

Abstract
Peroxisome proliferators are potent rodent liver carcinogens that act via a non-genotoxic mechanism. The mode of action of these agents in rodent liver includes increased cell proliferation, decreased apoptosis, secondary oxidative stress and other events; however, it is not well understood how peroxisome proliferators are triggering the plethora of the molecular signals leading to cancer. Epigenetic changes have been implicated in the mechanism of liver carcinogenesis by a number of environmental agents. Short-term treatment with peroxisome proliferators and other non-genotoxic carcinogens leads to global and locus-specific DNA hypomethylation in mouse liver, events that were suggested to correlate with a burst of cell proliferation. In the current study, we investigated the effects of long-term exposure to a model peroxisome proliferator WY-14,643 on DNA and histone methylation. Male SV129 mice were fed a control or WY-14,643-containing (1000 ppm) diet for 1 wk, 5 wks or 5 months. Treatment with WY-14,643 led to progressive global hypomethylation of liver DNA as determined by an HpaII-based cytosine extension assay with the maximum effect reaching over 200% at 5 months. Likewise, trimethylation of histone H4 lysine 20 and H3 lysine 9 was significantly decreased at all time points. The majority of cytosine methylation in mammals resides in repetitive DNA sequences. In view of this, we measured the effect of WY-14,643 on the methylation status of major and minor satellites, as well as in IAP, LINE1 and LINE2 elements in liver DNA. Exposure to WY-14,643 resulted in a gradual loss of cytosine methylation in major and minor satellites, IAP, LINE1 and LINE2 elements. The epigenetic changes correlated with the temporal effects of WY-14,643 on cell proliferation rates in liver, but no sustained effect on c-Myc promoter methylation was observed. Finally, WY-14,643 had no effect on DNA and histone methylation status in Pparα-null mice at any of the time points considered in this study. These data indicate the importance of epigenetic alterations in the mechanism of action of peroxisome proliferators and the key role of PPARα.
Introduction
Recent evidence shows that epigenetic changes play an important role in carcinogenesis induced by a variety of factors that act via either genotoxic or non-genotoxic mechanisms [1, 2]. Genotoxic carcinogens or products of their metabolic activation interact with DNA directly causing tumor formation [3], whereas non-genotoxic carcinogens are a diverse group of chemicals that induce neoplastic cell transformation by mechanisms other than direct DNA damage [4]. One of the most extensively studied classes of non-genotoxic carcinogens are peroxisome proliferators, a structurally diverse group of chemicals and therapeutic agents [5]. Long-term exposure to these chemicals results in the development of liver tumors in male and female mice and rats [6, 7]. The mode of action of peroxisome proliferators in rodent liver includes increased cell proliferation, decreased apoptosis, secondary oxidative stress, and other events [8]. It is widely recognized that the molecular target for their action in liver is the peroxisome proliferators-activated receptor (PPAR)α, and most of the effects, including the hepatocarcinogenic response, are the result of a PPARα-mediated mechanism [9, 10]. However, it is not well understood how peroxisome proliferators are triggering the plethora of molecular signals leading to cancer in rodent liver and why prolonged activation of PPARα-induced pathways leads to hepatocarcinogenesis.
Short-term treatment with non-genotoxic rodent carcinogens, including peroxisome proliferators, is known to lead to hypomethylation of DNA and proto-oncogenes in mouse liver, events that were suggested to correlate with a burst of cell proliferation [11-13]. Our previous studies using a methyl-deficient model of non-genotoxic hepatocarcinogenesis in rats showed that alteration of cellular epigenetic processes, such as DNA hypomethylation and loss of histone H4 lysine 20 trimethylation, are key steps in the carcinogenic process induced by methyl deficiency [14, 15]. Additionally, it has been demonstrated recently that global DNA hypomethylation was associated with the development of multiple liver tumors, while protecting from colon tumors, in Apc(Min/+)/Dnmt1(chip/c) mice providing additional evidence for the importance of epigenetic alterations in the origin of liver tumors [16]. These findings have led to a suggestion that sensitivity to tumorigenesis may be inversely related to the capacity to maintain normal patterns of the cellular epigenetic landscape [17].
In order to further characterize the long-term epigenetic effects of peroxisome proliferators in mouse liver and their dependence upon PPARα, we examined the hypothesis that epigenetic dysregulation is an important contributing factor to the mechanisms of carcinogenicity of these agents. The effects of WY-14,643, a model peroxisome proliferator, were examined in wild type or Pparα-null mice treated for up to 5 months. Global methylation of DNA, histone modification changes and other endpoints were evaluated.
Materials and Methods
Animals and Treatments
Pparα-null male mice (SV129 background; [18]), and corresponding wild type counterparts (6−8 weeks of age at the beginning of treatment) were used. Animals were housed in sterilized cages in a facility with a 12-hr night/day cycle. Temperature and relative humidity were held at 22±2°C and 50±5%, respectively. The UNC Division of Laboratory Animal Medicine maintains these animal facilities, and veterinarians were always available to ensure animal health. All animals were given humane care in compliance with NIH and institutional guidelines and studies were performed according to protocols approved by the appropriate institutional review board. Prior to experiments, animals were maintained on standard lab chow diet and purified water ad libitum. 4-Chloro-6-(2,3-xylidino)-pyrimidynylthioacetic acid (WY-14,643) was obtained from Aldrich (Milwaukee, WI). NIH-07 was used as the base for the pelleted diet (prepared by Harlan Teklad, Indianapolis, IN) containing either 0 ppm (control), or 1000 ppm of WY-14,643. Dietary concentration of WY-14,643 was measured by high performance liquid chromatography after the pellets were made and determined to be ±18% of the target concentration. Diet was administered ad libitum for 1 week, 5 weeks or 5 months. Animals had free access to water throughout the study and the health status of the animals was monitored every second day throughout the study. At sacrifice, mice were anesthetized with pentobarbital (100 mg/kg, i.p.) and following exsanguination livers were removed, weighed, placed in microcentrifuge tubes and snap frozen in liquid nitrogen. The samples were stored at −80°C until assayed.
Global DNA methylation analysis
The extent of the global DNA methylation was evaluated with a radiolabeled [3H]dCTP extension assay as described previously [19]. Briefly, 1 μg of genomic DNA was digested with 20 U of methylation-sensitive HpaII restriction endonuclease (New England Biolabs, Beverly, MA) for 16−18 h at 37°C. A second DNA aliquot (1 μg) was digested with methylation-insensitive iso-schizomer MspI, which cleaves CCGG sites in DNA regardless of CpG methylation status, to serve as a control for the digestion efficiency. Undigested DNA served as a background control. The single nucleotide extension reaction was performed in a 25 μl reaction mixture containing 1.0 μg DNA, 1X PCR buffer, 1.0 mM MgCl2, 0.25 U AmpliTaq DNA polymerase (Applied Biosystems, Foster City, CA), 0.1 μl of [3H]dCTP (57.4 Ci/mmol; Perkin Elmer, Boston, MA) and incubated at 56°C for 1 h. Samples were applied to DE-81 ion-exchange filters and washed three times with 0.5 M sodium phosphate buffer (pH 7.0) at room temperature. The filters were dried and processed for scintillation counting. [3H]dCTP incorporation into DNA is expressed as mean disintegrations per minute (dpm) per μg of DNA after subtraction of the dpm incorporation in undigested samples (background).
