

Abstract
Human serum albumin (HSA) is used as a resuscitation fluid in sepsis. This study investigated the potential protective properties of HSA on vascular function in a mouse endotoxic model in terms of oxidative and nitrosative stresses. Swiss mice were treated with either lipopolysaccharide (LPS) (50 mg/kg i.p.) or vehicle. One and five hours later, mice were infused with HSA (4%, 10 ml/kg), normal saline (0.9% NaCl, 30 ml/kg), or no fluid. Six hours after treatment, vascular reactivity was assessed on aortae and small mesenteric arteries. Measurements of NO and superoxide anion (O2−) by spin trapping and nuclear factor (NF)-κB, inducible NO synthase (iNOS), and peroxynitrite by Western blotting and immunohistochemical studies were conducted. HSA partially prevented the reduction of blood pressure induced by LPS and completely prevented both vascular hyporeactivity to phenylephrine and myogenic tone as well as endothelial dysfunction induced by the endotoxin. This was associated with a decreased up-regulation of NF-κ B, iNOS, and peroxynitrite in the vascular wall. LPS-induced tissue increases in both NO and O2− production was decreased by HSA. These data demonstrate the protective effect of HSA treatment in experimental endotoxic shock by reducing the inflammatory process leading to oxidative and nitrosative stresses and vascular hyporeactivity.
During severe sepsis, the cardiovascular system adopts a high cardiac output state with a low peripheral resistance hemodynamic profile that includes arterial dilatation.1,2 A prolonged or excessive drop of the peripheral resistance may cause progressive hypotension that is refractory to catecholamines, contributing to life-threatening cardiovascular failure.3,4 A release of reactive oxygen species by different pathways contributes to the failure of organs such as the lung, heart, brain, and liver.5,6 Data collected from clinical studies and different models of endotoxemia suggest that this phenomenon is related to the activation of the NF-κB/RelA pathway, enabling the expression of several specific genes involved in the pathogenesis of septic shock: cytokines, adhesion molecules, cyclooxygenase-2, and inducible nitric oxide synthase (iNOS).7,8 An induction of iNOS and an overproduction of nitric oxide (NO) play a major role in endotoxemia-induced vascular hyporeactivity in different experimental models9 as well as in the small vessels of patients with septic shock.10
Human serum albumin (HSA) is a multifunctional plasma protein, with ascribed ligand-binding, transport properties and enzymatic activities.11,12 HSA is involved in the control of oncotic pressure, the modulation of inflammatory pathways,13 and integrity of the microvasculature.14 Clinically, HSA has been used as a plasma expander in critically ill patients. However, evidence from meta-analysis and the recently published Saline versus Albumin Fluid Evaluation Study15 does not support a survival benefit of HSA compared to crystalloid solutions. Nevertheless, the Sepsis Occurrence in Acutely Ill Patients Study16 suggests that treatment with HSA reduces mortality and benefits survival in septic patients with hypoalbuminemia. Among the mechanisms explaining the protective effect of HSA, a modulation of cellular reduced glutathione as well as transcription of inflammatory genes and apoptosis have been reported.13,17 To our knowledge, a possible protective effect of HSA treatment against vascular disturbances and up-regulation of both NO and superoxide anion (O2−) production after endotoxic shock have not been investigated. This study investigated the potential protective anti-inflammatory and anti-oxidant properties of HSA on vascular dysfunction in an endotoxic model induced by the bacterial lipopolysaccharide (LPS).
Materials and Methods
Animals and Protocols: Experiments
All experiments were in accordance with the ethical regulations approved by the regional ethics committee for animal experimentation “CREMEAS” (Strasbourg, France). Male Swiss mice (35 to 40 g) were used. LPS (from Escherichia coli 055:B5; Sigma Aldrich, Saint Quentin Fallavier, France) was administered at a dose of 50 mg/kg i.p. Control mice received an equivalent volume of vehicle (0.9% NaCl solution) (sham group). One and five hours later, mice given LPS were randomized for intravenous resuscitation by using 10 ml/kg of 4% HSA (Vialebex; LFB, Paris, France), 30 ml/kg 0.9% saline, or no fluid resuscitation. Fluid resuscitation was infused intravenously via the tail vein under a lightly inhaled anesthetic (isoflurane 2%). Six hours after the intraperitoneal LPS injection, mice were anesthetized by intraperitoneal injection with the mixture of ketamine (100 mg/kg), medetomidine (50 μg/kg), and heparin (500 U/kg). The aorta, small mesenteric arteries (SMAs), heart, and lungs were harvested for different experiments.
We measured serum albumin, chloride, and total CO2 (as a surrogate of acid-base status) (Roche/Hitachi Modular P 800; Roche Diagnostics, Meylan, France) under basal conditions and after 6 hours of LPS in mice receiving either saline or HSA (n = 6 to 7 for each group of mice). LPS treatment did not significantly affect native albumin concentrations in the mouse groups (24 ± 0.9 g/L and 25 ± 0.8 g/L for control and LPS-treated mice, respectively). The plasma level of albumin was not modified by saline (25 ± 1.1 g/L). Although, the colorimetric quantitative determination of serum albumin did not allow differentiation between murine albumin and HSA, levels of total serum albumin were significantly enhanced after HSA treatment (32 ± 0.6 g/L). Furthermore, neither LPS treatment nor saline resuscitation affected plasma chloride levels (indirect potentiometry) (104 ± 0.6 mmol/L in control mice, 110 ± 0.5 mmol/L in LPS-treated mice, 102 ± 1.6 mmol/L in HSA treated mice, and 108 ± 1 mmol/L in saline resuscitation mice). Thus, the load of saline in the control group did not induce hyperchloremia under the experimental conditions used. As expected, LPS treatment reduced total CO2 level (23 ± 0.4 control mice versus 19 ± 0.7 mmol/L LPS-treated mice, *P < 0.05), as measured by enzymatic method. Such a reduction was not observed after either saline or HSA, being 21 ± 0.4 mmol/L and 22 ± 0.8 mmol/L, respectively.
