

Abstract
The misfolding and aggregation of normally soluble proteins has emerged as a key feature of several neurodegenerative diseases. In Parkinson’s disease, progressive loss of dopaminergic neurons is accompanied by polymerization of the cytoplasmic protein α-synuclein (αS) into filamentous inclusions found in neuronal somata (Lewy bodies) and dendrites (Lewy neurites). Similar αS aggregates occur in cortical neurons in dementia with Lewy bodies. Numerous reports now indicate that αS can interact with lipids. We previously found that treating dopaminergic cells expressing αS with polyunsaturated fatty acids (PUFAs) induced the formation of soluble, sodium dodecyl sulfate-stable oligomers whereas treatment with saturated fatty acids did not. Here, we examine the relevance of αS-PUFA interactions to the development of Parkinson’s disease-like cytopathology. Exposure of αS-overexpressing dopaminergic or neuronal cell lines to physiological levels of a PUFA induced the formation of proteinaceous inclusions in the cytoplasm. Kinetic experiments in-dicated that PUFA-induced soluble oligomers of αS precede these Lewy-like inclusions. Importantly, we found that αS oligomers were associated with cyto-toxicity, whereas the development of Lewy-like inclusions appeared to be protective. We conclude that alterations in PUFA levels can lead to aggregation of αS and subsequent deposition into potentially cyto-toxic oligomers that precede inclusions in dopaminergic cells.
α-Synuclein (αS) is a presynaptic protein implicated in Parkinson’s disease (PD) at the levels of cytopathology and genetics.1,2,3 In PD and the various clinically distinct but neuropathologically related neurodegenerative disorders in which αS also accumulates (the synucleinopathies), there appears to be a progressive conversion of the highly soluble αS protein into insoluble, β-sheet-rich filamentous assemblies, resulting in its intraneuronal deposition into Lewy bodies (LBs) and Lewy neurites, the cytopathological hallmarks of this group of disorders.
Biophysical studies have shown that αS is “natively unfolded”4; however, on it’s binding to acidic phospholipid vesicles in vitro, αS undergoes major conformational changes, resulting in an α-helical structure.5 The interactions of αS with phospholipids are mediated by its N-terminal region, which contains sequence homologies to the amphipathic, lipid-binding α-helices of class A2 apolipoproteins.5,6,7 The conserved structural similarity to the exchangeable apolipoproteins appears to explain the normal partitioning of αS between the aqueous and membranous compartments of the cytoplasm.5,8,9,10,11
αS-Lipid interactions can affect the kinetics of its aggregation in vitro.12,13,14,15,16,17 In particular, αS interactions with polyunsaturated fatty acids (PUFAs) can rapidly and dynamically affect its oligomerization and further aggregation.17,18,19,20,21 Given its primary structure, subcellular distribution to membranes, and conformational change on lipid binding, it is likely that αS interacts with lipids as part of its still undefined physiological function. For example, αS has been implicated by some studies in membrane lipid regulation and membrane trafficking.21,22,23,24,25,26
In accord with the initial discovery that αS expression regulates cytosolic and membrane PUFAs levels,20 several studies have recently shown that αS expression affects fatty acid (FA) uptake and metabolism.27,28,29 Specifically, decreases in certain PUFAs and increases in certain saturated fatty acid (SFA) levels were detected in phospholipids of αS−/− mouse brains. Moreover, the steady-state mass of neutral lipids is increased in brains of αS−/− mice.29 In addition to the evidences for a role for αS in FA regulation, αS gene expression was reported to be up-regulated in response to PUFA-enriched diets in rats.30,31 In the context of the fact that αS gene duplication or triplication and resultant αS overexpression causes familial PD,32 it is possible that qualitative or quantitative changes in PUFAs could serve as risk factors for PD through an effect on αS expression and/or aggregation.
