for localizing ARHGAP4 to the tips of both NIH/3T3 cells and axon growth cones, but that neither a functional GAP domain nor the C-terminal domain was important for this localization.
The Cytoskeleton and ARHGAP4 localiztion
The role of MTs and actin filaments in the localization of ARHGAP4 to the leading edge of NIH3T3 cells was examined. Nocodazole and Cytochalasin-D were used to destabilize MTs and actin filaments respectively. NIH/3T3 cells were transiently transfected to express FLAG-tagged full length ARHGAP4 (Fig. 4), the C-terminal truncation mutant 1-770 (Fig. 5) or the N-terminal truncation mutant 72-965 (data not shown). Fourty-eight hours after transfection, a confluent region in the culture was scratched/wounded to induce polarization of the cytoskeleton and directed movement of the NIH/3T3 cells towards the center of the wound. Three hours after wounding, the cells were treated with either 300 nM nocodazole or 1μM cytochalasin-D, which disrupted the normal pattern of microtubules and actin filaments in these cells (Supplemental Fig. 2). The cells were fixed 5 minutes later and the distribution of ARHGAP4 expression proteins were analyzed. The results show that full-length ARHGAP4 accumulates at the periphery of the leading edge of migrating NIH/3T3 cells (Fig. 4A–C), and that this localization is disrupted when actin filaments are destabilized (Fig. 4G–I) but not when MTs are destabilized (Fig. 4D–F). A similar result was observed for the 1-770 C-terminal truncation mutant protein (Fig. 5). However, as previously described in Figure 3, the 72-965 N-terminal truncation mutant was not localized to the leading edge of migrating NIH/3T3 cells and this pattern was not altered by either nocodazole or cytochalasinD (data not shown). These observations suggest that amino acids 1-71 are necessary for transport of ARHGAP4 to the cell periphery, and that localization of ARHGAP4 at the cell cortex appears to be dependent on its interaction with actin filaments.
ARGHAGP4 inhibits axon outgrowth
The function of ARHGAP4 on axon outgrowth was examined (Fig. 6). To determine whether or not ARHGAP4 regulates axon outgrowth, control dentate explants were transfected with FLAG empty vector (Fig. 6A1), or the ARHGAP4 proteins: full-length (1-965)-FLAG (Fig. 6A2), the R562A GAP-LOF mutant (Fig. 6A3), the C-terminal truncation mutant (1-770)-FLAG (Fig. A4), or the N-terminal truncation mutant (72-965)-FLAG (Fig. 6A5). Western analysis demonstrated the expression of EYFP-tagged proteins in homogenized extracts of dentate explants (Supplemental Fig. 1B), and immunofluorescence images showed that these proteins are strongly expressed in somata located in the granule cell layer of the explants (Supplemental Fig. 1C). MF axon and astrocyte outgrowth was measured as previously described (Butler et al., 2004) and outgrowth data were quantified in Figures 6B and 6C. Only expression of the full-length (1-965)-FLAG protein inhibited MF outgrowth (Fig. 6B), suggesting that both the N- and C-terminal regions of ARHGAP4 are important for regulating axon outgrowth. Similar results were observed in dissociated hippocampal neurons (Supplemental Fig. 3). ARHGAP4 specifically altered axon but not astrocyte migration from dentate explants (Fig. 6B, 6C). In view of the fact that only N-terminal truncation of ARHGAP4 altered its localization to the periphery of axons and fibroblasts, the N-terminal truncation data in Figure 6 suggest that ARHGAP4 must be properly localized in the growth cone to inhibit axon outgrowth. Results obtained with the C-terminal truncation mutant also show that even when ARHGAP4 is properly localized, the C-terminal region is necessary for inhibition of axon outgrowth, and suggest that ARHGAP4’s function is dependent on important interactions between its C-terminus and other proteins in growth cones.
Figure 6. Effects of mutant ARHGAP4 protein expression on MF axon outgrowth.

Dentate explants were transfected using ExGen 500 to express ARHGAP4 proteins as described in Methods. Representative images obtained using a SPOT CCD camera attached to a Nikon Optiphot-2 fluorescence microscope are shown. Explants were transfected with the FLAG parent vector as control (A1); full-length ARHGAP4 (1-965)-FLAG (A2); the GAP loss of function mutant (R562A)-FLAG (A3); the C-terminal truncation mutant (1-770)-FLAG (A4); or the N-terminal truncation mutant (72-965)-FLAG (A5). MF axons were visualized using an anti-β-tubulin III antibody and nuclei of granule cells seen at the edge of the explant were stained blue using DAPI. Mossy fiber axon outgrowth was analyzed as described in Methods. Results are shown as mean ± SEM, and ANOVA analysis was performed using SigmaStat 3.1 software (B). Astrocytes (immunopositive for GFAP) were visualized for outgrowth assays as described in Methods (not shown). Results of astrocyte outgrowth assays are shown as mean ± SEM, and ANOVA was performed as described in Methods (C).
The GAP function of ARHGAP4 is a potent regulator of axon outgrowth
ARHGAP4 has been shown to enhance GTP hydrolysis by Rho GTPases as shown by our data (Supplemental Fig. 4) and as previously reported (Foletta et al., 2002). However, it is unknown if its GAP function is important for the regulation of axon outgrowth or cell migration. In order to assess the importance of the GAP domain in axon outgrowth, the R562A mutant protein was expressed in granule cells. Residue 562 is the essential arginine in the arginine finger region of ARHGAP4. The standard method for examination of GAP domain function is to mutate this essential arginine to an alanine to generate a GAP LOF mutant. Although an R to A substitution mutant exhibits dramatically reduced GAP activity (Supplemental Fig. 4), it should retain all of the binding interactions for the unchanged FCH and SH3 regions. Because RhoGAPs with essential arginine mutations still bind their effector proteins, their lack of GAP activity can enable these mutant proteins to have a dominant-negative function. Indeed, the (R562A)-FLAG mutant protein is appropriately targeted to the peripheral tip of axons and NIH/3T3 cells (Fig. 3).
In contrast to the inhibitory effects of full-length ARHGAP4, MF explants and dissociated neurons expressing the (R562A)-FLAG protein showed significantly greater outgrowth (Fig. 6A3, 6B, Supplemental Fig. 3). Interestingly, MF outgrowth in (R562A)-expressing granule cells was not only longer, but appeared to be more branched and/or defasciculated (Fig. 6A3). These observations suggest that the GAP function of ARHGAP4 regulates several aspects of axon behavior.
