

Abstract
Mucin hypersecretion is a major pathological feature of many respiratory diseases, yet cellular mechanisms regulating secretion of mucin have not been fully elucidated. Previously, we reported that mucin hypersecretion induced by human neutrophil elastase involves activation of protein kinase C (PKC), specifically the δ-isoform (PKCδ). Here, we further investigated the role of PKCδ in mucin hypersecretion using both primary human bronchial epithelial cells and the human bronchial epithelial 1 cell line as in vitro model systems. Phorbol-12-myristate-13-acetate (PMA)-induced mucin hypersecretion was significantly attenuated by rottlerin, a PKCδ-selective inhibitor. Rottlerin also reduced PMA- or human neutrophil elastase-induced phosphorylation of myristoylated alanine-rich C kinase substrate (MARCKS) protein in these cells. Both secretion and MARCKS phosphorylation were significantly enhanced by the PKCδ activator bryostatin 1. A dominant-negative PKCδ construct (pEGFP-N1/PKCδK376R) transfected into human bronchial epithelial 1 cells significantly attenuated both PMA-induced mucin secretion and phosphorylation of MARCKS, whereas transfection of a wild-type construct increased PKCδ and enhanced mucin secretion and MARCKS phosphorylation. Similar transfections of a dominant-negative or wild-type PKCε construct did not affect either mucin secretion or MARCKS phosphorylation. The results suggest that PKCδ plays an important role in mucin secretion by airway epithelium via regulation of MARCKS phosphorylation.
Mucus produced by epithelium of respiratory, gastrointestinal, and reproductive tracts provides a barrier between the external environment and cellular components of the epithelial layer. Mucins, the glycoprotein component of mucus, constitute a family of large, highly glycosylated macromolecules that impart physical (aggregation, viscosity, viscoelasticity, and lubrication) and biological (protection) properties to mucus (reviewed in Ref. 1). Airway mucus is an integral component of the mucociliary clearance system in the trachea and bronchi and thus serves to protect the lower airways and alveoli from impingement of particulate matter and pathogens. However, mucin secretion is abnormally augmented in disease states, such as chronic bronchitis, asthma, and cystic fibrosis, increasing morbidity and mortality in these patients (reviewed in Refs. 1 and 2). Mucin hypersecretion is potentiated by many pathophysiological mediators, such as bacterial proteinases and endotoxin, adenine and guanine nucleotides, cytokines, inflammatory mediators, and eicosanoids (reviewed in Ref. 3). Intracellular mechanisms and signaling molecules involved in the secretory process have not been fully elucidated.
Protein kinase C (PKC) is a serine/threonine kinase involved in various exocytotic events in different cell types, including secretion of mucin,4,5 insulin,6 neurotransmitters,7 and platelet dense granules.8 Previously, we demonstrated that mucin secretion in airway epithelial cells is regulated by PKC via phosphorylation of the myristoylated alanine-rich C kinase substrate (MARCKS).9,10 In addition, we demonstrated that mucin hypersecretion in human airway epithelial cells in vitro in response to human neutrophil elastase (HNE) appears to be mediated by the δ-isoform of PKC (PKCδ).11 Not surprisingly, PKCδ, a novel PKC isoform, has a strong affinity for MARCKS and can phosphorylate MARCKS both in vitro and in vivo.12,13,14 Increasing evidence suggests that PKCδ mediates exocytotic secretion in several different cell types.4,8,15,16,17
Here, we further elucidate the role of PKCδ in the mucin secretory pathway in human airway epithelial cells in vitro. The mucin secretory response and phosphorylation of MARCKS were assessed after exposure of well differentiated normal human bronchial epithelial (NHBE) cells to phorbol-12-myristate-13-acetate (PMA), a general PKC activator, or bryostatin 1, a PKCδ/ε activator. In addition, we used the papilloma virus-transformed human bronchial epithelial 1 (HBE1) cell line for molecular manipulations. A dominant-negative PKCδ construct (K376R) transfected into HBE1 cells attenuated PMA-stimulated mucin secretion and MARCKS phosphorylation, whereas a similar dominant-negative PKCε construct was without effect. The results indicate that PKCδ is a key isoform regulating airway mucin secretion, and the mechanism of its action appears to involve phosphorylation of MARCKS protein.
Materials and Methods
Culture of Bronchial Epithelial Cells
Primary NHBE cells purchased from Cambrex Bioscience (Walkersville, MD) were expanded and maintained in a humidified air/5% CO2 incubator as described previously.9,11 Cells from at least three separate donors were used in these studies. Passage 31 or 32 of human papilloma virus-transformed human bronchial epithelial cells (HBE1)18 was seeded and maintained as described previously. Transient transfection of HBE1 cells was performed after 10 days of culture in air-liquid interface.
Exposure of Cells to Inhibitors or Secretagogues
Well differentiated NHBE cells were exposed to test agents both apically and basolaterally for 15 minutes (unless otherwise indicated). Transfected HBE1 cells were exposed to PMA (EMD Biosciences, La Jolla, CA) applied apically only. Cells were preincubated with the PKCδ-selective inhibitor, rottlerin (EMD Biosciences) for 20 minutes before PMA exposure. Initial stock solutions of rottlerin or PMA were prepared in dimethyl sulfoxide, kept at −20°C, and diluted in growth medium directly before use. When cells were exposed to PMA in the presence or absence of rottlerin, PMA was “spiked” into each well at the indicated concentration.
Measurement of Mucin Secretion
Mucin was collected both at baseline and after treatments as described previously.9 Baseline mucin secretion was used to normalize well-to-well variation. After baseline mucin samples were collected, cells were rested overnight and exposed to test reagents the next day for indicated periods of time. After each treatment period, secreted mucin was collected as the baseline sample and quantified by sandwich enzyme-linked immunosorbent assay using the 17Q2 antibody (Covance Research Products, Berkeley, CA), a monoclonal antibody that reacts specifically with a carbohydrate epitope on human airway mucins.19 The 17Q2 antibody was purified using an ImmunoPure(G) IgG purification kit (Pierce Biotechnology, Rockford, IL) following the manufacturer’s protocol and then conjugated with alkaline phosphatase (EMD Biosciences). To account for variability between cultures and experiments, levels of mucin secretion were reported as percentage of the nontreated control. Actual values for mucin released by control cell cultures in these experiments ranged from 40 to 70 ng/ml in NHBE cells and 7 to 10 ng/ml in HBE1 cells.