Continuous Measurements of Blood Pressure and Heart Rate by Telemetry
Mean blood pressure and heart rate were recorded using telemetry (Data Sciences, St. Paul, MN). This system consists of implantable radio frequency transmitters (TA11-PA20, 3.4 g) and a receiver placed under the holding cage of each animal. For the surgical procedure, mice were anesthetized with ketamine (100 mg/kg, i.p.) mixed with medetomidine (50 μg/kg, i.p.). The left carotid artery was isolated, the catheter linked to the transmitter was inserted into the carotid artery, and sutures were tied around the catheter and the artery. The body of the transmitter was placed into a subcutaneous pocket along the animal’s right flank. The skin incisions were then closed. Animals were kept under warming lights until they recovered. Animals were returned to their cages and allowed to recover from the surgery for 1 week. For data acquisition and analysis, mean blood pressure and heart rate were recorded for 30 seconds every 15 minutes on a daily basis. Data were averaged for each mouse.
Vascular Reactivity
Aortic Rings
Vascular reactivity of aortic rings was studied on a wire myograph (EMKA Technologies, Paris, France). The experiments were performed at 37°C in a physiological salt solution with the following composition: 119 mmol/L NaCl, 4.7 mmol/L KCl ,14.9 mmol/L NaHCO3, 1.2 mmol/L MgSO4.7H2O, 2.5 mmol/L CaCl2, 1.18 mmol/L KH2PO4, and 5.5 mmol/L glucose, continuously bubbled with 95% O2 and 5% CO2. After an equilibration period (at least 20 minutes) under the optimal passive tension, two successive contractions in response to the combination of KCl depolarization (100 mmol/L) and 10 μmol/L phenylephrine (Pe) (Sigma-Aldrich, Saint Quentin Falavier, France) were used to test the maximal contractile capacity of the vessels. After washing, endothelial function was assessed by testing the relaxing effect of acetylcholine (Ach 1 μmol/L) after precontraction by 1 μmol/L Pe. After a 20-minute washout period, concentration-response curves to Pe were elicited by cumulative administration of the vasoconstrictor agonist (1 nmol/L to 100 μmol/L) to vessels with endothelium in presence or absence of the NO-synthase inhibitor, NG-l-nitroarginine-methyl-ester (L-NAME, 100 μmol/L; Sigma-Aldrich). The inhibitor was added in the bath 30 minutes before the addition of Pe.
Isolated Mesenteric Arteries
We assessed myogenic tone in SMAs (≈130 to 160 μm diameter). Mesenteric arteries were cannulated at both ends in a video-monitored perfusion system (Living Systems Instrumentation, Burlington, VT). Arteries were bathed in physiological salt solution (pH 7.4; PO2, 160 mm Hg; PCO2, 37 mm Hg). Pressure was controlled by a servoperfusion system, and flow was generated by a peristaltic pump. Initially, diameter changes were measured by increasing the luminal pressure from 10 to 125 mmHg.
The presence of a functional endothelium was assessed by applying 10 μmol/L Ach in arteries precontracted with 1 μmol/L U46619. At the end of each experiment, to determine the passive diameter of the artery and to evaluate myogenic tone, arteries were perfused and superfused with a Ca2+-free physiological salt solution containing 2 mmol/L EGTA and 10 μmol/L sodium nitroprusside and pressure steps repeated. Myogenic tone was calculated at each pressure as follows: myogenic tone = passive diameter minus active diameter. In another set of experiments, the same protocol was used in presence of 100 μmol/L L-NAME.
Staining and Imaging by Confocal Microscopy
Vessels were frozen and cut into 10-μm sections. Fixed sections were incubated (2 hours at room temperature) in a blocking buffer (5% nonfat dry milk in phosphate-buffered saline) and then incubated overnight (4°C) with a monoclonal murine anti-iNOS antibody (1:50; Transduction Laboratories, Heidelberg, Germany) for iNOS immunostaining. A polyclonal NF-κB p65 antibody (1:100; Abcam, Cambridge, UK) or a mouse monoclonal anti-nitrotyrosine (clone 1A6) antibody (1:100; Upstate, Hampshire, UK) was used for NF-κB p65 or nitrotyrosine immunostaining, respectively. After three washes, the samples were incubated (1 hour, 37°C) with the secondary murine or rabbit fluorescence-labeled antibody Alexa Fluor 488 (1:100; Invitrogen Molecular Probes, Leiden, The Netherlands). After washing, vessel sections were mounted on glass slides. An Nikon Eclipse TE 300 (Nikon, Tokyo, Japan) inverted microscope with a MRC-1024ES scanner was used for the optical sectioning of tissues. Digital image records were performed using the LaseSharp Software. The Confocal Assistant (version 4.0.2) software was used for image analysis (TC Brelje, Department of Cell Biology and Neuroanatomy, University of Minnesota, Minneapolis, MN) and was expressed as a percentage (intensity of the studied sample versus intensity of control sample).