Methylation-sensitive arbitrarily primed PCR
The methylation status of GC-rich regions was determined by using the methylation-sensitive arbitrarily primed PCR (ms-AP-PCR) assay as described previously [20]. Briefly, 1 μg of genomic DNA was separately digested with 20 U each of methylation-insensitive endonuclease RsaI and methylation-sensitive endonuclease HpaII (New England Biolabs) at 37°C for 16−18 h. Restriction-digested DNA was amplified using AP-PCR with single MLG2 primer [19], which hybridized preferentially to GC-rich regions in the presence of [33P] dCTP (Perkin Elmer). PCR products were resolved on 5% polyacrylamide 7 M urea sequencing gel. Gel-resolution of amplified PCR products resulted in the generation of methylation-sensitive fingerprints. After electrophoresis, the gel was analyzed by Cyclone Storage Phosphor Screen system (Packard Instrument, Meriden, CT) for differences in the intensity of bands between control and WY-14,643-treated samples. Relative decrease in the intensity of a bands or the disappearance/appearance of a band in WY-14,643-treated samples as compared to control samples is interpreted as a loss of methylation/hypermethylation, respectively. The band intensities were quantified as described elsewhere [21].
The methylation changes within unmethylated DNA domains were determined by using the McrBC-msAP-PCR as described previously [22]. This technique allows detection of differentially methylated sites within unmethylated DNA domains enriched by regulatory sequences and CpG islands. Briefly, 2 μg of genomic DNA was digested overnight at 37°C in presence of 10 U/μg DNA of McrBC endonuclease (New England Biolabs). McrBC is a methylation-specific endonuclease, which, as opposed to methylation-sensitive restriction endonucleases, cleaves DNA containing 5-methylcytosine on one or both strands but does not act on unmethylated DNA. Additionally, it does not recognize HpaII sites (CCGG) in which the internal cytosine is methylated. McrBC-digested DNA fragments were separated on a 1% agarose gel, and DNA fragments larger than 1 kb were excised from the gel and purified by using a QIAquick Gel Extraction kit according to manufacturer's protocol (Qiagen, Valencia, CA). The fragments, which were enriched for unmethylated DNA, were then digested overnight with 20 U/μg DNA of methylation-sensitive restriction endonuclease SmaI (New England Biolabs), followed by digestion with 20 U/μg DNA of HpaII (New England Biolabs). The digested DNA was amplified by PCR and analyzed as detailed above.
Analysis of methylation status of DNA repetitive elements and c-Myc promoter
DNA methylation analysis of repetitive elements in mouse genome was performed by using the quantitative McrBC-PCR assay as described previously [23]. Cleavage of methylated DNA by McrBC induces DNA strand breaks and abrogates PCR amplification. Conversely, the presence of unmethylated cytosines in DNA prevents enzyme cleavage and can be detected by PCR amplification product recovery. Undigested DNA served as control. Following the McrBC treatment, subsequent PCR was used to amplify the intracisternal A particle (IAP) of long terminal repeats (LTR) retrotransposons and long interspersed nucleotide elements 2 (LINE2) representing the non-LTR retrotransposons. The quantitative aspect of the procedure was verified by a linear increase in PCR product recovery with increasing cycle number and DNA template concentration. The results were confirmed by quantitative real-time PCR using a SYBR GreenER SuperMix (Invitrogen, Carlsbad, CA) for iCycler (Bio-Rad, Hercules, CA). Methylated DNA sequences have decreased amount of PCR product after McrBC digestion. The results are presented as relative change in ratio of PCR product recovery after digestion of DNA with McrBC relative to undigested DNA.
The methylation status of LINE1 was determined by the COBRA assay [24], which consists of a standard bisulfite modification of genomic DNA, subsequent PCR amplification and digestion of the PCR product with the appropriate restriction endonuclease. The combination of sodium bisulfite treatment and PCR amplification results in methylation-dependent retention of preexisting restriction endonuclease sites, or methylation-dependent creation of new sites. Briefly, 2 μg of genomic DNA was treated with sodium bisulfite, and bisulfite-modified DNA was PCR amplified with primers corresponding to the regulatory region of mouse LINE1 sequence. The sense primer was 5'-GATTAAGACGTATTAAGAAAT-3', and the antisense primer was 5'-TTTGTAAGAAGATTTCGTTTAGTTA-3'. The PCR products were digested with 20 units of HpyCH4IV endonuclease (New England Biolabs). The digested PCR products were separated on a 3% high-resolution agarose gel (Sigma, St. Louis, MO), stained with ethidium bromide, photographed, and the band intensity was analyzed by ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
The methylation status of c-Myc promoter was determined by the quantitative McrBC-PCR assay [23] with primers corresponding to the regulatory region of mouse c-Myc gene. The sense primer was 5'-GGTGACTGATATACGCAGGGCAAGA-3', and the antisense primer was 5'-CCGGGGAAAGAGGAGGAGGAG-3'.
Analysis of histone H3 and histone H4 modifications
Acidic cell extracts were prepared from liver tissue as described previously [25]. Equal amount of total histones (40 μg) was mixed with two volumes of gel loading buffer (250 mM Tris-HCl (pH 8.0), 20% β-mercaptoethanol, 40% glycerol, 8% SDS, and 1.2 mg/ml bromophenol blue), heated for 5 min at 95°C, and resolved on 15% polyacrylamide gels. Proteins were transferred onto PVDF membranes (GE Healthcare Biosciences, Piscataway, NJ). The membranes were blocked for 4 h in Tris-buffered saline (TBS) containing 5% nonfat dry milk and 0.1% Tween-20. Primary antibodies against trimethyl-histone H3 lysine 9 (H3K9me3), and trimethyl-histone H4 lysine 20 (H4K20me3) were diluted 1:1000 and 1:2000, respectively, according to manufacturer's recommendations (Upstate, Charlottesville, VA). Primary antibody binding was performed at 4°C overnight with constant shaking. A secondary donkey anti-rabbit antibody, labeled with alkaline phosphatase (Santa Cruz Biotechnology, Santa Cruz, CA), was applied at 1:5000 dilutions and binding was carried out at room temperature for 1.5 h. Chemifluorescence detection was performed with the ECF Substrate for Western Blotting (GE Healthcare Biosciences) and measured directly by Storm Imaging System (Molecular Dynamics). Images are representative of three independent immunoblots and were analyzed by ImageQuant software (Molecular Dynamics). All membranes were stained with Coomassie Blue and with anti-histone H3 and anti-histone H4 antibodies to confirm equal protein loading.