Subcellular Localization of PKC Isoforms
Activation of PKCδ was assessed by subcellular fractionation following the protocol described by Kajstura et al20 and subsequent Western blot analysis using a PKCδ-specific antibody (Cell Signaling Technology, Inc., Danvers, MA). Briefly, cells were washed with cold PBS and scraped into lysis buffer [20 mmol/L Tris-Cl (pH 7.5), 1 mmol/L ethylenediamine tetraacetic acid, 100 mmol/L NaCl, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L dithiothreitol, 1% (v/v) protease inhibitor cocktail, and phosphatase inhibitor cocktail (Sigma, St. Louis, MO)]. The lysate was then sonicated and pelleted at 20,000 × g (Eppendorf 5417 centrifuge) for 40 minutes. The supernatant was collected and kept as the cytosolic fraction at −80°C until used. The remaining pellet was resuspended in lysis buffer containing 1% Triton X-100, sonicated, and centrifuged at 20,000 × g for 40 minutes. The supernatant membrane fraction was stored at −80°C until analyzed by Western blot.
Western Blot Analysis
Total MARCKS, phosphorylated MARCKS, PKCδ, and PKCε protein levels were measured via Western blot. The protein concentrations of cell lysates were quantified by a Bradford assay (Bio-Rad Laboratories, Hercules, CA). Sample lysates were prepared by boiling in 2× SDS sample buffer [125 mmol/L Tris-Cl (pH 6.8), 25% glycerol, 4% SDS, 10% β-mercaptoethanol, and 0.04% bromphenol blue] for 10 minutes. Sample lysates (30 to 60 μg) were loaded on 10 or 12% SDS-polyacrylamide gels and then transferred to a polyvinylidene difluoride membrane (Schleicher & Schuell BioScience, Inc., Keene, NH) following electrophoresis. Polyvinylidene difluoride membranes were blocked with 5% nonfat milk and then probed with an appropriate dilution of primary antibody followed by horseradish peroxidase-conjugated anti-mouse or anti-rabbit antibodies. Chemiluminescent detection was performed using ECL detection reagents (GE Health care Life Sciences, Piscataway, NJ) following the manufacturer’s protocol. Amounts of specific proteins in bands were quantified using Labworks image acquisition and analysis software 4.0. (Ultra Violet Products, Ltd., Upland, CA).
Antibodies against α-tubulin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and E-cadherin (BD Biosciences, San Jose, CA) were used as loading controls for cytosolic and membrane fractions, respectively. Phosphorylated MARCKS (at serine 152/156) was detected with a specific antibody (Cell Signaling Technology, Inc.). After detection, the membrane was stripped in 62.5 mmol/L Tris-Cl (pH 6.5), 10% SDS, and 100 mmol/L β-mercaptoethanol for 10 minutes at room temperature and reprobed with a monoclonal antibody against total MARCKS protein (clone no. 2F12; Upstate, Charlottesville, VA) to verify equal loading.
Transient Transfection of PKC Constructs
Transient transfection of vectors overexpressing wild-type or dominant-negative PKCδ and PKCε in HBE1 cells was performed using the FuGene 6 transfection reagent (Roche Applied Science, Indianapolis, IN) following the manufacturer’s protocol. The pEGFP-N1 vectors containing a wild-type PKCδ cDNA21 and a dominant-negative PKCδ mutant cDNA [lysine (AAG)→arginine (AGG) mutation, position 376]22 were generously provided by Dr. Arti Shukla (University of Vermont, Burlington, VT) and Dr. Peter Blumberg (National Cancer Institute, Bethesda, MD). The K376→R mutation in the ATP binding site of the catalytic domain has been demonstrated previously to inhibit PKCδ kinase activity.23 Briefly, HBE1 cells grown in air/liquid interface were dissociated in versene solution (Invitrogen, Carlsbad, CA) and re-seeded in 12-well culture plates at a density of 1 × 105 cells/cm2. After overnight incubation, cells were transfected with the pEGFP-N1 vector alone or the pEGFP-N1 vector containing either a wild-type or dominant-negative PKCδ cDNA (K376R). Isotype controls for the dominant-negative PKCδ consisted of transient transfection of both wild-type PKCε as well as a catalytically inactive dominant-negative PKCε (K437R) construct-tagged with hemagglutin24 (PKCε constructs were kindly provided by Dr. Jae-Won Soh, University of Inha, Inha, Republic of Korea). Cells were subsequently cultured for 48 hours to allow for detectable protein expression. Transfection of PKCδ constructs was confirmed by fluorescent microscopy and assessment of expression of green fluorescence protein (GFP)-tagged PKCδ assessed by Western blot analysis using monoclonal antibodies against PKCδ or GFP (Cell Signaling Technology, Inc.). Transfection of PKCε was confirmed by Western blot analysis using PKCε and HA monoclonal antibodies (Covance Research Products).
Cytotoxicity Assay
All treatments used were tested for cytotoxicity using a CytoTox 96 nonradioactive cytotoxicity assay kit (Promega, Madison, WI) according to the manufacturer’s instructions. The results were expressed as the ratio of released lactate dehydrogenase to total lactate dehydrogenase. Released lactate dehydrogenase never exceeded 10% of total lactate dehydrogenase with any of the treatments (data not shown).
Statistical Analysis
Data were expressed as the ratio of treatment to the corresponding vehicle (dimethyl sulfoxide or media) control. Results were evaluated using one-way analysis of variance with Dunnett’s test and a Bonferroni posttest correction for multiple comparisons.25 A P value of less than 0.05 was considered significant.
Results
Effect of Rottlerin on PMA-Induced Mucin Secretion and MARCKS Phosphorylation in NHBE Cells
To determine whether PKCδ is an important regulatory molecule in mucin secretion, we investigated the effect of rottlerin, a PKCδ-selective inhibitor, on PMA-induced mucin secretion in well differentiated NHBE cells. As illustrated in Figure 1A, 100 nmol/L PMA provoked translocation of PKCδ from cytosol to membrane in these cells. Inhibition of PKCδ activity by pretreatment of cells with rottlerin (1 to 10 μmol/L) for 20 minutes significantly attenuated PMA-induced mucin secretion in a concentration-dependent manner (Figure 1B). Phosphorylation of MARCKS mediated by PMA in the presence or absence of 15 μmol/L rottlerin was analyzed by Western blot. HNE, previously shown to stimulate mucin secretion in NHBE cells via a rottlerin-inhibitable mechanism,11 was used as an additional control. As shown in Figure 1C, both PMA- and HNE-induced phosphorylation of MARCKS were decreased by pretreatment with rottlerin.
Figure 1.