ARGHAGP4 inhibited cell motility and the GAP-LOF mutant enhanced motility
Since cytoskeletal elements must become organized in a polarized fashion to enable the directional migration of cells, the in vitro wound/scratch assay has become a useful method for studying the function of proteins that are involved in organizing the actin and MT cytoskeleton. Therefore, the wound assay was used to examine the effect of ARHGAP4 overexpression on NIH/3T3 cell migration. NIH/3T3 cells were transfected to express full-length EYFP-tagged ARHGAP4, and migration was quantified as described in Fig. 7A and Methods. When compared to control cells that expressed the EYFP vector alone (dashed line), a significantly smaller percentage of cells that expressed the full-length protein (1-965)-EYFP was observed at the forward edge of migrating cells at 2, 4 and 8 hours (Fig. 7B, black bars) (p ≤ 0.001).
Figure 7. Expression of the R562A GAP activity mutant in NIH/3T3 cells results in increased cell motility.
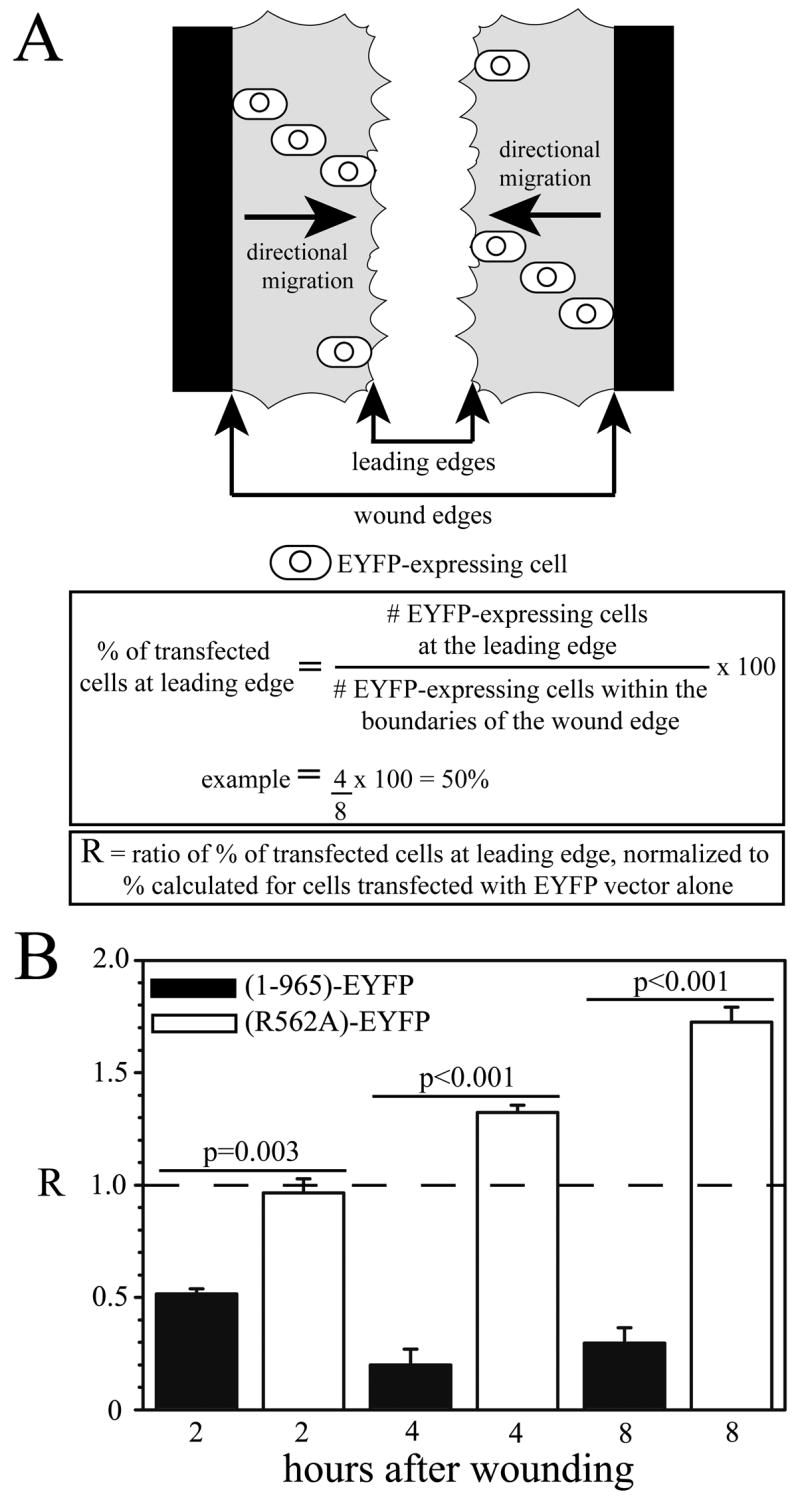
To assess the importance of GAP activity on cell motility, the (R562A)-EYFP mutant protein was expressed in NIH/3T3 cells. The parent EYFP, full-length (1-965)-EYFP, or (R562A)-EYFP vectors were transfected into NIH 3T3 cells using Lipofectamine 2000. Twenty-four hours after transfection, a scratch/wound was made in a confluent area of the culture plate and migration of transfected cells was assessed 2, 4 and 8 hours after the wound. The migration of transfected cells was quantified by counting the number of EYFP-positive cells at the line that represents the leading edge of migrating cells in the wound (fastest cells), and dividing by the total number of EYFP-positive cells between the wound edges (Fig. 9A). All percentage values were normalized to control values (cells expressing EYFP alone) and the ratio for control cells is represented by the horizontal dashed line (R=1.0, panel B). When compared to control cells expressing EYFP vector alone, significantly fewer cells expressing the full-length protein (1-965)-EYFP are observed at the leading edge of migrating cells at 2, 4 and 8 hours (Fig. 9B, black bars), while significantly more cells are observed at the leading edge if they express the (R562A)-EYFP mutant at 4 and 8 hours (Fig. 9B, white bars) (p ≤ 0.001). Importantly, there is a dramatic difference between the migration of cells expressing the full-length versus the R562A mutant proteins (p values shown in Fig. 9B). These observations suggest that the GAP function of ARHGAP4 is extremely important for regulation of cell motility. ANOVA analysis was performed using SigmaStat 3.1 software; n=3–5.