Statistical analysis
Results are presented as mean ± S.D. and were assessed by 2-way analysis of variance (ANOVA), using treatment and weeks as fixed factors, or one-way ANOVA, using treatment as the fixed factor. When necessary to maintain equal variance or normal data distribution, the results were ln transformed before conducting the ANOVA. p-Values <0.05 were considered significant.
Results
Chronic treatment with WY-14,643 causes a progressive PPARα-dependent loss of global DNA methylation in mouse liver
Continuous exposure to WY-14,643 at the dose level used in this study (1000 ppm) results in a 100% incidence of hepatocellular adenomas and carcinomas in wild type mice after about 11 months [9]. In this study, DNA methylation status was assessed in livers of wild type and Pparα-null mice fed WY-14,643-containing diet for up to 5 months. In wild type mice treated with WY-14,643 for 1 week, 5 weeks, or 5 months liver cell proliferation was elevated about 3-, 13- and 22-fold (as compared to time-matched mice fed control diet), respectively, and no liver tumors were detected [26]. WY-14,643 had no effect on liver cell proliferation in Pparα-null mice [26]. A cytosine extension assay that measures the proportion of unmethylated CpG sites in genomic DNA was used to assess effects of the treatment. The assay is based on the ability of the HpaII methylation-sensitive restriction enzyme to cleave unmethylated CCGG sequences and leave a 5' guanine overhang that can be used for the subsequent single nucleotide extension with labeled [3H]dCTP [19]. The extent of [3H]dCTP incorporation is directly proportional to the number of unmethylated CCGG sites. Figure 1 (top graph) shows that long-term administration of WY-14,643-containing diet led to a progressive demethylation of genomic DNA in wild type mice, evident from an increase in incorporation of [3H]dCTP into HpaII-digested DNA. In contrast, no treatment-related effects, except for non-significant transient DNA hypomethylation at 5 weeks, were evident in Pparα-null mice fed with WY-14,643 (Figure 1, bottom graph). While many factors may lead to ephemeral changes in DNA methylation, only sustained effects are generally considered as important in carcinogenesis [14].
Figure 1. Long-term treatment with WY-14,643 leads to progressive loss of global DNA methylation in the liver of wild type, but not Pparα-null mice.
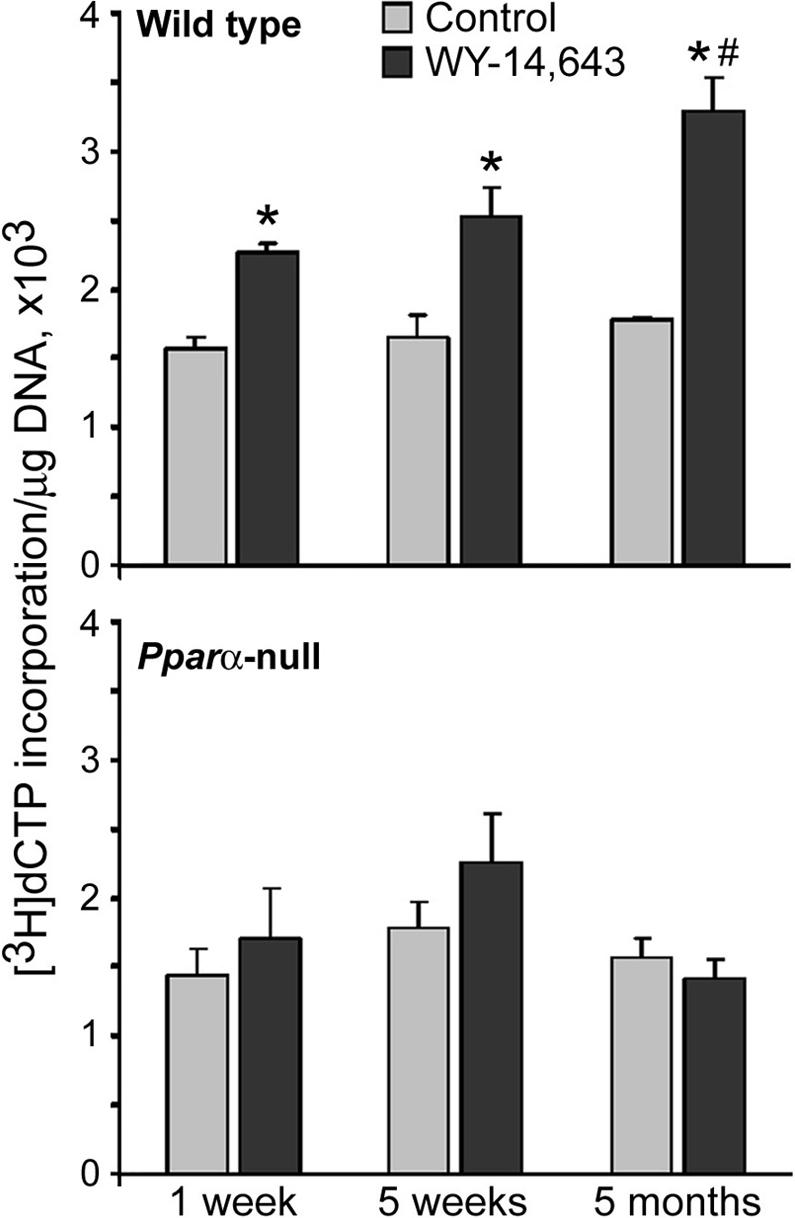
DNA methylation in liver of WY-14,643 (1000 ppm)-exposed wild type (top panel) and Pparα-null (bottom panel) mice and age-matched control mice was measured by the cytosine extension assay as detailed in Methods. The extent of [3H]dCTP incorporation is directly proportional to the number of unmethylated CCGG sites. Data is presented as mean±S.D. for each group (n=5) and asterisks (*) indicate significant difference from time/strain-matched control mice.
WY-14,643 induces region-specific DNA methylation changes in mouse liver
Mammalian genome consists of short (< 4kb) un-methylated domains embedded into longer methylated domains [25]. To determine whether the effects of WY-14,643 on global methylation in mouse liver are domain-specific, we measured methylation changes in methylated and largely un-methylated regions separately. First, we used ms-AP-PCR fingerprint analysis to determine methylation changes within methylated domains in the liver of wild type mice in response to feeding WY-14,643-containing diet. Progressive WY-14,643-induced hypomethylation occurs evenly within all methylated DNA domains, as evidenced by the decrease in the intensity of bands or the disappearance of a band compared to control samples (Figure 2, left panel). Quantification of band intensity for 15 regions with most prominent changes (Figure 2, left panel, arrows) indicated significant time-dependent hypomethylation in liver DNA at all locations evaluated by this assay (data not shown). The un-methylated domains in mammalian genome are frequently located in regulatory regions that are enriched in CpG islands [25]. To assess the potential effect of WY-14,643 on methylation in these regions, we used a modified (with a restriction enzyme McrBC) ms-AP-PCR. In contrast to significant changes in constitutively methylated DNA (Figure 2, left panel), the methylation status of un-methylated domains was not altered by WY-14,643 in any of the time points assessed (Figure 2, right panel).