Rottlerin, a selective PKCδ inhibitor, attenuates PMA-induced mucin secretion and phosphorylation of MARCKS in well differentiated NHBE cells. Cells were preincubated with 15 μmol/L rottlerin (or dimethyl sulfoxide solvent control) for 20 minutes before addition of 100 nmol/L PMA (B and C) or 500 nmol/L HNE (C). A: PKCδ translocates from cytosol to membrane in response to PMA (100 nmol/L) at 15 minutes. After detecting PKCδ, the polyvinylidene difluoride membrane was stripped and reprobed with antibodies against α-tubulin and E-cadherin, which were used as controls to detect the cytosolic and the membrane fraction, respectively. B: Rottlerin significantly attenuates PMA-induced mucin secretion in a concentration-dependent manner. *Significantly different from vehicle control (P < 0.05); †significantly different from PMA alone (P < 0.05). Data are presented as mean ± SEM (n = 4). C: Rottlerin attenuates phosphorylation of MARCKS in NHBE cells exposed to two different mucin secretagogues, HNE (500 nmol/L) or PMA (100 nmol/L). Phosphorylated MARCKS was detected by Western blot using a specific antibody against MARCKS phosphorylated at serine 152/156. After detecting phosphorylated MARCKS, the polyvinylidene difluoride membrane was stripped and reprobed with an antibody against total MARCKS protein as a loading control for each lane. Blots are representative of two replicate experiments. Densitometry measurements of the band intensities were quantified using LabWorks (BioImaging Systems, UVP, Inc., Cambridge, UK) and are shown below the blots.
Bryostatin 1 Provokes Mucin Secretion and MARCKS Phosphorylation in NHBE Cells
Bryostatin 1, a PKCδ/ε activator, was used to investigate further the relationship between PKCδ and mucin secretion in NHBE cells. A naturally occurring marine invertebrate-derived cyclic lactone, Bryostatin 1, causes rapid activation of PKCδ and prolonged protection of PKCδ against ubiquitination and proteolysis.26,27 Bryostatin 1 interacts with the diacylglycerol binding site on PKC,28 but its complete mode of action has not been fully elucidated, and it can affect PKCs in a cell-type specific manner.29 As illustrated in Figure 2, exposure of NHBE cells to bryostatin 1 over a range of concentrations (10 to 1000 nmol/L) for 15 minutes resulted in translocation of PKCδ from cytosol to membrane in response to all concentrations tested (Figure 2A). Mucin secretion was also significantly increased by bryostatin 1, with maximal stimulation at 100 nmol/L (Figure 2B). As illustrated in Figure 2C, phosphorylation of MARCKS also was induced in these cells in response to bryostatin 1 (from 10 to 1000 nmol/L) with maximal phosphorylation at 100 nmol/L. None of these treatments induced cytotoxicity as measured by lactate dehydrogenase release assay (data not shown).
Figure 2.

Effect of bryostatin 1, a PKCδ activator, on mucin secretion in well differentiated NHBE cells. NHBE cells were exposed to bryostatin 1 over a range of concentrations from 1 to 1000 nmol/L for 15 minutes. A: PKCδ translocates from cytosol to membrane in response to bryostatin 1. α-Tubulin and E-cadherin were used as controls for the cytosolic and membrane fractions, respectively. Blots are representative of three replicate experiments. B: Bryostatin 1 provokes mucin secretion by NHBE cells in a concentration-dependent manner. Significantly different from vehicle control: *P < 0.05; †P < 0.001; ‡P < 0.005. Data are presented as mean ± SEM (n = 4). C: Phosphorylation of MARCKS in NHBE cells is induced by bryostatin 1 in a concentration-dependent manner. Blots are representative of three replicate experiments.
PKC Activation Stimulates Mucin Secretion in HBE1 Cells
To investigate further a role for PKCδ as a regulator of mucin secretion using molecular manipulation of PKCδ activity, the HBE1 cell line was used. As illustrated in Figure 3, exposure of HBE1 cells to 500 nmol/L PMA for 15 minutes significantly increased mucin secretion (by ∼1.7-fold compared with medium vehicle control) and also induced phosphorylation of MARCKS in these cells.
Figure 3.
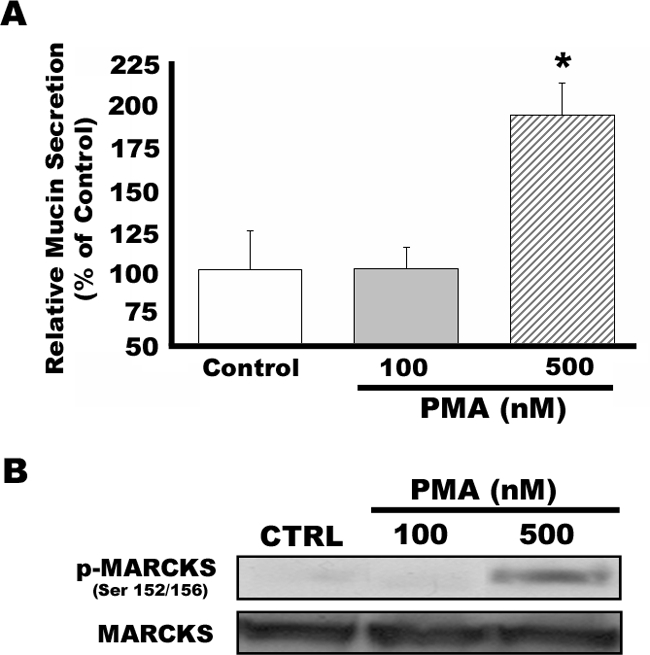
HBE-1 cells secrete mucin in response to PKC activation. HBE-1 cells maintained in air/liquid interface were exposed to 100 or 500 nmol/L PMA for 15 minutes. Mucin secretion and phosphorylation of MARCKS were assessed by enzyme-linked immunosorbent assay and Western blot analysis, respectively. A: Mucin secretion is significantly enhanced by PMA at 500 nmol/L (but not 100 nmol/L) in HBE1 cells. *Significantly different from vehicle control (P < 0.05). Data are presented as mean ± SEM (n = 4). B: Phosphorylation of MARCKS is increased by exposure of HBE1 cells to 500 nmol/L PMA. Blots are representative of three replicate experiments.
PKCδ Appears to Regulate Mucin Secretion in Airway Epithelial Cells
Transient transfection of the PKCδ and PKCε constructs into HBE1 cells was confirmed by fluorescent microscopy and Western blot analysis. After 48 hours of transfection, GFP expressed in the transfected cells was detected with a fluorescent microscope [Eclipse TE300 (Nikon, Tokyo, Japan) or Axiovert 35 (Zeiss, Welwyn Garden City, UK)] before PMA exposure (data not shown). After exposure to PMA, cells were lysed to detect expression of PKCδ protein fused with GFP via Western blot analysis using anti-PKCδ and -GFP antibodies (data not shown). Transfection of PKCε was confirmed by Western blot analysis using an antibody against PKCε and an HA tag. Transfection efficiency of all constructs and controls was about 20 to 25% as determined by quantification of GFP and X-gal assay for PKCδ and PKCε, respectively (data not shown).