Western Blotting
Lung tissue samples were crushed and homogenized and ∼50 μg of total protein from the supernatant fraction was loaded onto 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Blots were probed with a monoclonal mouse anti-iNOS (Transduction Laboratories). Proteins were transferred to nitrocellulose membranes and probed with a monoclonal mouse anti-α-actin antibody (Sigma-Aldrich). Bound antibodies were detected with a secondary peroxidase-conjugated anti-mouse IgG (Promega, Madison, WI). The blots were visualized using an enhanced chemiluminescence system (ECL Plus; Amersham, Buckinghamshire, UK).
Electronic Paramagnetic Resonance (EPR) Studies
NO Spin Trapping
After 5.5 hours, N,N-diethyldithiocarbamate (DETC) and Fe2+-citrate complex were separately injected into mice at the following doses: 400 mg/kg DETC (intraperitoneally) and 40 mg/kg FeSO4.7H2O in a solution containing 200 mg/kg of sodium citrate (s.c.). The animals were anesthetized and sacrificed 30 minutes later. Tissues (aorta, heart, and lung) and blood samples were harvested and flash-frozen in liquid nitrogen for subsequent EPR assay. These studies were performed on a tabletop x-band spectrometer miniscope (MS200; Magnettech, Berlin, Germany). Recordings were made at 77°K using a Dewar flask. The instrument settings were 10 mW of microwave power, 1 mT of amplitude modulation, 100 kHz of modulation frequency, 60 seconds of sweep time, and 5 numbers of scans.18 Levels of NO were expressed as amplitude of signal in unit per weight of dried sample.
Superoxide Anion Spin Trapping
Mice were sacrificed, and selected organs were dissected and placed in cold buffer. Freshly isolated samples were allowed to equilibrate in deferoxamine-chelated Krebs-HEPES solution containing 500 μmol/L 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidin (CMH), 25 μmol/L deferoxamine, and 5 μmol/L DETC at 37°C for 60 minutes. The reaction was stopped by placing samples on ice and then freezing in liquid nitrogen and analyzing in a Dewar flask by EPR spectroscopy. The instrument settings were as follows: temperature, 77° K; microwave power, 1 mW; amplitude modulation, 0.5 mT; sweep time, 60 seconds; field sweep, 60 G.
Data Analysis
Data obtained during measurements of arterial pressure and heart rate were compared using a nonparametric Wilcoxon signed rank test. Two-way analysis of variance for repeated measurements was used for comparison of vascular reactivity (data were tested for homogeneity of variance by Levene’s statistics), and nonparametric Kruskal-Wallis test or Mann-Whitney test were used for comparison Western blotting, NO, superoxide, and immunostaining signal measurements between the four groups. When a significant difference was found between groups, subsequent post hoc tests were performed. All these tests were performed with the Statview version 5.0 software (SAS Institute, Cary, NC). P < 0.05 was considered statistically significant. The effects of Ach were quantified as the percentage of relaxation in preparations previously contracted with Pe. All values are presented as mean ± SEM for n experiments. n represents the number of animals.
Results
Mean Arterial Pressure and Heart Rate in Mice
The MAP (Figure 1A) and heart rate were similar in all mouse groups before experimentation. In the sham group, MAP remained stable during the 6 hours of measurement. LPS treatment induced a significant decrease in MAP (P < 0.05). After LPS injection, saline and HSA partially reversed MAP to similar extents (P < 0.05) but remained low compared to the MAP of sham group. Compared to sham (431 ± 9 beats/minute), LPS raised heart rate (492 ± 11 beats/minute) (P < 0.05, n = 6 to 7). Saline and HSA did not alter heart rate (476 ± 10 and 481 ± 12 beats/minute).
Figure 1.

Telemetric blood pressure recordings. Mean arterial blood pressure (MAP) in sham group (•), HSA group (▪), saline group (▴), and no fluid resuscitation group (○) mice recorded during an 8-hour monitoring period. Data are expressed as means ± SE of n = 7 mice for each group. *P < 0.05, significantly different in sham group versus no fluid resuscitation group, HSA group versus no fluid resuscitation group, or saline group versus no fluid resuscitation group.
HSA Prevents Vascular Hyporeactivity and Improves Endothelial Dysfunction Induced by LPS
Aortae
Pe produced a concentration-dependent increase in contraction of intact aortic rings (Figure 2A). Compared to the sham group, the vascular responses to Pe were significantly decreased in aortae from LPS-treated mice (P < 0.01). Hence, LPS-induced hyporeactivity was not modified in the aorta harvested from animals resuscitated with saline (P < 0.01). Conversely, HSA prevented the vascular hyporeactivity elicited by LPS. Concerning endothelial function, Ach produced relaxation in a concentration-dependent manner (Figure 2B). Ach response was significantly reduced after LPS treatment (P < 0.05). Interestingly, HSA, but not saline resuscitation, improved relaxation to Ach (P < 0.05).
Figure 2.

HSA improves vascular contraction to Pe, myogenic tone, and endothelial function. A: Concentration-effect curves of aortae in sham group (•), HSA group (▪), saline group (▴), and no fluid resuscitation group (○), n = 5 for each group, exposed to Pe (3 nmol/L to 30 μmol/L). **P < 0.01, significantly different between HSA group and no fluid resuscitation group. Please note that there was no difference in contraction for Pe between aortae from HSA group and sham group. B: Concentration-effect curves of aortae harvested from sham group (•), HSA group (▪), saline group (▴), and no fluid resuscitation group (○) mice, n = 5 for each group of mice, exposed to Ach (1 nmol/L to 1 μmol/L). *P < 0.05, significantly different between HSA group and no fluid resuscitation group. Please note that there was no difference in percentage of relaxation to acetylcholine between aortae from HSA group and sham group. C: Diameter changes in response to increasing steps of pressure (myogenic tone) on mesenteric arteries isolated from sham group (•), HSA group (▪), saline group (▴), and no fluid resuscitation group (○) mice, n = 5 or 6 for each group of animals. **P < 0.01, significantly different between HSA group and no fluid resuscitation group. Please note that there was a difference in myogenic tone between aortae from HSA group and sham group.