Cells expressing the (R562A)-EYFP mutant represented a significantly higher percentage of cells at the forward edge at 4 and 8 hours (Fig. 7B, white bars) (p ≤ 0.001). Importantly, there was a dramatic difference between the migration of cells that expressed full-length versus R562A mutant proteins (p values shown in panel B), suggesting that ARHGAP4 inhibits cell motility. The fact that the (R562A)-EYFP expressing cells migrate faster than control cells (dashed line) also suggests that this mutant protein acts as a dominant-negative mutation. Reducing the level of endogenous ARHGAP4 by 40% using siRNA also enhanced cell motility (Fig. 8). These data provide strong evidence that endogenous ARHGAP4 functions as a potent inhibitor of NIH/3T3 cell migration.
Figure 8. Knockdown of endogenous ARHGAP4 increased motility of NIH/3T3 cells.

NIH/3T3 cells were co-transfected with the indicated siRNAs and an EYFP expression vector to identify transfected cells as previously described (Kanai et al., 2004). A: Cell motility was analyzed as described in Figure 7, in cells transfected with Control siRNA or ARHGAP4 siRNA. Results are shown as mean ± SEM, and statistical analysis was performed using SigmaStat software; n = 3. B: Western analysis (IB) was performed on cell lysates. Densitometric analysis of immunoreactive ARHGAP4 and actin bands was used to normalize endogenous ARHGAP4 expression, which corresponded to a 40% reduction in response to ARHGAP4 siRNA.
Discussion
This investigation examined the structure and function of ARHGAP4, a novel RhoGAP. RhoGAPs enhance the intrinsic GTPase activity of RhoGTPase, which turns off downstream signaling of these GTPases. The well-defined role of RhoGTPases in regulating actin dynamics suggests that RhoGAPs provide an important link in the pathways that control cellular movement. RhoGAPs localized to the leading edge of migrating cells and axonal growth cones are in the proper location to regulate cytoskeletal dynamics that control movement, and our study shows that ARHGAP4 is strongly localized to axons and growth cones in neurons and to the peripheral tips of NIH/3T3 cells (Figs. 2, 3, 4). ARHGAP4 appears to play a central role in the inhibition of both NIH/3T3 cell motility and axon outgrowth as demonstrated by the overexpression of full-length ARHGAP4. This inhibitory function is further supported by the enhancement of motility following a knockdown of endogenous ARHGAP4 expression using siRNA or endogenous ARHGAP4 function by expression of the dominant-negative R562A mutant protein. The significant enhancement of outgrowth produced by the dominant-negative R562A mutant protein also suggests that the GAP domain of ARHGAP4 plays an important role in its regulation of motility. This inhibition of motility is consistent with the ability of ARHGAP4 to inhibit the function of Rac1 and Cdc42 (Foletta et al., 2002), which are RhoGTPases that are typically associated with enhancing axon outgrowth and cell motility (Govek et al., 2005;Negishi and Katoh, 2002).
Truncated ARHGAP4 72-965 does not appear to be efficiently transported to the distal end of axons and growth cones or to the peripheral region of NIH/3T3 cells. These data show that the N-terminal FCH domain is critical for localizing ARHGAP4 to the leading edge of migrating NIH/3T3 cells and axon growth cones (Fig. 3). ARHGAP4 72-965 also loses its ability to regulate axon motility (Fig. 6), suggesting that ARHGAP4 must be properly localized at the leading edge to affect motility.
Accumulation at the leading edge appears to involve interactions with actin filaments rather than MTs (Figs. 4,5). When motile NIH/3T3 cells were treated with nocodazole to destabilize MTs, ARGAHP4 remained localized to the cell cortex (Figs. 4D–F), while destabilizing actin filaments with cytochalasin-D abolished its localization to the cortex (Figs. 4G–I). Unfortunately, we were not able to identify whether the N- or C-terminal regions of ARHGAP4 mediated this actin binding. This was because the N-terminal deletion mutant (72-965) does not traffic to the cell periphery, and although the 1-770 C-terminal deletion fragment appears to be localized to the periphery (Fig. 5C) it does not appear to be aligned as neatly with the cell cortex as the full-length protein (Fig. 4C). These observations are consistent with recently published data showing that the C-terminus SH3 domain of ARHGAP4 binds to the scaffolding protein Hem-1 (Weiner et al., 2006).
Weiner et al. showed that Hem-1 generated a complex of proteins that is targeted to the leading edge of polarized neutrophils. Several of these proteins, included Abi-1, Nck and ARHGAP4, appear to interact with Hem-1 through their homologous SH3 domains. The authors suggest that the Hem-1 leading edge complex plays a role in the Rac/actin/PIP3 positive feedback loop that modulates the linear cascades that connect chemoattractant receptors to the cytoskeleton in hematopoietic cells. Thus, Hem-1 and its associated protein complex appear to play an important role in organizing the leading edge of motile cells. According to this model, ARHGAP4’s GAP activity would be strategically localized to terminate the Rac-mediated actin polymerization at the leading edge of migrating cells. In addition, the increased axon outgrowth and/or cell migration produced by expression of dominant-negative (R562A) ARHGAP4 or by application of siRNA suggests that ARHGAP4 keeps these complexes inactive, presumably until activated by a chemoattractant signal. The chemoattractant signal could either inactivate ARHGAP4 or lead to its dissociation from the complex. Although this model does not preclude the possibility that ARHGAP4 also could be activated by direct binding to chemorepulsive receptors in a manner similar to the activation of slit-Robo GAP by Robo receptors (Peck et al., 2002;Wong et al., 2001), there currently is no evidence to support this model.
Unlike its restricted distribution in postnatal brain, ARHGAP4 is widely distributed in the CNS during embryonic development. This widespread distribution suggests that ARHGAP4 may play a central role in the regulation of cell and axon motility during embryonic development. For example, ARHGAP4 could be the inhibitory counterpart to p190GAP. Mice with p190 RhoGAP mutations exhibit defects in axon guidance and fasciculation (Brouns et al., 2001). In contrast to ARHGAP4, p190 RhoGAP overexpression leads to an increase in neurite outgrowth. Thus, proper neuronal development may require balanced regulation of ARHGAP4 and other GAPs. Reduction or inhibition of endogenous ARHGAP4 function using siRNA or by expression of the R562A dominant-negative mutant protein enhanced cell migration and axon outgrowth, suggesting that the inhibition of motility involves the activation of ARHGAP4. Thus, motile cells appear to regulate the rate of motility by removing some of the inhibitory or braking action mediated by ARHGAP4. These observations suggest that downregulation of ARHGAP4 function could have a significant impact on the repair of CNS injury and may provide an important therapeutic strategy for enhancing recovery from CNS injury or neurodegenerative diseases.