As illustrated in Figure 4A, mucin secretion by transfected HBE1 cells was stimulated by exposure to 500 nmol/L PMA. Transfection of HBE1 cells with the dominant-negative PKCδ construct (pEGFP-N1/PKCδK376R) resulted in significant reduction of PMA-induced mucin secretion (∼45%), whereas cells transfected with the wild-type PKCδ construct (pEGFP-N1/PKCδ) showed a significant enhancement of PMA-induced mucin secretion (∼40%) compared with control cells transfected with no DNA or empty vector (pEGFP-N1). In additional control studies, the effects of Bryostatin 1 on both mucin secretion and phosphorylation of MARCKS were markedly attenuated in cells transfected with the dnPKCδ construct (data not shown). PKCε constructs, either wild type or mutated, did not affect the secretory response to PMA. As shown in Figure 4B, phosphorylation of MARCKS in response to PMA was decreased in HBE1 cells transfected with the dominant-negative PKCδ construct (pEGFP-N1/PKCδK376R) but increased in cells transfected with the wild-type PKCδ construct (pEGFP-N1/PKCδ). PKCε constructs, either wild type or mutated, had no effect on PMA-induced MARCKS phosphorylation when transfected into HBE1 cells (Figure 4C).
Figure 4.

Transient transfection of HBE1 cells with a dominant-negative PKCδ construct results in reduction of mucin hypersecretion. HBE1 cells were transiently transfected with empty vector (pEGFP-N1), a wild-type PKCδ construct (pEGFP-N1/PKCδ), or a dominant-negative construct (pEGFP-N1/PKCδK376R) using the FuGene 6 transfection reagent as described in Materials and Methods. As an additional control, a wild-type (pHACE/PKCε) and a dominant-negative PKCε construct (pHACE/PKCεK437R) were also transfected. A: After 48 hours transfection, cells were exposed to 500 nmol/L PMA (lanes 2 to 8) or vehicle control (lane 1) for 15 minutes, at which time media were collected and mucin secretion assessed by enzyme-linked immunosorbent assay. Data are significantly different from media control (*P < 0.05; **P < 0.001); significantly different from cells transfected with no DNA or empty vector and exposed to PMA (†P < 0.05, ‡P < 0.001); and significantly different from cells transfected with the pEGFP-N1/PKCδK376R and exposed to PMA (§P < 0.05). Data are presented as mean ± SEM (n = 3∼4). After collecting media from transfected HBE1 cells exposed to PMA, cell lysates were analyzed by Western blot for MARCKS phosphorylated at Serine 152/156 residues (B and C). Blots are representative of three replicate experiments. B: Transfection of the dnPKCδ construct results in decreased phosphorylation of MARCKS in response to PMA, whereas overexpression of PKCδ via transfection of the wild-type construct enhances MARCKS phosphorylation after PMA exposure. Lane 1, control medium; lane 2, PMA plus control medium; lane 3, PMA plus mock transfection; lane 4, PMA plus empty vector (pEGFP-N1); lane 5, PMA plus wild-type PKCδ (pEGFP-N1/PKCδ); and lane 6, PMA plus dnPKCδ (pEGFP-N1/PKCδK376R). C: In a separate experiment, phosphorylation of MARCKS is attenuated by transfection of the dnPKCδ (pEGFP-N1/PKCδK376R; lane 2), whereas similar transfection of the dnPKCε (pHACE/PKCεK437R; lane 3) does not appear to decrease PMA-induced phosphorylation of MARCKS.
Discussion
Airway mucus is a heterogeneous mixture of water, mucins, enzymes and anti-enzymes, salts, endogenous and exogenous bacterial agents, organic compounds, and cell debris.30,31 Airway injury or disease often leads to alterations in mucin production and/or mucus composition. Despite recognition of mucin hypersecretion as a major pathophysiological feature in many airway diseases,2,32,33,34 relatively little is known about actual intracellular mechanisms that regulate secretion under normal or pathological conditions.
Previously, we reported that mucin secretion and PKCδ activity are both increased in normal human airway epithelial cells in response to HNE. In addition, pretreatment of cells with a PKCδ-selective inhibitor resulted in attenuation of HNE-induced mucin secretion.11 PKC is a serine/threonine kinase that has many roles in cell function. All PKCs contain both a regulatory and catalytic domain, each of which consists of four conserved and five variable regions. Two distinctive domains are separated by the V3 “hinge” region. Four conserved regions distributed in the regulatory domain (C1 and C2) and catalytic domain (C3 and C4) are responsible for activation. In addition, a pseudosubstrate or autoinhibitory domain is located in the amino terminus near the C1 domain.35 Enzymatic activity of PKC is derived from its catalytic domain on the binding of the C3 and C4 regions to ATP and substrate, respectively.
PKC isoforms have been classified into three subfamilies depending on their mode of activation: conventional (α, β, and γ) PKCs, which are activated by phosphatidylserine, diacylglycerol, or phorbol esters; novel (δ, ε, η, θ, and μ) PKCs, which are phosphatidylserine, diacylglycerol, or 12-O-tetradecanoylphorbol-13-acetate/PMA dependent but Ca2+ independent; and atypical (ζ and ι/λ) PKCs, which are Ca2+, phosphatidylserine, diacylglycerol, and 12-O-tetradecanoylphorbol-13-acetate/PMA independent for their activation.35,36 Involvement of PKC activation during airway mucin secretion in response to various secretagogues has been reported.9,37,38,39 However, involvement of any specific isoform(s) has not been fully characterized.
Among the PKC isoforms, PKCδ has been shown to regulate many aspects of lung/airway epithelial cell biology and function. Lungs of mice exposed to asbestos show increased membrane translocation of PKCδ and enhanced PKCδ protein expression, indicating PKCδ activation.40 Increased PKCδ in response to asbestos exposure is mainly detected in the membrane of proliferating cell nuclear antigen-positive cells, suggesting that asbestos fibers mediate proliferation of epithelial cells in a PKCδ-dependent manner. Interestingly, activation of PKCδ also is involved in asbestos-induced apoptosis in type 2-like epithelial cells (C10) via translocation of PKCδ to the mitochondria followed by activation of caspase-9 and -3 pathways.41 In the C5 human salivary epithelial cell line, PKCδ is also required for mitochondria-dependent apoptosis mediated by etoposide, UV radiation, or taxol.42 In that report, overexpression of wild-type PKCδ led to a robust induction of apoptosis (indicated by DNA fragmentation), whereas a dominant-negative PKCδ blocked apoptosis in response to diverse stimuli. Previous work has demonstrated that PKCδ is involved in regulating expression of inducible nitric oxide synthase, granulocyte colony stimulating factor, RANTES, and intercellular adhesion molecule-1 in airway epithelium.43,44
PKCδ has been implicated in secretory pathways of various cell types. It has recently been demonstrated to regulate secretion of neurotensin from human endocrine (BON) cells via phosphorylation of MARCKS protein.45 In human platelets, PKCδ is activated in response to protease-activated receptor agonist peptides (SFLLRN and AYPGKF), leading to dense granule release,8 a process attenuated by the PKCδ inhibitor, rottlerin, but not by Go6976 (a conventional PKC inhibitor). Ishikawa et al15 demonstrated that carbachol-stimulated insulin secretion in rat pancreatic islets is associated with translocation of PKCδ. In that study, carbachol-stimulated insulin secretion was not reduced by Go6976 but was significantly suppressed by an ambiguous PKC inhibitor, chelerythrine, suggesting that a novel PKC isoform, δ or ε, might play a regulatory role in the process. Cho et al17 demonstrated that PKCδ activation is involved in antigen-induced mast cell degranulation, which could subsequently be inhibited by rottlerin or transfection of a dominant-negative mutant of PKCδ. In contrast, Leitges et al16 reported that PKCδ is a negative regulator of antigen-induced mast cell degranulation.