SMAs
Compared to the sham group (70 ± 11 μm, n = 8), the contractile response, expressed as reduction of vessel diameter, to KCl depolarization was significantly decreased in SMAs taken from LPS-treated mice (29 ± 6 μm, n = 7) (P < 0.01). HSA (49 ± 9 μm, n = 8) but not saline (32 ± 7 μm, n = 6) partially prevented the hyporeactivity to KCl depolarization to the level of sham group (P < 0.01).
Compared to SMAs from the sham group, the contractile response to Pe was significantly decreased in SMAs from LPS-treated mice alone or after saline (31 ± 5 μm and 44 ± 6 μm versus 106 ± 13 μm) (P < 0.01). After HSA treatment, the hyporeactivity induced by LPS was partially prevented, and the contractile response was not significantly different to the sham group (70 ± 11 μm, n = 8). Myogenic tone, which was normalized to the corresponding passive arterial diameter, was significantly decreased in mice treated with LPS (P < 0.01, compared to sham). Most importantly, HSA but not saline resuscitation prevented the reduction of myogenic tone induced by LPS treatment (Figure 2C).
The Protective Effect of HSA on LPS-Induced Hyporeactivity Is Blunted by L-NAME
To investigate the mechanisms involved in the vascular hyporeactivity induced by LPS, the role of NO was evaluated by examining the effects of L-NAME on Pe-induced contractions. In the sham group, L-NAME did not modify either contraction to Pe (aorta) or myogenic tone (SMAs) (Figure 3, A and B). However, L-NAME abolished the LPS-induced hyporeactivity to Pe (P < 0.01) and myogenic tone (P < 0.01).
Figure 3.

HSA decreases NO-dependent vasorelaxation, NO release, and iNOS expression in aorta. A: Concentration-effect curves to Pe (3 nmol/L to 30 μmol/L) in absence (○, ▪) or in presence (♦, ⋄) of L-NAME (100 μmol/L) in mice aortae from no fluid resuscitation group (open symbols) and HSA group (filled symbols), respectively. B: Diameter change in response to increasing steps of pressure (myogenic tone) in the absence (○, ▪) or in the presence (♦, ⋄) of L-NAME (100 μmol/L) on mice mesenteric arteries isolated from no fluid resuscitation group (open symbols) and HSA group (filled symbols), respectively, **P < 0.01, significantly different between vessels from no fluid resuscitation group with and without the inhibitor. Please note that the inhibitor does not affect the contraction in vessels from HSA group. C: Quantification of the amplitude of NO-Fe(DETC)2 signal in unit/weight [mg of the dried sample A/W(ds), n = 6] in aorta from the four groups of mice. *P < 0.05, significantly different in sham group versus no fluid resuscitation group or in HSA group versus no fluid resuscitation group. D–F: Immunohistochemical staining for inducible NO synthase of aortae from sham group (D), no fluid resuscitation group (E), and HSA group mice (F). Green fluorescence was linked to secondary Alexa 488 anti-mouse-conjugated antibody. Scale bars = 150 μm.
HSA Prevents LPS-Dependent NO Overproduction and iNOS Expression in the Aorta
Compared to the sham group, LPS induced marked increase in NO production in the sham group (P < 0.05). HSA but not saline resuscitation prevented LPS-induced NO overproduction (P < 0.05) (Figure 3C). In agreement with these data, weak or no staining of iNOS was found in aortae from sham group (3.8 ± 0.3 arbitrary units) (Figure 3D), whereas LPS treatment induced marked iNOS labeling in the medial and adventitial layers of the aorta (32.3 ± 9.1 arbitrary units, P < 0.05) (Figure 3E). This increased iNOS staining was prevented by HSA treatment (6.4 ± 0.6 arbitrary units, P < 0.05) (Figure 3F). In separate experiments in cultured endothelial cells, we determined that LPS plus tumor necrosis factor-α enhanced NO through iNOS and O2− production; these effects were also prevented by HSA treatment (personal observations). The negative control obtained by incubation with a secondary murine fluorescent-labeled antibody did not display any staining (not shown).
HSA Prevents Tissue LPS-Induced NO Overproduction and Increase in iNOS Expression
Compared to the sham group, LPS induced significant increases in NO production as detected by the NO-Fe(DETC)2 EPR signal in the heart (P < 0.05) and lungs (P < 0.05) (Figure 4, A and B). Saline did not alter LPS-induced NO production. Conversely, HSA resuscitation reduced the increases in LPS-induced NO production in both tissues (P < 0.05). Western blot analysis confirmed a decrease in iNOS expression in the lungs of HAS-treated animals (Figure 4C).
Figure 4.