In summary, we have examined the structure and function of ARHGAP4, a novel RhoGAP molecule. Our results show that ARHGAP4 is a complex molecule whose N-terminus, GAP and C-terminus play different but essential roles in this proteins inhibition of cell and axon motility. The N-terminal amino acids 1-71 are important for the localization of ARHGAP4 to the cell periphery and axon growth cone, which appears be essential for its inhibition of cell motility. The C-terminus also appears to mediate protein-protein interactions. Together, these observations suggest that the function of ARHGAP4 depends on its interactions with protein complexes located in the periphery of motile cells and axon growth cones. Thus, this ARHGAP4 may be important not only for understanding neuronal development, but also for the repair of CNS damage following traumatic injury or disease.
Experimental Methods
Dentate explant cultures
Explant cultures of the dentate gyrus were prepared from 4-day old Sprague-Dawley rat pups as previously described (Butler et al., 2004). Rats were decapitated, and 200μm transverse hippocampal slices were prepared using a WPI Vibroslice. A cut was made along an axis perpendicular to the pyramidal cell layer and in front of the open end of the granule cell layer using a #15 scalpel, and the dentate was carefully removed from the hippocampus. The dentates were further processed to remove CA3c pyramidal cells and much of the hilus. Thus, dentate explants consisted almost entirely of the dentate granule cell layer and the molecular layers. Dentate explants were placed on 25mm Nunc Anopore Membranes coated with polylysine (Sigma, St. Louis, MO) and laminin (Invitrogen, Carlsbad, CA) and maintained in 6 well tissue culture plates containing 1.5ml Neurobasal/B27 medium (Invitrogen).
Dissociated Dentate Cultures
The dentate gyrus was microdissected from 200 μm hippocampal slices prepared from postnatal day 5–7 rats as previously described (Butler et al., 2004). The dentates were rinsed once in Neurobasa/B27 and then dissociated in Neurobasal/B27 with papain (2 ug/ml at 15–23 U/μg) for 30 minutes at RT. After incubation, dentates were rinsed and triturated gently 3 times, and recovered material was centrifuged at 1000 x g for ten minutes. The pellet was resuspended in Neurobasal/B27 and seeded at a concentration of about 50–100 cells/mm2 onto polylysine/laminin coated coverslips. Neurons were transfected at 3 DIV and analyzed at 5 DIV.
NIH/3T3 cell culture
NIH/3T3 cells were obtained from the American Type Culture Collection (ATCC, cell line CRL-1658) and were cultured according to ATCC protocol using Dulbecco’s Modified Eagle’s medium plus 10% bovine serum (DMEM+10% bovine serum).
Construction of ARHGAP4 cDNA expression vectors
ARHGAP4 cDNA was generated by polymerase chain reaction (PCR) using the pεMTH-ARHGAP4 plasmid as a template (Foletta et al. 2002). Specific primers (5′-end primer: 5′-CTAGGCGGCCGCATGGCGGCGCACGGGAAGTTGCGG-3′; 3′-end primer: 5′-GCTAGAATTCGACTGGCTTGCGAGTTGAATCTGG-3′) were used to insert Not I and Eco RI restriction sites into the 5′ and 3′ ends, respectively, of the ARHGAP4 cDNA. The Not I/Eco RI fragment was then subcloned into the multiple cloning site of the pCMV-Tag 4A vector plasmid (Stratagene, La Jolla, CA) to drive expression of a full-length ARHGAP4 (encoding amino acids 1-965) fusion protein with a carboxy-terminal FLAG epitope: (1-965)-FLAG.
To generate the vector encoding a truncated ARHGAP4 fusion protein (72-965)-FLAG, specific PCR primers (5′-end primer: 5″-CATAGCGGCCGCATGGAACGCTTTACTAG-3′; 3′-end primer: 5′-GCTAGAATTCGACTGGCTTGCGAGTTGAATCTGG-3′) were used to generate Not I/Eco RI fragments that were then subcloned into pCMV-Tag 4A, as above. To generate the vector encoding a truncated ARHGAP4 fusion protein (1-770)-FLAG, specific PCR primers (5′-end primer: 5′-CTAGGCGGCCGCATGGCGGCGCACGGGAAGTTGCGG-3′; 3′-end primer: 5′-GCTAGAATTCAGCCTCCACAACTCCCTCG-3′) were used to generate Not I/Eco RI fragments that were then subcloned into pCMV-Tag 4A, as above.
To generate the vector encoding a full-length ARHGAP4-carboxy terminal EYFP fusion protein (1-965)-EYFP, PCR primers (5′-end primer: 5′-AGAAAGATCTATGGCGGCGCACGGG-3′; 3′-end primer: 5′-CTAGAATTCCGACTGGCTTGCGAGTTGAATCTG-3′) were used to introduce Bgl II and Eco RI restriction sites into the ARHGAP4 cDNA for subcloning into the pEYFP-N1 vector (Clontech, Mountainview, CA). The same strategy was used to generate a vector encoding an ARHGAP4 fragment as the fusion protein (1-71)-EYFP, using PCR primers (5′-end primer: 5′-AGAAAGATCTATGGCGGCGCACGGG-3′; 3′-end primer: 5′-GGACGAATTCAGTATGTTCCAGCAGCAT-3′).
The ARHGAP4 vector encoding (1-965)-FLAG was used as a template for site-directed mutagenesis to create cDNA that encoded an arginine→alanine substitution at amino acid 562 (R562A) in the full-length protein. The Stratagene QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene #200514) was used according to the manufacturer’s protocol, and mutant cDNA was generated using the 5′-phosphorylated primer (5′-CTGCAACATGAAGGCATCTTCGCGGTATCAGGTGCCCAGG-3′, base pair changes shown in bold). Reaction products were incubated with Dpn I to digest parental cDNA templates and were then transformed into XL-10-Gold Ultracompetent bacteria. The vector encoding an ARHGAP4 fusion protein, (R562A)-EYFP, was generated using the same PCR primers that were used to make the vector encoding ARHGAP4 (1-965)-EYFP. All plasmids were purified using the Qiagen Maxi Plasmid Kit (Qiagen, Inc., Valencia, CA) and modifications were verified by sequencing. In transfected NIH/3T3 cells (see below), protein expression driven by these vectors resulted in immunoreactive proteins of the expected apparent molecular weights by Western Analysis (see below) with the anti-FLAG antibody described above (Supplemental Figure 1).