PKCδ also has been recognized as a regulator of mucin production and secretion in airway epithelial cells. Our previous report suggested that the PKCδ isoform serves as a regulatory molecule in HNE-induced airway mucin secretion from NHBE cells.11 Specific involvement of PKCδ also has been shown in rat secretory cells (SPOC) in response to purinergic agonists (ATPγS) and PMA.4 Recently, Wu et al46 demonstrated that expression of a major gel-forming mucin, MUC5B, is regulated in a PKCδ-dependent manner in human airway epithelial cells (NHBE primary cells and the HBE1 and A549 cell lines).
Another isoform that potentially could be involved in mucin secretion is PKCε. PKCε translocates from cytosol to membrane in response to PMA stimulation of NHBE cells, similar to that observed for PKCδ.11 Activation of PKCε appears to contribute to cAMP-stimulated bicarbonate secretion from mouse duodenal mucosa in vitro,47 and a role for PKCε as a negative regulator has been suggested in epidermal growth factor- and carbachol-mediated Cl− secretion from human intestinal epithelial cells (T84).48,49 Recently, Suzuki et al50 suggested that PKCε plays a dominant role in glucagon-like peptide-1-induced insulin secretion by rat islet cells. In their studies, they observed that glucagon-like peptide-1 mediates translocation of both PKCα and PKCε from cytosol to membrane and also, interestingly, translocation of MARCKS from the plasma membrane to the cytosol. However, a relationship between PKCε activation and MARCKS translocation in their study was not clear, because a PKCε inhibitor peptide attenuated glucagon-like peptide-1-mediated insulin secretion but did not affect MARCKS translocation.
Previously, we demonstrated that MARCKS protein is a key molecule in the mucin secretory pathway in airway epithelium.9,51 MARCKS is phosphorylated by PKC at serine residues 152, 156, and 163 located in its phosphorylated site domain.13 Although MARCKS is a prominent PKC substrate, its phosphorylation is differentially regulated depending on the specificity of each PKC isoform. Herget et al13 demonstrated the differential efficiency of each PKC isoform to phosphorylate MARCKS by performing PKC phosphorylation assays in vitro. It was shown that PKCs α, β1, β2, γ, δ, and ε, but not ζ, can phosphorylate MARCKS, with PKCδ being the most potent isoform, followed by ε and β1, as determined by Vmax to Km catalytic efficiency ratios.13 This finding was supported by results of studies by Fujise et al12 showing that PKCδ has the highest affinity for MARCKS among the isoforms.
The results of the studies reported here indicate a relationship between airway mucin secretion and PKCδ. This is supported by several lines of evidence. First, we demonstrate that mucin secretion correlates well with PKCδ-mediated phosphorylation of MARCKS in both NHBE and HBE1 cells. Two mucin secretagogues (HNE and PMA) (Figure 1) and the PKCδ activator bryostatin 1 (Figure 2) induce mucin secretion and phosphorylation of MARCKS. In addition, pretreatment of cells with the selective PKCδ inhibitor rottlerin decreases phosphorylation of MARCKS in response to HNE or PMA and also attenuates mucin secretion (Figure 1).
Rottlerin is often referred to as a selective inhibitor of PKCδ, and it has been shown to inhibit activity of this isoform by competing with ATP for binding to the enzyme molecule with a median inhibitory concentration (IC50) of 3 to 6 μmol/L.52 Gschwendt et al53 showed that rottlerin has a specific inhibitory effect on PKCδ rather than on other PKC isoforms (IC50; 30 to 42 μmol/L for α, β, and γ and 80 to 100 μmol/L for ε, η, and ζ) via in vitro experiments using purified proteins of each. Specific inhibition of PKCδ by rottlerin at 3 to 6 μmol/L also has been confirmed by Keenan et al.54 Rottlerin has been widely used for selective inhibition of PKCδ kinase activity in numerous in vitro systems.11,46,55,56,57
However, the specificity and exact targets of many pharmacological and chemical inhibitors can be variable, and rottlerin also has been shown to possibly affect a number of other enzymes and molecules, including, for example, calmodulin-kinase III.55 In a cell-free system, 20 μmol/L rottlerin down-regulated activity of MAPKAP-K2, PRAK, and GSKβ while being essentially ineffective against PKCδ.58 However, other evidence seems to support an inhibitory effect of rottlerin on PKCδ activity in epithelial cells. Page et al44 actually measured the effect of 2 μmol/L rottlerin on recombinant PKCδ activity in a cell-free system and showed, via phosphorylation of MBP, that rottlerin substantially reduced PKCδ activity. Similarly, Song et al59 demonstrated a direct inhibitory effect of 4 μmol/L rottlerin on PKCδ activation by the nitric oxide donor NOR-1 in airway epithelial cells.
In a previous study, we showed that rottlerin at concentrations used here significantly decreased PKC activity in airway epithelial cells.11 Similar to our findings reported here, the effects of rottlerin often parallel those of transfection of dnPKCδ constructs into epithelial cells.41,44 In addition, we show here that that rottlerin, at concentrations that attenuate mucin secretion in response to PMA, also attenuates phosphorylation of MARCKS in human airway epithelial cells (Figure 1). Because calmodulin-kinase III, MAPKAP-K2, PRAK, or GSKβ does not phosphorylate MARCKS protein directly and because the time course of the study (30 minutes) essentially precludes synthesis of new proteins, the evidence suggests that the effects of rottlerin seen here relate to attenuation of PKCδ activity. We do recognize, however, that there could be other nonspecific effects of rottlerin that might influence the results.
Transfection of HBE1 cells with a dominant-negative PKCδ allowed targeted investigation of PKCδ mediation of the mucin secretion process. Competition of endogenous PKCδ kinase activity with overexpression of the dominant-negative construct resulted in significant reduction of both PMA-induced mucin secretion and phosphorylation of MARCKS (Figure 4). Additionally, overexpression of wild-type PKCδ significantly enhanced both mucin secretion and phosphorylation of MARCKS in PMA-stimulated cells. Because PKCε can also phosphorylate MARCKS, we transfected either a wild-type or a dominant-negative PKCε construct (generated in the same manner as the PKCδ constructs) as an additional control. The results indicated that PKCε was not involved in the MARCKS-mediated mucin secretion pathway (Figure 4).