Here, we report that PUFA-induced soluble oligomers precede the formation of proteinaceous cytoplasmic inclusions in neuronal cell lines. The resultant Lewy-like inclusions react with antibodies to αS, phosphorylated αS, ubiquitin, and HSP-70. Further, we provide evidence that PUFA-induced soluble oligomers confer cytotoxicity, whereas PUFA-induced inclusions may be protective. We discuss the implications of these new findings for the mechanism of neuronal dysfunction in PD and other synucleinopathies.
Materials and Methods
Cell Cultures, Western Blotting, Immunoprecipitation
The mesencephalic cell lines MES 23.5 and MN9D, which have dopaminergic properties,33,34 and the neuronal cell line SK-N-SH were stably transfected with wild-type human αS cDNA in the pCDNA 3.1 vector using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). A reported feature of αS-transfected cells is that all stable clones gradually lose αS expression after being continuously passaged for 2 to 3 months or more.9 To overcome this technical problem and increase consistency of results, we kept frozen aliquots of αS-overexpressing clones. The clones were frozen at 55 to 65 days after DNA transfection, and fresh aliquots were thawed routinely every 4 to 8 weeks. We kept track of the time that a specific clone was maintained in culture from thawing: young clones 2 to 4 weeks, intermediate clones 4 to 6 weeks, and old clones 6 to 8 weeks. Conditioning living cells with FAs and cell fractionation were as described previously.17 Protein samples of high-speed supernatant (after 280,000 × g) were incubated at 65°C for 16 to 18 hours17 before loading on an 8 to 16% NuPAGE Bis-Tris (Invitrogen) or 14% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Immunoblots reacted with H3C anti-αS antibody (Ab; gift from Julia George, University of Illinois, Urbana-Champaign, IL),6 anti-ubiquitin Ab (Stressgen Bioreagents, Ann Arbor, MI) or anti-phospho Ser129 αS Ab (Wako-Chem, Osaka, Japan). Immunoprecipitation from high-speed cytosols was conducted as previously reported35 with anti-ubiquitin Abs (MBL, Nagoya, Japan; and Stressgen Bioreagents, MI) and anti-αS antibodies, H3C6 and Syn-1 (Transduction Laboratories, Lexington, KY).
Primary Neuronal Culture
Cortical cell cultures were prepared as described previously.36 Briefly, the cortex region was dissected from 1- to 2-day-old C57BL/6 mice obtained from the Jackson Laboratories (Bar Harbor, ME), dissociated by trypsin treatment, followed by trituration with a siliconized Pasteur pipette, and then plated in 60-mm dishes coated with poly-d-lysine (Sigma, St. Louis, MO). Culture medium consisted of minimal essential medium (Invitrogen), 0.6% glucose, 0.1 g/L bovine transferrin (Calbiochem, La Jolla, CA), 0.25 g/L insulin (Sigma), 0.3 g/L glutamine, 5 to 10% fetal calf serum (Sigma), 2% B-27 supplement (Invitrogen). To eliminate the glia cells, 8 μmol/L cytosine b-d-arabinofuranoside (Sigma) were added to the culture 3 days after preparation and removed after an additional 3 to 4 days. Cultures were maintained at 37°C in a 95% air/5% CO2 humidified incubator, and culture medium was replaced every 4 to 7 days. Experiments were performed on cultures grown for ∼14 days.
Immunocytochemistry (ICC)
For immunostaining, cells were prepermeabilized with 0.002% Triton X-100 in phosphate-buffered saline (PBS) for 1 minute. Cells were then fixed with 4% paraformaldehyde for 10 minutes on ice and permeabilized with 0.2% Triton X-100 in PBS and 1% goat serum for 5 minutes at room temperature. The slides were next treated with 70% formic acid for 15 minutes at room temperature followed by extensive washes and blocking with 1.5% goat serum in PBS. Slides were then reacted with primary Abs: anti-αS monoclonal Ab (LB509 1:100; Zymed, South San Francisco CA); anti-ubiquitin polyclonal Ab (1:100; DAKO, Glostrup, Denmark), anti-HSP 70 polyclonal Ab (1:100; Medical and Biological Laboratories, Nagoya, Japan) or anti-phospho Ser129 monoclonal Ab (1:100, Wako-Chem) and secondary Ab at 1:200, anti-mouse Alexa flour 488 (Molecular Probes, Eugene, OR) and anti-rabbit-Cy5 (Jackson Laboratory). Slides were sealed with mounting medium (catalog no. M1289; Sigma, Rehovot, Israel). Slides were analyzed by confocal microscopy with laser argon 488 (filter BA 510 IF dichroic mirror filter) and laser helium neon 633 (filter BA 660 IF) (laser-scanning microscope 410, Zeiss, Oberkochen, Germany). Inclusions were counted and sized independently by two investigators blinded to the experimental conditions, using Image Pro/Image J softwares (Media Cybernetics Inc., Silver Spring, MD). The average number of the two counts is reported.