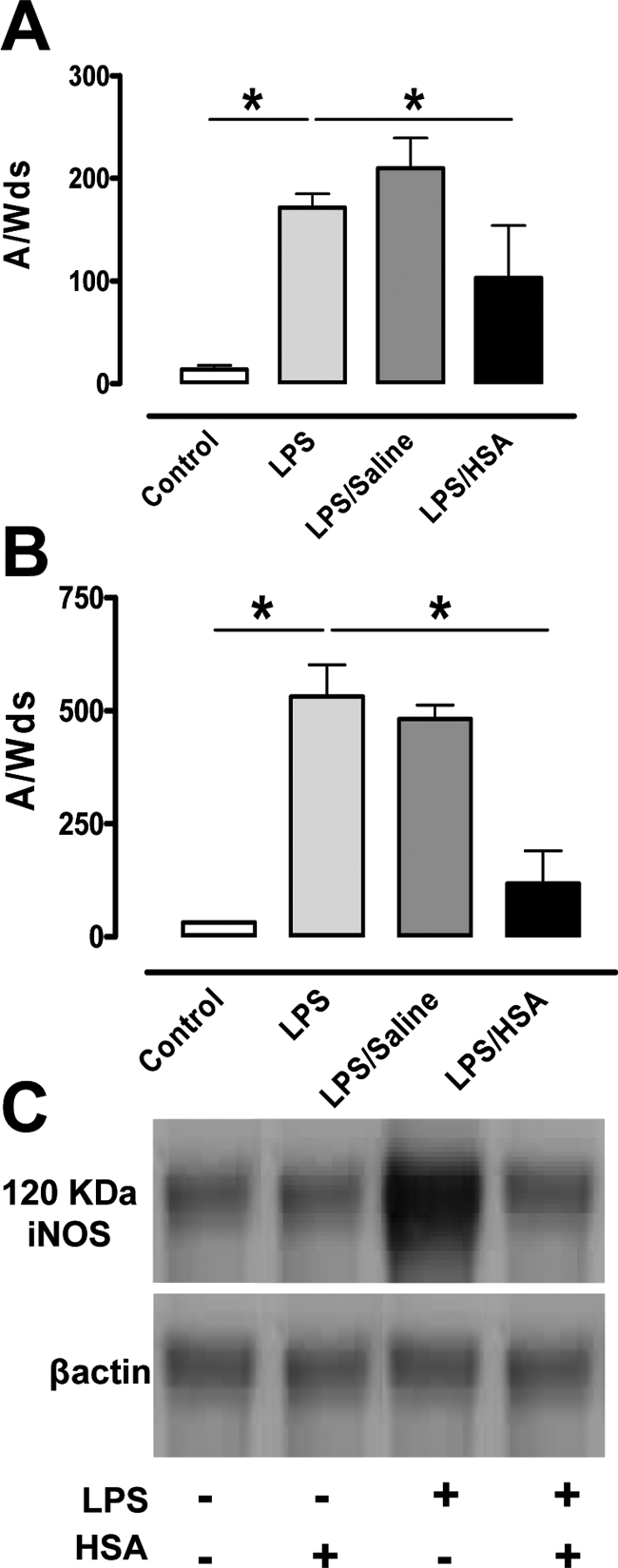
HSA reduces NO overproduction and iNOS expression in heart and lung. A and B: Quantification of the amplitude of NO-Fe(DETC)2 signal in unit/weight [mg of the dried sample A/W(ds), n = 6] in heart (A) and lung (B) from the four groups of mice. *P < 0.05, significantly different in sham group versus no fluid resuscitation group and in HSA group versus no fluid resuscitation group. C: Representative Western blots of three experiments, revealing reduced iNOS expression in lung from HSA group mice.
HSA Reduced the LPS-Induced Up-Regulation of NF-κB
The NF-κB heterodimer is co-localized in the cytoplasm with the inhibitory protein IκB. On cell stimulation, IκB is phosphorylated, removed, and degraded, allowing NF-κB to induce transcription. Immunohistochemical staining of the p65 subunit of NF-κB was used as an index of activation of NF-κΒ (Figure 5). No specific staining occurred in aortae from the sham group (4.2 ± 1.5 arbitrary units) (Figure 5A). Treatment with LPS induced marked staining of the p65/RelA subunit of NF-κB in the medial and in adventitial layers of the aorta (19.4 ± 3.1 arbitrary units, P < 0.05) (Figure 5B). This staining was almost abolished in aortic rings taken from LPS-treated mice receiving HSA (8 ± 1 arbitrary units) (Figure 5C). Negative controls obtained by incubation with the secondary rabbit fluorescence-labeled antibody did not display any staining (not shown).
Figure 5.

HSA reduces the activation of NF-κB RelA/p65 in aortic wall. A–C: Immunohistochemical staining for NF-κB p65 subunit in aortae from sham group (A), no fluid resuscitation group (B), and HSA group (C) of mice (n = 3). Green fluorescence was linked with secondary Alexa 488 anti-rabbit-conjugated antibody. The corresponding phase contrast pictures are also shown (D–F). Scale bars = 150 μm.
HSA Reduced LPS-Induced Oxidative and Nitrosative Stresses
To investigate the role of LPS in oxidative stress, we evaluated the tissue production of O2− by EPR spin trapping. LPS treatment induced an increase in O2− levels in aorta, heart, and lung (P < 0.05) (Figure 6, A–C). Saline treatment did not modify the LPS-enhanced O2− production (P < 0.05), whereas HSA attenuated the oxidative stress induced by LPS as the O2− levels were similar to those in the sham group. It is known that NO can react with O2− leading to the formation of peroxynitrite anion (ONOO−). Weak or no staining of ONOO− was found in aortae from sham group (2.8 ± 0.3 arbitrary units) (Figure 6D). LPS treatment induced a marked increase in ONOO− labeling in the medial layer (12.2 ± 1.2 arbitrary units, P < 0.05) (Figure 6E), and HSA suppressed ONOO− staining (4.6 ± 0.7 arbitrary units) (Figure 6F). The negative control obtained by incubation with the secondary murine fluorescent-labeled antibody did not display any staining (not shown).