Collectively, the results suggest that active PKCδ, via phosphorylation of MARCKS, appears to be a key regulatory molecule in the mucin secretion pathway in airway epithelial cells in vitro. PKCδ appears to be a highly important PKC isoform in airway epithelium, influencing cell growth and proliferation,40 apoptosis,41,42 and inflammation43,44 Our results indicate another important role for PKCδ in airway epithelial function, regulation of mucin secretion, and this seems to occur via a mechanism involving PKCδ-induced phosphorylation of MARCKS protein. These findings fit in well with our previous reports that MARCKS is a key molecule regulating mucin secretion, and the dynamics of its phosphorylation and dephosphorylation appear to be important in this function. The precise mechanism of how MARCKS acts to regulate the secretory pathway, as well as other proteins and molecules involved, remain to be determined.
Acknowledgments
We thank Dr. Linda Martin and Dr. Jorge Piedrahita (College of Veterinary Medicine, North Carolina State University) for offering generous access to fluorescence microscopes. We also thank Dr. Arti Shukla (University of Vermont), Drs. Stuart Yuspa and Peter Blumberg (National Cancer Institute), Dr. Jae-Won Soh (University of Inha), Dr. Atsuko Yoneda (Imperial College, London, UK), and Dr. Deanne Hyrciw (University of Queensland, Queensland, Australia) for supplying various PKC constructs.
Footnotes
Address reprint requests to Dr. Kenneth B. Adler, Department of Molecular Biomedical Sciences, College of Veterinary Medicine, North Carolina State University, 4700 Hillsborough St., Raleigh, NC 27606. E-mail: kenneth_adler@ncsu.edu.
Supported by Grant R37 HL36982 from the NIH.
K.B.A. serves on the scientific advisory board of Sepracor; has received a research grant from AstraZeneca, Inc., and research grants from Sepracor; holds founders shares of BioMarck; and serves as a scientific consultant and member of the scientific advisory board without monetary compensation.
References
- Rose MC. Mucins: structure, function, and role in pulmonary diseases. Am J Physiol. 1992;263:L413–L429. doi: 10.1152/ajplung.1992.263.4.L413. [DOI] [PubMed] [Google Scholar]
- Rogers DF, Barnes PJ. Treatment of airway mucus hypersecretion. Ann Med. 2006;38:116–125. doi: 10.1080/07853890600585795. [DOI] [PubMed] [Google Scholar]
- Adler KB, Li Y. Airway epithelium and mucus: intracellular signaling pathways for gene expression and secretion. Am J Respir Cell Mol Biol. 2001;25:397–400. doi: 10.1165/ajrcmb.25.4.f214. [DOI] [PubMed] [Google Scholar]
- Abdullah LH, Bundy JT, Ehre C, Davis CW. Mucin secretion and PKC isoforms in SPOC1 goblet cells: differential activation by purinergic agonist and PMA. Am J Physiol Lung Cell Mol Physiol. 2003;285:L149–L160. doi: 10.1152/ajplung.00359.2002. [DOI] [PubMed] [Google Scholar]
- Plaisancie P, Ducroc R, El Homsi M, Tsocas A, Guilmeau S, Zoghbi S, Thibaudeau O, Bado A. Luminal leptin activates mucin-secreting goblet cells in the large bowel. Am J Physiol Gastrointest Liver Physiol. 2006;290:G805–G812. doi: 10.1152/ajpgi.00433.2005. [DOI] [PubMed] [Google Scholar]
- Yaney GC, Fairbanks JM, Deeney JT, Korchak HM, Tornheim K, Corkey BE. Potentiation of insulin secretion by phorbol esters is mediated by PKC-alpha and nPKC isoforms. Am J Physiol Endocrinol Metab. 2002;283:E880–E888. doi: 10.1152/ajpendo.00474.2001. [DOI] [PubMed] [Google Scholar]
- Shoji-Kasai Y, Itakura M, Kataoka M, Yamamori S, Takahashi M. Protein kinase C-mediated translocation of secretory vesicles to plasma membrane and enhancement of neurotransmitter release from PC12 cells. Eur J Neurosci. 2002;15:1390–1394. doi: 10.1046/j.1460-9568.2002.01972.x. [DOI] [PubMed] [Google Scholar]
- Murugappan S, Tuluc F, Dorsam RT, Shankar H, Kunapuli SP. Differential role of protein kinase C delta isoform in agonist-induced dense granule secretion in human platelets. J Biol Chem. 2004;279:2360–2367. doi: 10.1074/jbc.M306960200. [DOI] [PubMed] [Google Scholar]
- Li Y, Martin LD, Spizz G, Adler KB. MARCKS protein is a key molecule regulating mucin secretion by human airway epithelial cells in vitro. J Biol Chem. 2001;276:40982–40990. doi: 10.1074/jbc.M105614200. [DOI] [PubMed] [Google Scholar]
- Singer M, Martin LD, Vargaftig BB, Park J, Gruber AD, Li Y, Adler KB. A MARCKS-related peptide blocks mucus hypersecretion in a mouse model of asthma. Nat Med. 2004;10:193–196. doi: 10.1038/nm983. [DOI] [PubMed] [Google Scholar]
- Park JA, He F, Martin LD, Li Y, Chorley BN, Adler KB. Human neutrophil elastase induces hypersecretion of mucin from well-differentiated human bronchial epithelial cells in vitro via a protein kinase C{delta}-mediated mechanism. Am J Pathol. 2005;167:651–661. doi: 10.1016/s0002-9440(10)62040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujise A, Mizuno K, Ueda Y, Osada S, Hirai S, Takayanagi A, Shimizu N, Owada MK, Nakajima H, Ohno S. Specificity of the high affinity interaction of protein kinase C with a physiological substrate, myristoylated alanine-rich protein kinase C substrate. J Biol Chem. 1994;269:31642–31648. [PubMed] [Google Scholar]
- Herget T, Oehrlein SA, Pappin DJ, Rozengurt E, Parker PJ. The myristoylated alanine-rich C-kinase substrate (MARCKS) is sequentially phosphorylated by conventional, novel and atypical isotypes of protein kinase C. Eur J Biochem. 1995;233:448–457. doi: 10.1111/j.1432-1033.1995.448_2.x. [DOI] [PubMed] [Google Scholar]
- Cabell CH, Verghese GM, Rankl NB, Burns DJ, Blackshear PJ. MARCKS phosphorylation by individual protein kinase C isozymes in insect Sf9 cells. Proc Assoc Am Physicians. 1996;108:37–46. [PubMed] [Google Scholar]