Figure 2. Region-specific effects of WY-14,643 on DNA methylation status in mouse liver.
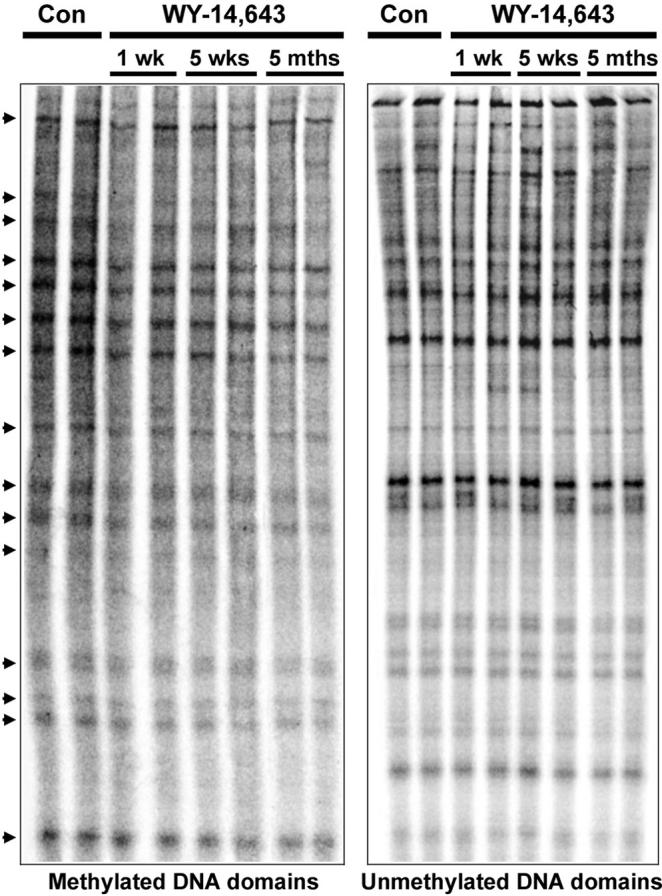
Representative ms-AP-PCR (left panel) and McrBC-ms-AP-PCR (right panel) fingerprints of methylation status of highly methylated (left) and unmethylated (right) DNA regions from livers of control (one sample each from 1 week and 5 month groups) and WY-14,643-fed mice. Relative decrease in the intensity or disappearance of each band (arrows), as compared to corresponding control, is an evidence for loss of methylation. The results were reproduced in two independent experiments with all samples. Two samples from each time point are shown.
Effect of WY-14,643 on methylation status of DNA repetitive elements in mouse liver
The majority of CpG methylation in mammalian cells is located in repetitive DNA elements [27] and one of the main functions of DNA methylation proteins in somatic cells is to maintain stability of the genome by silencing the expression of these repetitive sequences [28]. It has been suggested that genome-wide DNA hypomethylation in cancer cells and during carcinogenesis largely affect methylation status of these repetitive sequences [28]. The mouse genome consists of almost 19% of non-long terminal repeat (LTR) retrotransposons or LINE1 elements, which form the single largest fraction of interspersed repeats [23]. Furthermore, LTR retrotransposons, including the highly active intracisternal A-particle (IAP) elements, account for over 10% of all spontaneous mutations in mice [23]. Thus, we measured the effect of long term exposure to WY-14,643 on methylation status of these repetitive elements in mouse liver. Treatment with WY-14,643 resulted in a progressive decrease of LINE1, LINE2 and LTR IAP methylation (Figure 3). In addition, hypomethylation of major and minor satellites was also observed in liver of WY-14,643-treated wild type mice (data not shown). Interestingly, methylation of c-Myc gene was not affected by long-term dietary treatment with WY-14,643 even though WY-14,643-related hypomethylating effect on c-Myc gene early after a single dose of WY-14,643 has been observed [11].
Figure 3. Effects of WY-14,643 on methylation status of LINE1, LINE2, and LTR IAP retrotransposons and c-Myc gene in mouse liver.
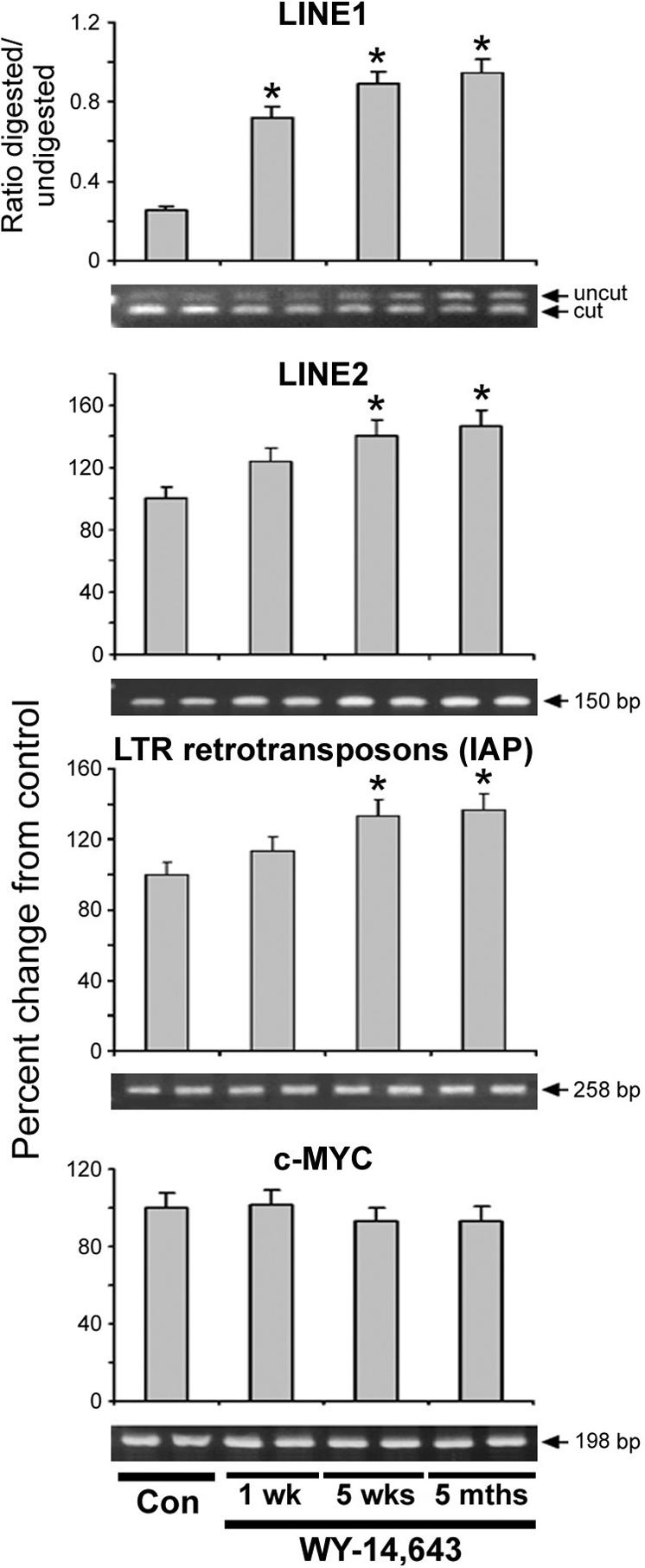
The status of LINE1 methylation (top panel) was evaluated using COBRA assay with the HpyCH4IV restriction endonuclease. The data is presented as ratio between uncut and HpyCH4IV-cut fragments in liver of WY-14,643-fed mice and the age-matched control mice. The increased ratio between undigested and HpyCH4IV -digested PCR products rats indicates the loss of LINE-1 methylation. The status of LINE2 and LTR IAP (middle two panels), and c-Myc promoter (lower panel) methylation was evaluated by quantitative McrBC-PCR assay. The results are presented as ratio of PCR product recovery between McrBC-digested DNA and undigested DNA. Increase in McrBC-digested DNA/undigested DNA ratio is indicative of hypomethylation status. Data is presented as mean±S.D. for controls (all time points, n=15) and each treatment group (n=5). Asterisks (*) indicate significant difference from time/strain-matched control mice. Representative PCR images are shown.