Figure 6.

HSA reduces LPS-induced oxidative stress. A–C: Quantification of the amplitude of O2−-CMH signal in unit/weight [mg] of the dried sample A/W(ds), n = 6] in aorta (A), heart (B), and lung (C) from the four groups of mice. *P < 0.05, significantly different in sham group versus no fluid resuscitation group and in HSA group versus no fluid resuscitation group. D–F: Immunohistochemical staining for nitrotyrosine of aortae from sham group (D), no fluid resuscitation group (E), and HSA group (F) (n = 3). Green fluorescence was linked with secondary Alexa 488 anti-mouse-conjugated antibody. Scale bars = 150 μm.
Discussion
In this study, we provide evidence for a protective role of HSA on vascular function during LPS-induced shock. HSA improves LPS-induced endothelial dysfunction and reduces LPS-induced vascular hyporeactivity. This effect was associated with a reduced iNOS expression in the vascular wall as well as reduced NO production in aorta and organs such as lung and heart. Moreover, HSA reduced in situ activation of the transcriptional factor NF-κB, thus reducing oxidative and nitrosative stresses in aorta, heart, and lung.
Prompt restoration of intravascular volume is an early goal in the treatment of patients with septic shock, and although the use of crystalloids or colloids for volume replacement in critically ill patients is still debated, both fluids are widely used, and both reduce mortality to the same extent.19 In our study, mice did not receive catecholamines. Nevertheless, we found that early volume replacement with either HSA or saline solution yielded similar hemodynamic improvements in MAP after LPS treatment although both solutions possess different volumic expanding properties.
Other studies have already suggested a beneficial role of HSA in sepsis-induced organ dysfunction,14,20 but the mechanism remains unclear, especially with respect to cardiovascular aspects. Thus, HSA has been reported to improve heart function in endotoxic shock in rat21 in which HSA, but not other resuscitation fluids, reduced iNOS mRNA transcription and subsequent expression of the enzyme in cardiomyocytes. These results suggest a role for HSA in the reduction of sepsis-induced cardiac failure.21
A major finding of our work was that HSA, but not saline, improved endothelial dysfunction and prevented the vascular hyporeactivity to Pe (aorta) and myogenic tone (resistance arteries). To our knowledge, this is the first report to shed new light on the beneficial effect of HSA treatment on vascular dysfunction induced by LPS. This property of HSA probably plays a major role in controlling the process of refractory vasoplegia in patients with septic shock.22 It is well documented, including in human arteries, that the vascular hyporeactivity to different vasoconstrictors after LPS administration is linked with overproduction of NO because of iNOS activation within the vascular wall.10,23 In line with this, we found that vascular hyporeactivity induced by LPS is prevented after NO synthase inhibition. We extended these findings to the LPS-induced reduction of myogenic tone, which plays a significant role in the low peripheral resistance profile observed in septic shock patients. In addition, the LPS-enhanced in situ NO production was associated with increased iNOS expression in the vascular wall. It is of particular interest that the potentiating effect of L-NAME is blunted on HSA administration in both Pe-induced contraction in the aorta and myogenic tone in SMAs. Furthermore, HSA, but not saline, prevented the ability of LPS to increase NO production and iNOS expression in the aorta. Moreover, HSA prevented the overproduction of NO and iNOS expression in target organs such as the heart and the lung. Altogether, we demonstrate that HSA overcomes the deleterious effect of an overproduction of NO that is associated with an induction of iNOS in vascular and target organs after LPS treatment without any deleterious effect of the use of nonselective NOS inhibitor such as pulmonary hypertension associated with concomitant endothelial NO synthase blockade. In contrast, we found that HSA tended to prevent LPS-induced endothelial dysfunction. The latter is linked to reduced endothelial NO release in the aorta. Sepsis is also associated with increased oxidative stress, marked by increased circulating and tissue O2− production. It is possible that the beneficial effects of HSA are probably linked to its anti-oxidant properties, probably because of its ability either to scavenge free radicals as suggested by Quinlan and colleagues24 or to interact with circulating molecules or cells such as polymorphonuclear cells involved in the process leading to enhanced reactive oxygen species production.25 Moreover, HSA might also have a direct beneficial effect on the vascular wall, endothelium, and glycocalyx.26 Indeed, HSA can bind with gp60 cell membrane receptors,27 allowing its internalization and alteration of biochemical intracellular pathways leading to oxidative stress. Thus, it is likely that the anti-oxidant properties of HSA is potentially able to reduce circulating and tissue O2− production and thus prevent the endothelial dysfunction and vascular hyporeactivity. Indeed, the reduction of O2− production leads to a decrease in its interaction with NO and thus in the production of the highly toxic ONOO−. Peroxynitrite is produced in large amounts during septic shock and has harmful effects on various tissues,28 especially in blood vessels as reported by Szabo and colleagues.29 Furthermore, O2− is involved in leukocyte infiltration, induces DNA damage, worsens lipid peroxidation, and promotes the synthesis of proinflammatory cytokines. The mechanism whereby HSA decreases oxidative stress has not been assessed previously, but in this study we demonstrate that HSA, but not saline, prevents the increases in O2− production in aorta and end target organs such as heart and lung. Moreover, HSA also attenuated LPS-induced ONOO− production in the aortic wall. This property of HSA is essential in preventing vascular hyporeactivity, endothelial dysfunction, and end organ damage in endotoxic animals and probably in septic shock patients.