ARHGAP4 gene silencing with small interfering RNA
Small interfering RNAs (siRNAs) were generated using the Silencer siRNA Construction Kit (Ambion, Inc.)(Chen et al., 2005). Nucleotides 203–223 of mouse ARHGAP4 mRNA were chosen as targets for silencing using the Ambion siRNA Target Finder program. Briefly, complementary DNA oligonucleotides containing this region of ARHGAP4 cDNA and a complementary region of the 3′ end of the T7 promoter were annealed to a T7 promoter primer and filled in with Klenow DNA polymerase to generate double-stranded templates. ARHGAP4 oligonucleotides: 5′-ACAAGTTGGCTGAACGCTTTACCTGTCTC-3′, and 5′-TAAAGCGTTCAGCCAACTTGTCCTGTCTC-3′. Control oligonucleotides: 5′-ACAAATTAGCGGAGCGATTCACCTGTCTC-3′, and 5′-TGAATCGCTCCGCTAATTTGTCCTGTCTC-3′ (6 underlined nucleotides encode silent mutations). The resulting double-stranded DNA templates were used to generate siRNA strands in in vitro transcription reactions using T7 RNA polymerase. ARHGAP4 or Control reaction products were combined to permit annealing of the siRNA strands, and DNA template removal, single-stranded RNA removal, and purification, were done according to the manufacturer’s protocol. siRNAs were transfected into NIH/3T3 cells using Lipofectamine 2000 at a final concentration of 2nM. After transfection, cells were cultured an additional 2 days prior to Western analysis or immunohistochemistry for wound assays.
Transfection methods for dentate explants
The manufacturer’s in vivo transfection protocol was used to transfect dentate explants using ExGen 500. Briefly, 20μg of plasmid expression vectors were added to 50μl of sterile 5% glucose and 3.6μl ExGen 500 in vivo transfection reagent (#R0521, Fermentas, Hanover, MD). The DNA was condensed for 10 minutes at room temperature with the ExGen 500. Then, 1μl of the transfection solution was applied to each explant using a micropipette, and cultures were returned to the incubator for 48 hours. These interface cultures are not submerged in medium, therefore transfection reagents stay in contact with cultures until they eventually become diluted by the growth medium located below the porous culture insert. Transfection reagent volumes were sufficient to cover entire explants.
Transfection methods for dissociated neurons and NIH/3T3 cells
Lipofectamine 2000 (Invitrogen) was used to transfect NIH/3T3 cells and dissociated hippocampal neurons according to the manufacturers’ protocols.
Immunostaining
Immunostaining of NIH/3T3 cells and dissociated neurons was performed as previously described (Orellana and Marfella-Scivittaro, 2000;Marfella-Scivittaro et al., 2002;Orellana et al., 2003); (Butler et al., 2004). Cells or explants were incubated with 0.1% Triton X-100 for 1-5 min. and then incubated in block (10% serum in PBS) for 30 min. at room temperature, washed 3 times for 5 min. each with phosphate-buffered saline (PBS), and incubated with the primary antibody diluted in block for 1 hour at 37°C. Samples were washed 3 times for 5 min. each with block, incubated with secondary antibody for 1 hour at 37°C, and washed 3 times for 5 min. each with PBS. Primary antibodies used in this study were anti-GFAP (1:200, MP Biomedicals, Irvine, CA; or 1:1,000, Sigma, St. Louis, MO), rabbit polyclonal antibody anti-ARHGAP4 (1:1,000, (Foletta et al., 2002)), anti-β-Tubulin III (1:1,000, Sigma), M2 anti-FLAG (1:250-1:1000, Sigma), and JL8 anti-GFP (1: 1,000, BD Clontech). The cell adhesion molecule L1 was identified using a rabbit polyclonal antibody (1:1,000, gift from Dr. Vance. The polyclonal antibody against ARHGAP4 was generated by Foletta et al. using amino acids 4–20, which a BLAST search showed to be unique to ARHGAP4. The antibody was affinity purified using amino acids 4–20 (Foletta et al. Methods), and the authors demonstrated that presabsorption of the antibody with this unique sequence of amino acids eliminated specific immunoreactivity in rat hippocampal sections (Foletta et al., Fig 8B). Secondary antibodies used were obtained from Molecular Probes (Eugene, OR.), and are noted in the Figure legends. F-actin was detected using fluorescent phalloidin conjugates from Molecular Probes. Photomicrographs were obtained using a Leica TCS confocal microscope or a SPOT Digital Camera attached to a Nikon Optiphot-2 Fluorescence Microscope.
SDS-PAGE and Western analysis
Equal quantities of experimental protein samples from cosedimentation assays and from tissue and cell homogenates were subjected to SDS-PAGE, and transferred to a solid support for Western analysis and ECL (Amersham Biosciences, Piscataway, NJ) signal detection, as described (Orellana and Marfella-Scivittaro, 2000;Marfella-Scivittaro et al., 2002;Orellana et al., 2003). To confirm equal sample loading, blots were stripped and reprobed for immunoreactive α-tubulin expression (anti-α-tubulin, 1:200, Molecular Probes/Invitrogen, Carlsbad, CA) or using the C2 anti-Actin antibody (1:500, Santa Cruz Biotechnology, Santa Cruz, CA). Densitometric quantitation of immunoreactive signals on exposed films was performed using Kodak 1D software (Eastman Kodak Co., Rochester, NY).
Axon and astrocyte outgrowth assay in dentate explants
Mossy fiber axon and astrocyte outgrowth was analyzed as previously described by our lab (Butler et al., 2004). Briefly, the granule cell layer was dissected from P4 rat hippocampi, plated on laminin-coated tissue culture inserts, and transfected as described above. An almost pure population of MF axons emerges from the edges of these round explants and extends onto the laminin substrate. Axons were visualized using an anti-β-tubulin III antibody. Explant images were obtained using the SPOT Digital Camera attached to a Nikon Optiphot-2 Fluorescence Microscope. The area of axon outgrowth was measured using the software supplied with the SPOT CCD camera. Total area (axons + explant) was obtained by drawing a circle that touched the distal tips of the elongated axons emanating from around the explant. The area of axon outgrowth was obtained by subtracting the area of the DAPI-stained explant from the Total area. The area of axon outgrowth was then divided by the area of the explant to normalize for the size of each individual explant. All values were then normalized for outgrowth with the parent vector (FLAG). These area measurements represent the outgrowth of a large population of axons rather than the length of a single axon. Astrocytes were visualized using an anti-GFAP antibody, and outgrowth was measured as for axons.