Effect of WY-14,643 on histones in mouse liver
Histone methylation is an important component of maintaining genome stability. To determine the effects of long-term dietary exposure to WY-14,643 on methylation status of histones H3 and H4, we assessed the content of trimethylated histones in livers of wild type and Pparα-null mice treated for up to 5 months. A rapid and sustained loss of histone H3K9 and histone H4K20 trimethylation was observed in the wild type mice fed WY-14,643-containing diet (Figure 4, top panel). The level of histone H3K9me3 and H4K20me3 in liver of the wild type mice after only 1 week of treatment was ∼55% and 63% lower, respectively, than in control group, and remained suppressed for up to 5 months. In contrast, the level of H3K9me3 and H4K20me3 were not affected in Pparα-null mice at any of the time points (Figure 4, bottom panel). To determine the possible mechanism for the loss of H3K9 and H4K20 trimethylation in the liver of wild type mice fed WY-14,643-containing diet, we assessed the protein levels of the major histone methyltransferases Suv39h1, PRDM/Riz1 (both responsible for histone H3K9 trimethylation [29]), and Suv4−20h2 (histone H4K20 trimethylation [30]). WY-14,643 had no effect on Suv39h1, while expression of PRDM/Riz1 increased significantly as early as 1 week of treatment and remained elevated for up to 5 months (Figure 5). The effect on expression of Suv420h2 was more gradual and the amounts of this protein in livers of mice fed WY-14,643 was lower than in control animals.
Figure 4. Long-term treatment with WY-14,643 leads to progressive loss of histone H4K20me3 and H3K9me3 in the liver of wild type, but not Pparα-null mice.
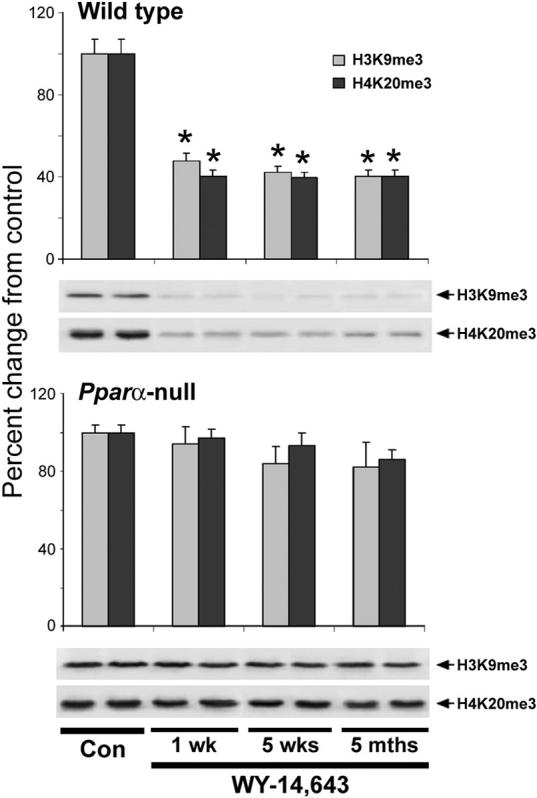
Acid extracts of total histones prepared from livers of wild type (top panel) or Pparα-null (bottom panel) mice were separated by SDS-PAGE and subjected to immunoblotting using specific antibodies against histone H3K9me3 and histone H4K20me3 as detailed in Methods. Results (mean±S.D.) are presented as change in methylation in each treatment group (n=5) relative to control mice (all time points, n=15). Equal sample loading was confirmed by immunostaining against histone H3 and histone H4. Representative immunoblot images are shown. Asterisks (*) indicate significant difference from time/strain-matched control mice.
Figure 5. Effects of WY-14,643 on expression of Suv39h1, Suv4−20h2, and PRDM/Riz1 histone methyltransferases in mouse liver.
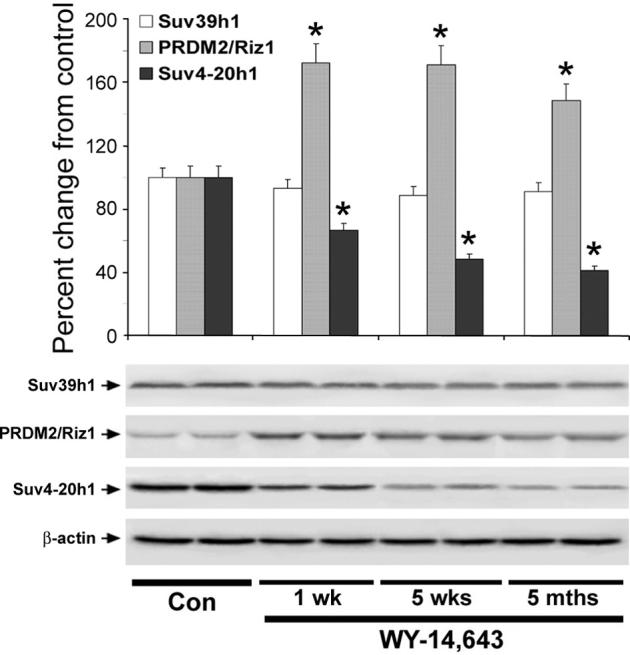
Liver tissue lysates were separated by SDS-PAGE and subjected to immunoblotting using specific antibodies against Suv39h1, Suv4−20h2, and PRDM/Riz1 histone methyltransferases. Equal sample loading was confirmed by immunostaining against β-actin. Bar graph shows quantitative evaluation (percent change, mean±S.D.) of the Suv39h1, Suv4−20h2, and PRDM/Riz1 expression in livers of WY-14,643-treated mice (n=5 for each time point) relative to those in control animals (all time points, n=15). Representative Western immunoblot images from two independent experiments are shown for each protein. Asterisks (*) indicate significant difference from time/strain-matched control mice.