Finally, the mechanism whereby LPS induces shock is associated with its ability to activate the NF-κB/Rel transcription family, enabling the expression of several critical genes involved in the pathogenesis of septic shock. We report that HSA reduces NF-κBA activation in the vascular wall, which in turn will reduce oxidative and nitrosative stresses induced by LPS in blood vessels and so prevents vascular hyporeactivity while also improving endothelial dysfunction, as reported in this study.
Our study was designed to better understand the mechanisms of the protective effects of HSA on vascular dysfunction during endotoxemia. The model that we used reproduced hemodynamics of septic patients, and HSA was infused early in this mouse endotoxic shock model. Thus, it can be speculated that the anti-inflammatory and anti-oxidant effects of HSA were not overwhelmed, and the results from a more aggressive LPS challenge or a delayed HSA infusion may have been different. Such considerations may explain some discrepancies with data reported in human clinical trials.30 In conclusion, our study demonstrates a protective effect of HSA treatment in endotoxic shock, a condition that is associated with inflammatory processes that lead to oxidative and nitrosative stresses and vascular hyporeactivity in animal models. Our data provide a rationale for HSA treatment in patients with septic shock.
Acknowledgments
We thank Dr. Gallois for help in quantitative determination of mouse serum albumin, chloride, and total CO2; and Prof. Ismael Laher for a careful reading of the manuscript.
Footnotes
Address reprint requests to Dr. Ramaroson Andriantsitohaina, INSERM UMR 771, CNRS UMR, 6214, Faculté de Médecine, rue Haute de Reculée, 49000 Angers, France. E-mail: ramaroson.andriantsitohaina@univ-angers.fr.
Supported by the Fonds Européen de Développement Régional (FEDER no. 8891).
References
- Parrillo JE. Pathogenetic mechanisms of septic shock. N Engl J Med. 1993;328:1471–1477. doi: 10.1056/NEJM199305203282008. [DOI] [PubMed] [Google Scholar]
- Russell JA. Management of sepsis. N Engl J Med. 2006;355:1699–1713. doi: 10.1056/NEJMra043632. [DOI] [PubMed] [Google Scholar]
- Court O, Kumar A, Parrillo JE, Kumar A. Clinical review: myocardial depression in sepsis and septic shock. Crit Care. 2002;6:500–508. doi: 10.1186/cc1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers EP, McIntyre L, Morro DC, Rivers KK. Early and innovative interventions for severe sepsis and septic shock: taking advantage of a window of opportunity. CMAJ. 2005;173:1054–1065. doi: 10.1503/cmaj.050632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor VM, Rocha M, De la Fuente M. Immune cells: free radicals and antioxidants in sepsis. Int Immunopharmacol. 2004;4:327–347. doi: 10.1016/j.intimp.2004.01.020. [DOI] [PubMed] [Google Scholar]
- Clapp BR, Hingorani AD, Kharbanda RK, Mohamed-Ali V, Stephens JW, Vallance P, MacAllister RJ. Inflammation-induced endothelial dysfunction involves reduced nitric oxide bioavailability and increased oxidant stress. Cardiovasc Res. 2004;64:172–178. doi: 10.1016/j.cardiores.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Peters K, Unger RE, Brunner J, Kirkpatrick CJ. Molecular basis of endothelial dysfunction in sepsis. Cardiovasc Res. 2003;60:49–57. doi: 10.1016/s0008-6363(03)00397-3. [DOI] [PubMed] [Google Scholar]
- Orfanos SE, Mavrommati I, Korovesi I, Roussos C. Pulmonary endothelium in acute lung injury: from basic science to the critically ill. Intensive Care Med. 2004;30:1702–1714. doi: 10.1007/s00134-004-2370-x. [DOI] [PubMed] [Google Scholar]
- Stoclet JC, Muller B, Andriantsitohaina R, Kleschyov A. Overproduction of nitric oxide in pathophysiology of blood vessels. Biochemistry (Mosc) 1998;63:826–832. [PubMed] [Google Scholar]
- Stoclet JC, Martinez MC, Ohlmann P, Chasserot S, Schott C, Kleschyov AL, Schneider F, Andriantsitohaina R. Induction of nitric oxide synthase and dual effects of nitric oxide and cyclooxygenase products in regulation of arterial contraction in human septic shock. Circulation. 1999;100:107–112. doi: 10.1161/01.cir.100.2.107. [DOI] [PubMed] [Google Scholar]
- Quinlan GJ, Martin GS, Evans TW. Albumin: biochemical properties and therapeutic potential. Hepatology. 2005;41:1211–1219. doi: 10.1002/hep.20720. [DOI] [PubMed] [Google Scholar]
- Sakurai Y, Ma SF, Watanabe H, Yamaotsu N, Hirono S, Kurono Y, Kragh-Hansen U, Otagiri M. Esterase-like activity of serum albumin: characterization of its structural chemistry using p-nitrophenyl esters as substrates. Pharm Res. 2004;21:285–292. doi: 10.1023/b:pham.0000016241.84630.06. [DOI] [PubMed] [Google Scholar]