Axon length measurement in Dissociated Dentate Cultures
The cells were transfected at 3 DIV and fixed at 5 DIV. Cultures were transfected with ARHGAP4-FLAG cDNA expression vectors and visualized using FLAG immunocytochemistry. Control cultures were transfected using the pEYFP-N1 vector (Clontech, Mountainview, CA) and visualized using native EYFP fluorescence. Measurements were conducted as described (Kawano et al., 2005), and only neurons that were not in contact with other cells or processes were analyzed.
NIH/3T3 migration assay
A typical wound assay was used to measure NIH/3T3 cell migration. Briefly, a scratch/wound was made in a confluent area of the culture plate and the rate of cell migration towards the center of the wound was measured. Transfected cells were assessed 2, 4 and 8 hours after wounding. Migration of transfected cells was quantified by counting the number of EYFP-positive cells at the line that represents the leading edge of migrating cells in the wound (fastest cells), and dividing by the total number of EYFP-positive cells between the wound edges (shown in Figure 7 Panel A). All percentage values were normalized to control values (cells expressing EYFP alone) and the ratio for control cells is equal to 1.0 (represented by the horizontal dashed line shown in Figure 7 Panel B).
Recombinant GST-fusion proteins
The GAP domains (amino acids 474–743) of wild type ARHGAP4 (GST-WT-GAP) and R562A (GST-R562A-GAP) were generated by pcr using (1-965)-FLAG and (R562A)-FLAG as templates. Primers 5′ GCAGAATTCCTGCAGGCCAAGCATGAAAAGCTCCAG, and 3′ CATAAGCTTCTAGCTCTCCAACTGGCCATCCCCCAG introduced EcoRI and HindIII sites (in bold) to clone into the GST-fusion expression vector pET-41a (Novagen).
Protein purification was performed as previously described (Tong et al. 2005). Vectors encoding GST-WT-GAP and GST-R562A-GAP were transformed into One Shot® BL21(DE3) bacterial cells (Invitrogen) and screened according to the manufacturer’s protocol. Positive colonies were inoculated in LB + 50 μg/ml Kanamycin and grown overnight at 225 rpms, 37°C. Cultures were diluted 1:100 in fresh LB + Kanamycin, grown for 3 hours, then induced with 0.1 mM IPTG and grown overnight at 225 rpms, 30°C. The cells were pelleted and resuspended in GST-binding buffer (50 mM phosphate buffer pH 6.8, 100 mM NaCl, 4 mM DTT, 4 mM MgCl2) + sigma protease inhibitor cocktail, then sonicated on ice. Insoluble debris was cleared by centrifugation and the supernatant fractions were filtered through a 0.22 micron filter. GST-Bind™ Resin (Novagen) was equilibrated in GST-binding buffer then combined with the resulting lysates for 20 min. at RT. Lysates were removed by gravity filtration on a column and the beads were washed 3x in GST-binding buffer. GST-fusion proteins were eluted off the beads with (100 mM Tris pH 8.5, 20 mM glutathione), and verified by SDS-PAGE and coommassie staining.
In Vitro GAP assay
The RhoGAP Assay Biochem kit #BK105 (Cytoskeleton) was used to determine the relative GAP activity of both GST-WT-GAP and GST-R562A-GAP recombinant proteins corresponding to amino acids 474–743 of ARHGAP4. Briefly, GST-WT-GAP, GST-R562A-GAP, RhoA-His, Rac1-His, and Cdc42-His proteins were diluted to 50 μM in nanopure water at 4°C. Reactions were set up in triplicate to examine GST-WT-GAP or GST-R562A-GAP + RhoA-His, Rac1-His or Cdc42-His, as well as reactions with either GTPase alone or the GST-WT-GAP or GST-R562A-GAP alone. Reactions were combined with 1x Reaction Buffer in a 96 well plate on ice, and GTP was added to each well at a final concentration of 200 μM. The plate was shaken at 200 rpm, 5 sec. and then incubated at 37°C for 20 min. At the end of the reaction, 120 μl of Cytophos reagent was added to each well. The reactions were incubated for 10 min. at RT and then the absorbance was read at 650 nm to assay the level of GTP hydrolysis. Reactions containing 1x Reaction buffer + Cytophos reagent only were used as background controls.
Statistical determinations
Statistical determinations were made using SigmaStat V3.01 software (Systat Inc., Point Richmond, CA).
Supplementary Material
ARHGAP4 full-length and mutant proteins were expressed in NIH/3T3 cells transfected using Lipofectamine 2000 (Panel A) and in hippocampal explant cultures transfected using ExGen 500 (Panel B). Cell lysates were subjected to SDS-PAGE and Western analysis (IB) using the M2 anti-FLAG antibody (Sigma) or JL-8 anti-GFP antibody (Clontech). Apparent molecular weight of the full-length protein is indicated. C) Immunocytochemical evidence for ARHGAP4 expression in dentate explants was obtained using the M2 anti-FLAG antibody (Sigma) and biotinylated secondary (Vector Labs). The secondary antibody was complexed with avidin alexa 594 (Molecular Probes) to visualize immunofluorescence. Explants treated with ExGen 500 alone (Control) showed very weak immunostaining in the granule cell body layer (white outline), while those treated with the ARHGAP-FLAG cDNA expression vector complexed with ExGen 500 exhibited strong FLAG immunostaining that was primarily localized in the granule cell body layer (white outline). Cultures were counterstained with DAPI to visualize the total area of the explants. Scale bar = 100 μm.
Wound assays were performed as described in Methods. Three hours after wounding, the NIH 3T3 cells were treated with vehicle alone (DMSO) (A–C, G–I), 300 nM nocodazole (D–F), or 1 μM cytochalasin-D (J–L) and fixed 5 minutes later. Panels D–F and J–L demonstrate that microtubules and microfilaments were extensively disrupted by these concentrations of nocodazole and cytochalasin-D, respectively. Images were acquired using a Leica confocal microscope. Fluorescent images were merged with brightfield images (C,F,I,L) to show leading edge boundaries.
Dissociated dentate granule cells were transfected at 3DIV and fixed at 5DIV. Neurons transfected with ARHGAP4-FLAG cDNA expression vectors were visualized by FLAG immunostaining, while native EYFP fluorescence was observed in control cells transfected with pEYFP-N1 cDNA expression vectors. The longest axon lengths from transfected neurons were measured and quantified. Neurons expressing (1-965)-FLAG had shorter axons compared to EYFP controls, while those expressing (R562A)-FLAG had longer axons. Axon outgrowth was not altered by expression of 1-770-FLAG or 72-965-FLAG proteins. Data are representative of at least 50 neurons from 3 independent experiments and are presented as ± SEM, (* p<0.05).