Discussion
This study shows that long-term dietary administration of WY-14,643 leads to prominent epigenetic changes in mouse liver. These effects included progressive loss of global DNA methylation, hypomethylation of mobile repetitive DNA elements, and loss of histone H3K9 and H4K20 trimethylation accompanied by diminished protein levels of Suv4−20h2 histone methyltransferase. In contrast, WY-14,643 exposure had no effect on global DNA methylation or trimethylation of histones in liver of Pparα-null mice indicating that epigenetic effects of peroxisome proliferators, like many other liver-specific molecular changes caused by these agents in rodents, are mediated through a PPARα-dependent mechanism.
It is well established that tumorigenesis is associated with alterations in cellular epigenetic status, especially with hypomethylation of DNA [31, 32]. Genome-wide loss of DNA methylation have been regarded as a common event in cancer cells [32]. Recent studies have suggested a link between long-term exposure to carcinogens, epigenetic alterations, and the ensuing malignant transformation [1, 2]. Specifically, studies show that a sustained loss of DNA methylation in liver is an early and indispensable event in hepatocarcinogenesis induced by long-term exposure to both genotoxic and non-genotoxic agents in rodents [14, 17, 21, 25]. Furthermore, it has been hypothesized that sensitivity to tumorigenesis may be inversely related to the capacity to maintain normal patterns of DNA methylation [17]. In view of this, the progressive hypomethylation of DNA in the liver of WY-14,643-exposed wild type mice may be considered an important factor in mechanism of PP-induced rodent carcinogenesis.
It has been suggested that peroxisome proliferator-induced increases in liver cell proliferation would prevent the methylation of the newly synthesized strands of DNA [11]. This hypothesis was supported by the fact that after a single administration of WY-14,643 to mice the temporal relationship between increased cell proliferation and DNA hypomethylation of the c-Myc gene was observed. Our data shows that long-term treatment with WY-14,643 produces a pattern of gradually worsening dysregulation of normal methylation patterns in genomic DNA and histones and that these effects correlate with constitutively increasing cell proliferation in liver at these time points [26]. Indeed, it was shown that the ability of DNA methyltransferase 1, the most abundant DNA methyltransferase in somatic cells, to maintain sufficient methylation status of CpG-rich domains is limited under conditions of such elevated rates of DNA synthesis [33]. Several possible alternative explanations exist for the mechanism of DNA hypomethylation after exposure to WY-14,643. First, long-term exposure to peroxisome proliferators leads to secondary oxidative stress in liver [34, 35] which may result in oxidative damage of methylated cytosine residues and depletion in the level of 5-methylcytosine in DNA [36]. Second, formation of high levels of oxidative stress-induced DNA adducts like 8-hydroxydeoxyguanosine and 5-hydroxymethylcytosine is known to facilitate DNA hypo-methylation by preventing methylation of the target cytosine by DNA methyltransferase 1 [37, 38], or by significantly inhibiting protein binding to the methylated CpG islands [36]. Third, activation of base excision repair, especially long-patch base excision DNA repair, which plays a predominant role in the removal of oxidative stress-induced DNA lesions and was shown to be induced following long-term treatment with peroxisome proliferators [39] could also lead to genome-wide hypomethylation.
We observed no effect on c-Myc promoter methylation with continuous exposure for 1 week or more, in contrast to the effects of a single dose of WY-14,643 [11]. Thus, alterations in the genome methylation patterns with continuous exposure to non-genotoxic liver carcinogens, such as WY-14,643, may not be confined to specific cell proliferation-related genes. It has been shown that genome-wide DNA hypomethylation in cancer, including liver cancer, largely affects repetitive DNA elements [28, 40, 41]. Additionally, recent evidence indicates that hypomethylation of retroelements occurs even in precancerous stages of development of human hepatocellular carcinoma [41, 42], as well as at early stages of non-genotoxic and genotoxic rodent hepatocarcinogenesis [43, 44]. Loss of cytosine methylation at repetitive DNA sequences is associated with increased transcription of these elements. In view of this, our observation of hypomethylation of LTR IAP, LINE1 and LINE2 retrotrasposons following long-term exposure to WY-14,643 may be an evidence of their reactivation, which in turn would have an effect on the stability of the genome. Furthermore, emerging evidence suggests a crucial role of histone H3 lysine 9 and H4 lysine 20 trimethylation in the maintenance of genomic stability [30]. One of the primary functions of H3K9me3 and H4K20me3 is the formation of constitutive heterochromatin and trimethylation of H3K9 and H4K20 is considered a prominent epigenetic mark of silenced heterochromatin. Major and minor satellites and LINE1 retrotransposons in mouse genome are predominantly enriched with H3K9me3 and H4K20me3, and trimethylation of H4K20 is the sole prominent mark for LTR IAP [23]. Loss of H3K9 and H4K20 trimethylation is known to be associated with activation of LTR IAP and LINE1 transcription [23], increased genomic and chromosomal instability, and increased tumor incidence [40, 45]. Additionally, loss of H4K20me3 compromises the ability of the DNA damage checkpoint control [46], and loss of H3K9me3 compromises the balance between cell proliferation and differentiation processes favoring cell proliferation [47]. In addition, considering the critical role of histone methyltransferases in the preservation of chromatin structure [30], our observations of the retained expression of Suv39h1 and increased levels of PRDM/Riz protein under conditions of the loss in histone H3K9 and H4K20 trimethylation and diminished Suv4−20h2 expression may be interpreted as a cellular defense mechanism for maintenance of heterochromatin organization and liver cell viability.
In conclusion, WY-14,643-induced loss of global and repeat-associated DNA hypomethylation, loss of H3K9 and H4K20 trimethylation, and diminished expression of Suv4−20h2 histone methyltransferase in liver of wild type mice may be evidence of compromised genomic integrity via chromatin decondensation, activation of mobile repetitive DNA elements resulting in a variety of genomic instability events including cis- and trans-insertional mutagenesis, unequal homologous recombination, rearrangements, and segmental duplications leading to deletions and duplications [48]. The causal role of these lesions as an integral part of neoplastic transformation in the etiology of cancer is now commonly accepted [49]. Furthermore, lack of effect of WY-14,643 on DNA and histone methylation in Pparα-null mice suggests that methylation changes are dependent upon PPAPα-mediated events in rodent liver, such as cell proliferation and oxidative stress to DNA. Since it is widely believed that humans are not at risk for PPARα-mediated liver cancer [8] this study provides important information that distinguishes the relevance and the potential role of epigenetic factors in the mechanism of action of peroxisome proliferators in mice, species which are susceptible to hepatocarcinogenesis by these agents, and humans.