- Ishikawa T, Iwasaki E, Kanatani K, Sugino F, Kaneko Y, Obara K, Nakayama K. Involvement of novel protein kinase C isoforms in carbachol-stimulated insulin secretion from rat pancreatic islets. Life Sci. 2005;77:462–469. doi: 10.1016/j.lfs.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Leitges M, Gimborn K, Elis W, Kalesnikoff J, Hughes MR, Krystal G, Huber M. Protein kinase C-delta is a negative regulator of antigen-induced mast cell degranulation. Mol Cell Biol. 2002;22:3970–3980. doi: 10.1128/MCB.22.12.3970-3980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SH, Woo CH, Yoon SB, Kim JH. Protein kinase Cdelta functions downstream of Ca2+ mobilization in FcepsilonRI signaling to degranulation in mast cells. J Allergy Clin Immunol. 2004;114:1085–1092. doi: 10.1016/j.jaci.2004.07.035. [DOI] [PubMed] [Google Scholar]
- Yankaskas JR, Haizlip JE, Conrad M, Koval D, Lazarowski E, Paradiso AM, Rinehart CA, Jr, Sarkadi B, Schlegel R, Boucher RC. Papilloma virus immortalized tracheal epithelial cells retain a well-differentiated phenotype. Am J Physiol. 1993;264:C1219–C1230. doi: 10.1152/ajpcell.1993.264.5.C1219. [DOI] [PubMed] [Google Scholar]
- Lin H, Carlson DM, St George JA, Plopper CG, Wu R. An ELISA method for the quantitation of tracheal mucins from human and nonhuman primates. Am J Respir Cell Mol Biol. 1989;1:41–48. doi: 10.1165/ajrcmb/1.1.41. [DOI] [PubMed] [Google Scholar]
- Kajstura J, Cigola E, Malhotra A, Li P, Cheng W, Meggs LG, Anversa P. Angiotensin II induces apoptosis of adult ventricular myocytes in vitro. J Mol Cell Cardiol. 1997;29:859–870. doi: 10.1006/jmcc.1996.0333. [DOI] [PubMed] [Google Scholar]
- Mischak H, Pierce JH, Goodnight J, Kazanietz MG, Blumberg PM, Mushinski JF. Phorbol ester-induced myeloid differentiation is mediated by protein kinase C-alpha and -delta and not by protein kinase C-beta II, -epsilon, -zeta, and -eta. J Biol Chem. 1993;268:20110–20115. [PubMed] [Google Scholar]
- Li L, Lorenzo PS, Bogi K, Blumberg PM, Yuspa SH. Protein kinase Cdelta targets mitochondria, alters mitochondrial membrane potential, and induces apoptosis in normal and neoplastic keratinocytes when overexpressed by an adenoviral vector. Mol Cell Biol. 1999;19:8547–8558. doi: 10.1128/mcb.19.12.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Yu JC, Shin DY, Pierce JH. Characterization of a protein kinase C-delta (PKC-delta) ATP binding mutant: an inactive enzyme that competitively inhibits wild type PKC-delta enzymatic activity. J Biol Chem. 1995;270:8311–8318. doi: 10.1074/jbc.270.14.8311. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Soh JW, Jeoung DI, Cho CK, Jhon GJ, Lee SJ, Lee YS. PKC epsilon -mediated ERK1/2 activation involved in radiation-induced cell death in NIH3T3 cells. Biochim Biophys Acta. 2003;1593:219–229. doi: 10.1016/s0167-4889(02)00392-0. [DOI] [PubMed] [Google Scholar]
- Kleinbaum DG, Kupper LL, Muller KE. Boston, MA: PWS-Kent Publishing Co.; Applied Regression Analysis and Other Multivariable Methods. 1988 [Google Scholar]
- Szallasi Z, Denning MF, Smith CB, Dlugosz AA, Yuspa SH, Pettit GR, Blumberg PM. Bryostatin 1 protects protein kinase C-delta from down-regulation in mouse keratinocytes in parallel with its inhibition of phorbol ester-induced differentiation. Mol Pharmacol. 1994;46:840–850. [PubMed] [Google Scholar]
- Heit I, Wieser RJ, Herget T, Faust D, Borchert-Stuhltrager M, Oesch F, Dietrich C. Involvement of protein kinase Cdelta in contact-dependent inhibition of growth in human and murine fibroblasts. Oncogene. 2001;20:5143–5154. doi: 10.1038/sj.onc.1204657. [DOI] [PubMed] [Google Scholar]
- Mutter R, Wills M. Chemistry and clinical biology of the bryostatins. Bioorg Med Chem. 2000;8:1841–1860. doi: 10.1016/s0968-0896(00)00150-4. [DOI] [PubMed] [Google Scholar]
- Szallasi Z, Smith CB, Pettit GR, Blumberg PM. Differential regulation of protein kinase C isozymes by bryostatin 1 and phorbol 12-myristate 13-acetate in NIH 3T3 fibroblasts. J Biol Chem. 1994;269:2118–2124. [PubMed] [Google Scholar]
- Rose MC, Voynow JA. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol Rev. 2006;86:245–278. doi: 10.1152/physrev.00010.2005. [DOI] [PubMed] [Google Scholar]
- Williams OW, Sharafkhaneh A, Kim V, Dickey BF, Evans CM. Airway mucus: from production to secretion. Am J Respir Cell Mol Biol. 2006;34:527–536. doi: 10.1165/rcmb.2005-0436SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voynow JA, Young LR, Wang Y, Horger T, Rose MC, Fischer BM. Neutrophil elastase increases MUC5AC mRNA and protein expression in respiratory epithelial cells. Am J Physiol. 1999;276:L835–L843. doi: 10.1152/ajplung.1999.276.5.L835. [DOI] [PubMed] [Google Scholar]
- Ordoñez CL, Khashayar R, Wong HH, Ferrando R, Wu R, Hyde DM, Hotchkiss JA, Zhang Y, Novikov A, Dolganov G, Fahy JV. Mild and moderate asthma is associated with airway goblet cell hyperplasia and abnormalities in mucin gene expression. Am J Respir Crit Care Med. 2001;163:517–523. doi: 10.1164/ajrccm.163.2.2004039. [DOI] [PubMed] [Google Scholar]
- Kirkham S, Sheehan JK, Knight D, Richardson PS, Thornton DJ. Heterogeneity of airways mucus: variations in the amounts and glycoforms of the major oligomeric mucins MUC5AC and MUC5B. Biochem J. 2002;361:537–546. doi: 10.1042/0264-6021:3610537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Rocha AB, Mans DR, Regner A, Schwartsmann G. Targeting protein kinase C: new therapeutic opportunities against high-grade malignant gliomas? Oncologist. 2002;7:17–33. doi: 10.1634/theoncologist.7-1-17. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- Ko KH, Jo M, McCracken K, Kim KC. ATP-induced mucin release from cultured airway goblet cells involves, in part, activation of protein kinase C. Am J Respir Cell Mol Biol. 1997;16:194–198. doi: 10.1165/ajrcmb.16.2.9032127. [DOI] [PubMed] [Google Scholar]