- Cantin AM, Paquette B, Richter M, Larivee P. Albumin-mediated regulation of cellular glutathione and nuclear factor kappa B activation. Am J Respir Crit Care Med. 2000;162:1539–1546. doi: 10.1164/ajrccm.162.4.9910106. [DOI] [PubMed] [Google Scholar]
- Margarson MP, Soni NC. Effects of albumin supplementation on microvascular permeability in septic patients. J Appl Physiol. 2002;92:2139–2145. doi: 10.1152/japplphysiol.00201.2001. [DOI] [PubMed] [Google Scholar]
- Finfer SBR, Boyce N, French J, Myburgh J, Norton R. SAFE Study Investigators: a comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med. 2004;350:2247–2256. doi: 10.1056/NEJMoa040232. [DOI] [PubMed] [Google Scholar]
- Dubois MJ, Orellana-Jimenez C, Melot C, De Backer D, Berre J, Leeman M, Brimioulle S, Appoloni O, Creteur J, Vincent JL. Albumin administration improves organ function in critically ill hypoalbuminemic patients: a prospective, randomized, controlled, pilot study. Crit Care Med. 2006;34:2536–2540. doi: 10.1097/01.CCM.0000239119.57544.0C. [DOI] [PubMed] [Google Scholar]
- Powers KA, Kapus A, Khadaroo RG, He R, Marshall JC, Lindsay TF, Rotstein OD. Twenty-five percent albumin prevents lung injury following shock/resuscitation. Crit Care Med. 2003;31:2355–2363. doi: 10.1097/01.CCM.0000084846.45830.AA. [DOI] [PubMed] [Google Scholar]
- Chalupsky K, Lobysheva I, Nepveu F, Gadea I, Beranova P, Entlicher G, Stoclet JC, Muller B. Relaxant effect of oxime derivatives in isolated rat aorta: role of nitric oxide (NO) formation in smooth muscle. Biochem Pharmacol. 2004;67:1203–1214. doi: 10.1016/j.bcp.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Vincent JL, Gerlach H. Fluid resuscitation in severe sepsis and septic shock: an evidence-based review. Crit Care Med. 2004;32:S451–S454. doi: 10.1097/01.ccm.0000142984.44321.a4. [DOI] [PubMed] [Google Scholar]
- Vincent JL, Sakr Y, Reinhart K, Sprung CL, Gerlach H, Ranieri VM. Is albumin administration in the acutely ill associated with increased mortality? Results of the SOAP study. Crit Care. 2005;9:R745–R754. doi: 10.1186/cc3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walley KR, McDonald TE, Wang Y, Dai S, Russell JA. Albumin resuscitation increases cardiomyocyte contractility and decreases nitric oxide synthase II expression in rat endotoxemia. Crit Care Med. 2003;31:187–194. doi: 10.1097/00003246-200301000-00029. [DOI] [PubMed] [Google Scholar]
- Landry DW, Oliver JA. The pathogenesis of vasodilatory shock. N Engl J Med. 2001;345:588–595. doi: 10.1056/NEJMra002709. [DOI] [PubMed] [Google Scholar]
- Hernanz R, Alonso MJ, Zibrandtsen H, Alvarez Y, Salaices M, Simonsen U. Measurements of nitric oxide concentration and hyporeactivity in rat superior mesenteric artery exposed to endotoxin. Cardiovasc Res. 2004;62:202–211. doi: 10.1016/j.cardiores.2004.01.014. [DOI] [PubMed] [Google Scholar]
- Quinlan GJ, Margarson MP, Mumby S, Evans TW, Gutteridge JM. Administration of albumin to patients with sepsis syndrome: a possible beneficial role in plasma thiol repletion. Clin Sci. 1998;95:459–465. [PubMed] [Google Scholar]
- Kouoh F, Gressier B, Luyckx M, Brunet C, Dine T, Cazin M, Cazin JC. Antioxidant properties of albumin: effect on oxidative metabolism of human neutrophil granulocytes. Il Farmaco. 1999;54:695–699. doi: 10.1016/s0014-827x(99)00082-8. [DOI] [PubMed] [Google Scholar]
- Jacob M, Bruegger D, Rehm M, Stoeckelhuber M, Welsch U, Conzen P, Becker BF. The endothelial glycocalyx affords compatibility of Starling’s principle and high cardiac interstitial albumin levels. Cardiovasc Res. 2007;73:575–586. doi: 10.1016/j.cardiores.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Tiruppathi C, Song W, Bergenfeldt M, Sass P, Malik AB. Gp60 activation mediates albumin transcytosis in endothelial cells by tyrosine kinase-dependent pathway. J Biol Chem. 1997;272:25968–25975. doi: 10.1074/jbc.272.41.25968. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Mazzon E, Di Paola R, Esposito E, Macarthur H, Matuschak GM, Salvemini D. A role for nitric oxide-mediated peroxynitrite formation in a model of endotoxin-induced shock. J Pharmacol Exp Ther. 2006;319:73–81. doi: 10.1124/jpet.106.108100. [DOI] [PubMed] [Google Scholar]
- Szabó C, Zingarelli B, Salzman AL. Role of poly-ADP ribosyltransferase activation in the vascular contractile and energetic failure elicited by exogenous and endogenous nitric oxide and peroxynitrite. Circ Res. 1996;78:1051–1063. doi: 10.1161/01.res.78.6.1051. [DOI] [PubMed] [Google Scholar]
- Alderson P, Bunn F, Lefebvre C, Li WP, Li L, Roberts I, Schierhout G: Human albumin solution for resuscitation and volume expansion in critically ill patients. Cochrane Database Syst Rev 2004, CD001208pp 1–28 [DOI] [PubMed] [Google Scholar]