Acknowledgments
Grant Support: This study was supported, in part, by National Institutes of Health grants ES12686, ES11391, ES11660 and ES13342.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Note: The views expressed in this paper do not necessarily represent those of the U.S. Food and Drug Administration.
Reference List
- 1.Jaffe LF. Epigenetic theories of cancer initiation. Adv. Cancer Res. 2003;90:209–230. doi: 10.1016/s0065-230x(03)90007-8. [DOI] [PubMed] [Google Scholar]
- 2.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat. Rev Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 3.Shuker DE. The enemy at the gates? DNA adducts as biomarkers of exposure to exogenous and endogenous genotoxic agents. Toxicol Lett. 2002;134:51–56. doi: 10.1016/s0378-4274(02)00162-5. [DOI] [PubMed] [Google Scholar]
- 4.Silva LB, Van der Laan JW. Mechanisms of nongenotoxic carcinogenesis and assessment of the human hazard. Regul. Toxicol Pharmacol. 2000;32:135–143. doi: 10.1006/rtph.2000.1427. [DOI] [PubMed] [Google Scholar]
- 5.Peraza MA, Burdick AD, Marin HE, Gonzalez FJ, Peters JM. The toxicology of ligands for peroxisome proliferator-activated receptors (PPAR) Toxicol Sci. 2006;90:269–295. doi: 10.1093/toxsci/kfj062. [DOI] [PubMed] [Google Scholar]
- 6.Reddy JK, Rao MS, Azarnoff DL, Sell S. Mitogenic and carcinogenic effects of a hypolipidemic peroxisome proliferator, [4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio]acetic acid (Wy-14, 643), in rat and mouse liver. Cancer Res. 1979;39:152–161. [PubMed] [Google Scholar]
- 7.Rusyn I, Peters JM, Cunningham ML. Modes of action and species-specific effects of di-(2-ethylhexyl)phthalate in the liver. Crit Rev. Toxicol. 2006;36:459–479. doi: 10.1080/10408440600779065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klaunig JE, Babich MA, Baetcke KP, Cook JC, Corton JC, David RM, Deluca JG, Lai DY, McKee RH, Peters JM, Roberts RA, Fenner-Crisp PA. PPARalpha agonist-induced rodent tumors: modes of action and human relevance. Crit Rev. Toxicol. 2003;33:655–780. doi: 10.1080/713608372. [DOI] [PubMed] [Google Scholar]
- 9.Peters JM, Cattley RC, Gonzalez FJ. Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis. 1997;18:2029–2033. doi: 10.1093/carcin/18.11.2029. [DOI] [PubMed] [Google Scholar]
- 10.Peters JM, Cheung C, Gonzalez FJ. Peroxisome proliferator-activated receptor-alpha and liver cancer: where do we stand? J Mol. Med. 2005;83:774–785. doi: 10.1007/s00109-005-0678-9. [DOI] [PubMed] [Google Scholar]
- 11.Ge R, Wang W, Kramer PM, Yang S, Tao L, Pereira MA. Wy-14,643-induced hypomethylation of the c-myc gene in mouse liver. Toxicol Sci. 2001;62:28–35. doi: 10.1093/toxsci/62.1.28. [DOI] [PubMed] [Google Scholar]
- 12.Tao L, Wang W, Li L, Kramer PK, Pereira MA. DNA hypomethylation induced by drinking water disinfection by-products in mouse and rat kidney. Toxicol Sci. 2005;87:344–352. doi: 10.1093/toxsci/kfi257. [DOI] [PubMed] [Google Scholar]
- 13.Bachman AN, Phillips JM, Goodman JI. Phenobarbital induces progressive patterns of GC-rich and gene-specific altered DNA methylation in the liver of tumor-prone B6C3F1 mice. Toxicol Sci. 2006;91:393–405. doi: 10.1093/toxsci/kfj155. [DOI] [PubMed] [Google Scholar]
- 14.Pogribny IP, Ross SA, Wise C, Pogribna M, Jones EA, Tryndyak VP, James SJ, Dragan YP, Poirier LA. Irreversible global DNA hypomethylation as a key step in hepatocarcinogenesis induced by dietary methyl deficiency. Mutat. Res. 2006;593:80–87. doi: 10.1016/j.mrfmmm.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 15.Pogribny IP, Tryndyak VP, Muskhelishvili L, Rusyn I, Ross SA. Methyl deficiency, alterations in global histone modifications, and carcinogenesis. J Nutr. 2007;137:216S–222S. doi: 10.1093/jn/137.1.216S. [DOI] [PubMed] [Google Scholar]
- 16.Yamada Y, Jackson-Grusby L, Linhart H, Meissner A, Eden A, Lin H, Jaenisch R. Opposing effects of DNA hypomethylation on intestinal and liver carcinogenesis. Proc. Natl. Acad Sci U. S. A. 2005;102:13580–13585. doi: 10.1073/pnas.0506612102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodman JI, Watson RE. Altered DNA methylation: a secondary mechanism involved in carcinogenesis. Annu Rev Pharmacol Toxicol. 2002;42:501–525. doi: 10.1146/annurev.pharmtox.42.092001.141143. [DOI] [PubMed] [Google Scholar]
- 18.Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol. Cell Biol. 1995;15:3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pogribny I, Yi P, James SJ. A sensitive new method for rapid detection of abnormal methylation patterns in global DNA and within CpG islands. Biochem Biophys. Res Commun. 1999;262:624–628. doi: 10.1006/bbrc.1999.1187. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalgo ML, Liang G, Spruck CH, III, Zingg JM, Rideout WM, III, Jones PA. Identification and characterization of differentially methylated regions of genomic DNA by methylation-sensitive arbitrarily primed PCR. Cancer Res. 1997;57:594–599. [PubMed] [Google Scholar]
- 21.Watson RE, Curtin GM, Doolittle DJ, Goodman JI. Progressive alterations in global and GC-rich DNA methylation during tumorigenesis. Toxicol Sci. 2003;75:289–299. doi: 10.1093/toxsci/kfg190. [DOI] [PubMed] [Google Scholar]
- 22.Tryndyak V, Kovalchuk O, Pogribny IP. Identification of differentially methylated sites within unmethylated DNA domains in normal and cancer cells. Anal Biochem. 2006;356:202–207. doi: 10.1016/j.ab.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 23.Martens JH, O'Sullivan RJ, Braunschweig U, Opravil S, Radolf M, Steinlein P, Jenuwein T. The profile of repeat-associated histone lysine methylation states in the mouse epigenome. EMBO J. 2005;24:800–812. doi: 10.1038/sj.emboj.7600545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25:2532–2534. doi: 10.1093/nar/25.12.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tryndyak VP, Muskhelishvili L, Kovalchuk O, Rodriguez-Juarez R, Montgomery B, Churchwell MI, Ross SA, Beland FA, Pogribny IP. Effect of long-term tamoxifen exposure on genotoxic and epigenetic changes in rat liver: implications for tamoxifen-induced hepatocarcinogenesis. Carcinogenesis. 2006;27:1713–1720. doi: 10.1093/carcin/bgl050. [DOI] [PubMed] [Google Scholar]