- Abdullah LH, Conway JD, Cohn JA, Davis CW. Protein kinase C and Ca2+ activation of mucin secretion in airway goblet cells. Am J Physiol. 1997;273:L201–L210. doi: 10.1152/ajplung.1997.273.1.L201. [DOI] [PubMed] [Google Scholar]
- Scott CE, Abdullah LH, Davis CW. Ca2+ and protein kinase C activation of mucin granule exocytosis in permeabilized SPOC1 cells. Am J Physiol. 1998;275:C285–C292. doi: 10.1152/ajpcell.1998.275.1.C285. [DOI] [PubMed] [Google Scholar]
- Lounsbury KM, Stern M, Taatjes D, Jaken S, Mossman BT. Increased localization and substrate activation of protein kinase C delta in lung epithelial cells following exposure to asbestos. Am J Pathol. 2002;160:1991–2000. doi: 10.1016/s0002-9440(10)61149-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla A, Stern M, Lounsbury KM, Flanders T, Mossman BT. Asbestos-induced apoptosis is protein kinase C delta-dependent. Am J Respir Cell Mol Biol. 2003;29:198–205. doi: 10.1165/rcmb.2002-0248OC. [DOI] [PubMed] [Google Scholar]
- Matassa AA, Carpenter L, Biden TJ, Humphries MJ, Reyland ME. PKCdelta is required for mitochondrial-dependent apoptosis in salivary epithelial cells. J Biol Chem. 2001;276:29719–29728. doi: 10.1074/jbc.M100273200. [DOI] [PubMed] [Google Scholar]
- Chorley BN, Li Y, Fang S, Park JA, Adler KB. (R)-albuterol elicits antiinflammatory effects in human airway epithelial cells via iNOS. Am J Respir Cell Mol Biol. 2006;34:119–127. doi: 10.1165/rcmb.2005-0338OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page K, Li J, Zhou L, Iasvovskaia S, Corbit KC, Soh JW, Weinstein IB, Brasier AR, Lin A, Hershenson MB, Iasvoyskaia S. Regulation of airway epithelial cell NF-kappa B-dependent gene expression by protein kinase C delta. J Immunol. 2003;170:5681–5689. doi: 10.4049/jimmunol.170.11.5681. [DOI] [PubMed] [Google Scholar]
- Li J, O’Connor KL, Greeley GH, Jr, Blackshear PJ, Townsend CM, Jr, Evers BM. Myristoylated alanine-rich C kinase substrate-mediated neurotensin release via protein kinase C-delta downstream of the Rho/ROK pathway. J Biol Chem. 2005;280:8351–8357. doi: 10.1074/jbc.M409431200. [DOI] [PubMed] [Google Scholar]
- Wu DY-C, Wu R, Reddy SP, Lee YC, Chang MM. Distinctive epidermal growth factor receptor/extracellular regulated kinase-independent and -dependent signaling pathways in the induction of airway mucin 5B and mucin 5AC expression by phorbol 12-myristate 13-acetate. Am J Pathol. 2007;170:20–32. doi: 10.2353/ajpath.2007.060452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuo BG, Chow JY, Barrett KE, Isenberg JI. Protein kinase C potentiates cAMP-stimulated mouse duodenal mucosal bicarbonate secretion in vitro. Am J Physiol Gastrointest Liver Physiol. 2004;286:G814–G821. doi: 10.1152/ajpgi.00251.2003. [DOI] [PubMed] [Google Scholar]
- Chow JY, Uribe JM, Barrett KE. A role for protein kinase cepsilon in the inhibitory effect of epidermal growth factor on calcium-stimulated chloride secretion in human colonic epithelial cells. J Biol Chem. 2000;275:21169–21176. doi: 10.1074/jbc.M002160200. [DOI] [PubMed] [Google Scholar]
- Song JC, Hanson CM, Tsai V, Farokhzad OC, Lotz M, Matthews JB. Regulation of epithelial transport and barrier function by distinct protein kinase C isoforms. Am J Physiol Cell Physiol. 2001;281:C649–C661. doi: 10.1152/ajpcell.2001.281.2.C649. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Zhang H, Saito N, Kojima I, Urano T, Mogami H. Glucagon-like peptide 1 activates protein kinase C through Ca2+-dependent activation of phospholipase C in insulin-secreting cells. J Biol Chem. 2006;281:28499–28507. doi: 10.1074/jbc.M604291200. [DOI] [PubMed] [Google Scholar]
- Krunkosky TM, Fischer BM, Martin LD, Jones N, Akley NJ, Adler KB. Effects of TNF-alpha on expression of ICAM-1 in human airway epithelial cells in vitro: signaling pathways controlling surface and gene expression. Am J Respir Cell Mol Biol. 2000;22:685–692. doi: 10.1165/ajrcmb.22.6.3925. [DOI] [PubMed] [Google Scholar]
- Woo C-H, Lim J-H, Kim J-H. VCAM-1 upregulation via PKCδ-p38 kinase-linked cascade mediates the TNF-α-induced leukocyte adhesion and emigration in the lung airway epithelium. Am J Physiol. 2005;288:L307–L316. doi: 10.1152/ajplung.00105.2004. [DOI] [PubMed] [Google Scholar]
- Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–98. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- Keenan C, Goode N, Pears C. Isoform specificity of activators and inhibitors of protein kinase C gamma and delta. FEBS Lett. 1997;415:101–108. doi: 10.1016/s0014-5793(97)01104-6. [DOI] [PubMed] [Google Scholar]
- Wang QJ, Acs P, Goodnight J, Giese T, Blumberg PM, Mischak H, Mushinski JF. The catalytic domain of protein kinase C-delta in reciprocal delta and epsilon chimeras mediates phorbol ester-induced macrophage differentiation of mouse promyelocytes. J Biol Chem. 1997;272:76–82. doi: 10.1074/jbc.272.1.76. [DOI] [PubMed] [Google Scholar]
- Clark AS, West KA, Blumberg PM, Dennis PA. Altered protein kinase C (PKC) isoforms in non-small cell lung cancer cells: pKCdelta promotes cellular survival and chemotherapeutic resistance. Cancer Res. 2003;63:780–786. [PubMed] [Google Scholar]
- Nabha SM, Glaros S, Hong M, Lykkesfeldt AE, Schiff R, Osborne K, Reddy KB. Upregulation of PKC-delta contributes to antiestrogen resistance in mammary tumor cells. Oncogene. 2005;24:3166–3176. doi: 10.1038/sj.onc.1208502. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JS, Kang CM, Yoo MB, Kim SJ, Yoon HK, Kim YK, Kim KH, Moon HS, Park SH. Nitric oxide induces MUC5AC mucin in respiratory epithelial cells through PKC and ERK dependent pathways. Respir Res. 2007;8:28. doi: 10.1186/1465-9921-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]