The baseline GTPase activity of purified recombinant RhoA-His, Rac1-His, or Cdc42-His was quantified using a spectrophotometer to measure the change in absorbance at 650nm corresponding to released phosphate resulting from the hydrolysis of GTP to GDP after 20 minutes. The change in GTPase activity was then measured in the presence of recombinant GST-GAP(WT) or GST- GAP(R562A) recombinant protein to determine the activity of the GAP domain for the different GTPases and to demonstrate that the R562A mutation resulted in significantly lower enhancement of GTP hydrolysis than WT GAP. Results are expressed as the mean ± SEM, and statistical analysis was performed using SigmaStat software; n = 3.
Acknowledgments
This work was supported by National Institutes of Health Grant NS 41383 (ATM) and by NIMH ZO1-MH-002498-16 (WSY).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Aspenstrom P. A Cdc42 target protein with homology to the non-kinase domain of FER has a potential role in regulating the actin cytoskeleton. Curr Biol. 1997;7:479–487. doi: 10.1016/s0960-9822(06)00219-3. [DOI] [PubMed] [Google Scholar]
- Brouns MR, Matheson SF, Settleman J. p190 RhoGAP is the principal Src substrate in brain and regulates axon outgrowth, guidance and fasciculation. Nat Cell Biol. 2001;3:361–367. doi: 10.1038/35070042. [DOI] [PubMed] [Google Scholar]
- Butler CD, Schnetz SA, Yu EY, Davis JB, Temple K, Silver J, Malouf AT. Keratan sulfate proteoglycan phosphacan regulates mossy fiber outgrowth and regeneration. J Neurosci. 2004;24:462–473. doi: 10.1523/JNEUROSCI.3040-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Dai JC, Orellana SA, Greenfield EM. Endogenous protein kinase inhibitor gamma terminates immediate-early gene expression induced by cAMP-dependent protein kinase (PKA) signaling: termination depends on PKA inactivation rather than PKA export from the nucleus. J Biol Chem. 2005;280:2700–2707. doi: 10.1074/jbc.M412558200. [DOI] [PubMed] [Google Scholar]
- Dent EW, Gertler FB. Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron. 2003;40:209–227. doi: 10.1016/s0896-6273(03)00633-0. [DOI] [PubMed] [Google Scholar]
- Dickson BJ. Rho GTPases in growth cone guidance. Curr Opin Neurobiol. 2001;11:103–110. doi: 10.1016/s0959-4388(00)00180-x. [DOI] [PubMed] [Google Scholar]
- Fankhauser C, Reymond A, Cerutti L, Utzig S, Hofmann K, Simanis V. The S. pombe cdc15 gene is a key element in the reorganization of F-actin at mitosis. Cell. 1995;82:435–444. doi: 10.1016/0092-8674(95)90432-8. [DOI] [PubMed] [Google Scholar]
- Foletta VC, Brown FD, Young WS., 3 Cloning of rat ARHGAP4/C1, a RhoGAP family member expressed in the nervous system that colocalizes with the Golgi complex and microtubules. Brain Res Mol Brain Res. 2002;107:65–79. doi: 10.1016/s0169-328x(02)00448-5. [DOI] [PubMed] [Google Scholar]
- Fuchs U, Rehkamp G, Haas OA, Slany R, Konig M, Bojesen S, Bohle RM, Damm-Welk C, Ludwig WD, Harbott J, Borkhardt A. The human formin-binding protein 17 (FBP17) interacts with sorting nexin, SNX2, and is an MLL-fusion partner in acute myelogeneous leukemia. Proc Natl Acad Sci U S A. 2001;98:8756–8761. doi: 10.1073/pnas.121433898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamblin SJ, Smerdon SJ. GTPase-activating proteins and their complexes. Curr Opin Struct Biol. 1998;8:195–201. doi: 10.1016/s0959-440x(98)80038-9. [DOI] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Van AL. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- Greer P. Closing in on the biological functions of Fps/Fes and Fer. Nat Rev Mol Cell Biol. 2002;3:278–289. doi: 10.1038/nrm783. [DOI] [PubMed] [Google Scholar]
- Huber AB, Kolodkin AL, Ginty DD, Cloutier JF. Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Annu Rev Neurosci. 2003;26:509–563. doi: 10.1146/annurev.neuro.26.010302.081139. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron. 2004;43:513–525. doi: 10.1016/j.neuron.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Kawano Y, Yoshimura T, Tsuboi D, Kawabata S, Kaneko-Kawano T, Shirataki H, Takenawa T, Kaibuchi K. CRMP-2 is involved in kinesin-1-dependent transport of the Sra-1/WAVE1 complex and axon formation. Mol Cell Biol. 2005;25:9920–9935. doi: 10.1128/MCB.25.22.9920-9935.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Zhang B, Zheng Y. Structural determinants required for the interaction between Rho GTPase and the GTPase-activating domain of p190. J Biol Chem. 1997;272:32830–32835. doi: 10.1074/jbc.272.52.32830. [DOI] [PubMed] [Google Scholar]
- Linder S, Hufner K, Wintergerst U, Aepfelbacher M. Microtubule-dependent formation of podosomal adhesion structures in primary human macrophages. J Cell Sci 113 Pt. 2000;23:4165–4176. doi: 10.1242/jcs.113.23.4165. [DOI] [PubMed] [Google Scholar]
- Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 2000;1:173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- Marfella-Scivittaro C, Quiñones A, Orellana SA. Cyclic AMP-dependent protein kinase and proliferation differ in normal and polycystic kidney epithelia. American Journal of Physiology: Cell Physiology. 2002;282:C693–C707. doi: 10.1152/ajpcell.00122.2001. [DOI] [PubMed] [Google Scholar]
- Modregger J, Ritter B, Witter B, Paulsson M, Plomann M. All three PACSIN isoforms bind to endocytic proteins and inhibit endocytosis. J Cell Sci 113 Pt. 2000;24:4511–4521. doi: 10.1242/jcs.113.24.4511. [DOI] [PubMed] [Google Scholar]
- Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003;13:13–22. doi: 10.1016/s0962-8924(02)00004-1. [DOI] [PubMed] [Google Scholar]
- Nassar N, Hoffman GR, Manor D, Clardy JC, Cerione RA. Structures of Cdc42 bound to the active and catalytically compromised forms of Cdc42GAP. Nat Struct Biol. 1998;5:1047–1052. doi: 10.1038/4156. [DOI] [PubMed] [Google Scholar]
- Negishi M, Katoh H. Rho family GTPases as key regulators for neuronal network formation. J Biochem (Tokyo) 2002;132:157–166. doi: 10.1093/oxfordjournals.jbchem.a003205. [DOI] [PubMed] [Google Scholar]