- 26.Woods CG, Burns AM, Bradford BU, Ross PK, Kosyk O, Swenberg JA, Cunningham ML, Rusyn I. WY-14,643-induced cell proliferation and oxidative stress in mouse liver are independent of NADPH oxidase. Toxicol. Sci. In Press. 2007 doi: 10.1093/toxsci/kfm104. [DOI] [PubMed] [Google Scholar]
- 27.Rollins RA, Haghighi F, Edwards JR, Das R, Zhang MQ, Ju J, Bestor TH. Large-scale structure of genomic methylation patterns. Genome Res. 2006;16:157–163. doi: 10.1101/gr.4362006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schulz WA, Steinhoff C, Florl AR. Methylation of endogenous human retroelements in health and disease. Curr. Top. Microbiol. Immunol. 2006;310:211–250. doi: 10.1007/3-540-31181-5_11. [DOI] [PubMed] [Google Scholar]
- 29.Santos-Rosa H, Caldas C. Chromatin modifier enzymes, the histone code and cancer. Eur. J Cancer. 2005;41:2381–2402. doi: 10.1016/j.ejca.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 30.Kourmouli N, Jeppesen P, Mahadevhaiah S, Burgoyne P, Wu R, Gilbert DM, Bongiorni S, Prantera G, Fanti L, Pimpinelli S, Shi W, Fundele R, Singh PB. Heterochromatin and tri-methylated lysine 20 of histone H4 in animals. J Cell Sci. 2004;117:2491–2501. doi: 10.1242/jcs.01238. [DOI] [PubMed] [Google Scholar]
- 31.Xie W, Radominska-Pandya A, Shi Y, Simon CM, Nelson MC, Ong ES, Waxman DJ, Evans RM. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc. Natl. Acad Sci U. S. A. 2001;98:3375–3380. doi: 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ehrlich M. Cancer-linked DNA hypomethylation and its relationship to hypermethylation. Curr. Top. Microbiol. Immunol. 2006;310:251–274. doi: 10.1007/3-540-31181-5_12. [DOI] [PubMed] [Google Scholar]
- 33.Liang G, Chan MF, Tomigahara Y, Tsai YC, Gonzales FA, Li E, Laird PW, Jones PA. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol. Cell Biol. 2002;22:480–491. doi: 10.1128/MCB.22.2.480-491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rusyn I, Rose ML, Bojes HK, Thurman RG. Novel role of oxidants in the molecular mechanism of action of peroxisome proliferators. Antiox Redox Signal. 2000;2:607–621. doi: 10.1089/15230860050192350. [DOI] [PubMed] [Google Scholar]
- 35.O'Brien ML, Spear BT, Glauert HP. Role of oxidative stress in peroxisome proliferator-mediated carcinogenesis. Crit Rev Toxicol. 2005;35:61–88. doi: 10.1080/10408440590905957. [DOI] [PubMed] [Google Scholar]
- 36.Valinluck V, Tsai HH, Rogstad DK, Burdzy A, Bird A, Sowers LC. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2) Nucleic Acids Res. 2004;32:4100–4108. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cerda S, Weitzman SA. Influence of oxygen radical injury on DNA methylation. Mutat. Res. 1997;386:141–152. doi: 10.1016/s1383-5742(96)00050-6. [DOI] [PubMed] [Google Scholar]
- 38.Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67:946–950. doi: 10.1158/0008-5472.CAN-06-3123. [DOI] [PubMed] [Google Scholar]
- 39.Rusyn I, Denissenko MF, Wong VA, Butterworth BE, Cunningham ML, Upton PB, Thurman RG, Swenberg JA. Expression of base excision repair enzymes in rat and mouse liver is induced by peroxisome proliferators and is dependent upon carcinogenic potency. Carcinogenesis. 2000;21:2141–2145. doi: 10.1093/carcin/21.12.2141. [DOI] [PubMed] [Google Scholar]
- 40.Peters AH, O'Carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A, Opravil S, Doyle M, Sibilia M, Jenuwein T. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107:323–337. doi: 10.1016/s0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- 41.Chalitchagorn K, Shuangshoti S, Hourpai N, Kongruttanachok N, Tangkijvanich P, Thong-ngam D, Voravud N, Sriuranpong V, Mutirangura A. Distinctive pattern of LINE-1 methylation level in normal tissues and the association with carcinogenesis. Oncogene. 2004;23:8841–8846. doi: 10.1038/sj.onc.1208137. [DOI] [PubMed] [Google Scholar]
- 42.Saito Y, Kanai Y, Sakamoto M, Saito H, Ishii H, Hirohashi S. Overexpression of a splice variant of DNA methyltransferase 3b, DNMT3b4, associated with DNA hypomethylation on pericentromeric satellite regions during human hepatocarcinogenesis. Proc. Natl. Acad Sci U. S. A. 2002;99:10060–10065. doi: 10.1073/pnas.152121799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Asada K, Kotake Y, Asada R, Saunders D, Broyles RH, Towner RA, Fukui H, Floyd RA. LINE-1 Hypomethylation in a Choline-Deficiency-Induced Liver Cancer in Rats: Dependence on Feeding Period. J Biomed Biotechnol. 2006;2006:17142-. doi: 10.1155/JBB/2006/17142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tryndyak VP, Kovalchuk O, Muskhelishvili L, Montgomery B, Rodriguez-Juarez R, Melnyk S, Ross SA, Beland FA, Pogribny IP. Epigenetic reprogramming of liver cells in tamoxifen-induced rat hepatocarcinogenesis. Mol. Carcinog. 2007;46:187–197. doi: 10.1002/mc.20263. [DOI] [PubMed] [Google Scholar]
- 45.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 46.Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, Kouzarides T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119:603–614. doi: 10.1016/j.cell.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 47.Ait-Si-Ali S, Guasconi V, Fritsch L, Yahi H, Sekhri R, Naguibneva I, Robin P, Cabon F, Polesskaya A, Harel-Bellan A. A Suv39h-dependent mechanism for silencing S-phase genes in differentiating but not in cycling cells. EMBO J. 2004;23:605–615. doi: 10.1038/sj.emboj.7600074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kazazian HH., Jr. Mobile elements: drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- 49.Coleman WB, Tsongalis GJ. Molecular mechanisms of human carcinogenesis. EXS. 2006:321–349. doi: 10.1007/3-7643-7378-4_14. [DOI] [PubMed] [Google Scholar]