- Orellana SA, Marfella-Scivittaro C. Distinctive cyclic AMP-dependent protein kinase subunit localization is associated with cyst formation and loss of tubulogenic capacity in Madin Darby Canine Kidney cell clones. J Biol Chem. 2000;275:21233–21240. doi: 10.1074/jbc.M001964200. [DOI] [PubMed] [Google Scholar]
- Orellana SA, Quiñones AM, Mandapat ML. Ezrin Distribution is Abnormal in Principal Cells from a Murine Model of Autosomal Recessive Polycystic Kidney Disease. Pediatric Research. 2003;54:406–412. doi: 10.1203/01.PDR.0000077480.82519.E1. [DOI] [PubMed] [Google Scholar]
- Peck J, Douglas G, Wu CH, Burbelo PD. Human RhoGAP domain-containing proteins: structure, function and evolutionary relationships. FEBS Lett. 2002;528:27–34. doi: 10.1016/s0014-5793(02)03331-8. [DOI] [PubMed] [Google Scholar]
- Qualmann B, Kelly RB. Syndapin isoforms participate in receptor-mediated endocytosis and actin organization. J Cell Biol. 2000;148:1047–1062. doi: 10.1083/jcb.148.5.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Nelson DL, Stewart DM. Cdc42-interacting protein 4 mediates binding of the Wiskott-Aldrich syndrome protein to microtubules. J Biol Chem. 2000;275:7854–7861. doi: 10.1074/jbc.275.11.7854. [DOI] [PubMed] [Google Scholar]
- Tribioli C, Droetto S, Bione S, Cesareni G, Torrisi MR, Lotti LV, Lanfrancone L, Toniolo D, Pelicci P. An X chromosome-linked gene encoding a protein with characteristics of a rhoGAP predominantly expressed in hematopoietic cells. Proc Natl Acad Sci U S A. 1996;93:695–699. doi: 10.1073/pnas.93.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner OD, Rentel MC, Ott A, Brown GE, Jedrychowski M, Yaffe MB, Gygi SP, Cantley LC, Bourne HR, Kirschner MW. Hem-1 complexes are essential for Rac activation, actin polymerization, and myosin regulation during neutrophil chemotaxis. PLoS Biol. 2006;4:e38. doi: 10.1371/journal.pbio.0040038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann T, Bokoch GM, Waterman-Storer CM. Regulation of leading edge microtubule and actin dynamics downstream of Rac1. J Cell Biol. 2003;161:845–851. doi: 10.1083/jcb.200303082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K, Ren XR, Huang YZ, Xie Y, Liu G, Saito H, Tang H, Wen L, Brady-Kalnay SM, Mei L, Wu JY, Xiong WC, Rao Y. Signal transduction in neuronal migration: roles of GTPase activating proteins and the small GTPase Cdc42 in the Slit-Robo pathway. Cell. 2001;107:209–221. doi: 10.1016/s0092-8674(01)00530-x. [DOI] [PubMed] [Google Scholar]
- Yeung YG, Soldera S, Stanley ER. A novel macrophage actin-associated protein (MAYP) is tyrosine-phosphorylated following colony stimulating factor-1 stimulation. J Biol Chem. 1998;273:30638–30642. doi: 10.1074/jbc.273.46.30638. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ARHGAP4 full-length and mutant proteins were expressed in NIH/3T3 cells transfected using Lipofectamine 2000 (Panel A) and in hippocampal explant cultures transfected using ExGen 500 (Panel B). Cell lysates were subjected to SDS-PAGE and Western analysis (IB) using the M2 anti-FLAG antibody (Sigma) or JL-8 anti-GFP antibody (Clontech). Apparent molecular weight of the full-length protein is indicated. C) Immunocytochemical evidence for ARHGAP4 expression in dentate explants was obtained using the M2 anti-FLAG antibody (Sigma) and biotinylated secondary (Vector Labs). The secondary antibody was complexed with avidin alexa 594 (Molecular Probes) to visualize immunofluorescence. Explants treated with ExGen 500 alone (Control) showed very weak immunostaining in the granule cell body layer (white outline), while those treated with the ARHGAP-FLAG cDNA expression vector complexed with ExGen 500 exhibited strong FLAG immunostaining that was primarily localized in the granule cell body layer (white outline). Cultures were counterstained with DAPI to visualize the total area of the explants. Scale bar = 100 μm.
Wound assays were performed as described in Methods. Three hours after wounding, the NIH 3T3 cells were treated with vehicle alone (DMSO) (A–C, G–I), 300 nM nocodazole (D–F), or 1 μM cytochalasin-D (J–L) and fixed 5 minutes later. Panels D–F and J–L demonstrate that microtubules and microfilaments were extensively disrupted by these concentrations of nocodazole and cytochalasin-D, respectively. Images were acquired using a Leica confocal microscope. Fluorescent images were merged with brightfield images (C,F,I,L) to show leading edge boundaries.
Dissociated dentate granule cells were transfected at 3DIV and fixed at 5DIV. Neurons transfected with ARHGAP4-FLAG cDNA expression vectors were visualized by FLAG immunostaining, while native EYFP fluorescence was observed in control cells transfected with pEYFP-N1 cDNA expression vectors. The longest axon lengths from transfected neurons were measured and quantified. Neurons expressing (1-965)-FLAG had shorter axons compared to EYFP controls, while those expressing (R562A)-FLAG had longer axons. Axon outgrowth was not altered by expression of 1-770-FLAG or 72-965-FLAG proteins. Data are representative of at least 50 neurons from 3 independent experiments and are presented as ± SEM, (* p<0.05).
The baseline GTPase activity of purified recombinant RhoA-His, Rac1-His, or Cdc42-His was quantified using a spectrophotometer to measure the change in absorbance at 650nm corresponding to released phosphate resulting from the hydrolysis of GTP to GDP after 20 minutes. The change in GTPase activity was then measured in the presence of recombinant GST-GAP(WT) or GST- GAP(R562A) recombinant protein to determine the activity of the GAP domain for the different GTPases and to demonstrate that the R562A mutation resulted in significantly lower enhancement of GTP hydrolysis than WT GAP. Results are expressed as the mean ± SEM, and statistical analysis was performed using SigmaStat software; n = 3.