Cell Viability Assay
To determine cell viability, cells were plated in 96-well plates in standard medium 1 day before they were treated with specific media for the time indicated. Two cell-viability assay kits were used to determine the metabolic activity of the cells: XTT and WST-1 (Roche Diagnostics, Mannheim, Germany). The assay kits were used according to the manufacturer’s recommendations and yielded similar results.
Results
PUFAs Induce Endogenous αS Oligomerization and Aggregation in Primary Neurons
We first asked whether PUFAs could induce the oligomerization of αS at endogenous levels in primary neurons. For this aim, we treated primary cortical neurons of normal mouse brains (14 days in culture) in serum-free medium supplemented with a relatively low physiological concentration (50 μmol/L) of α-linolenic acid (ALA, 18:3) or oleic acid (OA, 18:1) for 18 hours, together with bovine serum albumin (BSA) as a well-studied FA-carrier protein (at a concentration of 10 μmol/L; see Materials and Methods). In parallel, we maintained sister cultures with FA-free BSA to control for basal αS oligomer levels. We probed for the appearance of monomer and soluble oligomers by Western blotting with H3C Ab. Although levels of the αS monomer (∼17 kDa) were not altered by the FA treatments, enhanced levels of αS oligomers, including dimers (∼35 kDa), trimers (∼53 kDa), and higher species, were readily detected in the PUFA-treated neurons versus levels in the OA- or BSA-treated control neurons (Figure 1a). Therefore, in agreement with our previous results,17 18:3 PUFA but not 18:1 monounsaturated fatty acid (MUFA) induces αS oligomerization of endogenous αS in primary neurons.
Figure 1.

α-Linolenic acid, an 18:3 PUFA but not oleic acid an 18:1 MUFA, induces the oligomerization and aggregation of endogenous αS in primary cortical neurons. Cortical primary cultures (at 10 to 14 days in vitro) from normal mouse brains were conditioned for 18 hours in serum-free medium supplemented with BSA with or without 50 μmol/L 18:1 or 18:3. a: Samples of high-speed cytosols (after 280,000 × g) were incubated at 65°C overnight (see Materials and Methods), loaded on 14% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and blotted with H3C αS antibody. b: ICC of sister primary cultures treated with BSA with or without FAs or standard serum-supplemented medium as above and reacted with H3C anti-αS Ab. Scale bars = 20 μm.
We next performed ICC to analyze FA’s potential effect on αS cytopathology in primary cortical cultures (13 days in culture) of normal mouse brain. We treated the primary cultures with 50 μmol/L of 18:3, 18:1, or with BSA only (as above) and compared it to a sister culture, conditioned in parallel, in standard serum-containing medium. The cultures were probed with the anti-αS Ab, H3C. Although αS distribution in the BSA and 18:1-treated cultures was diffused, a more punctuate appearance of αS was observed in the 18:3 PUFA and standard serum culture (Figure 1b). The 18:3 effect was indistinguishable from standard serum-supplemented medium, indicating that 18:3 at 50 μmol/L acted in the physiological range. We concluded that the FA effect on αS cytopathology corresponds with the effect on αS oligomerization. Because we wished to assess the cytopathological effects of the PUFA-induced accumulation of soluble oligomers, but endogenous mouse αS does not accumulate into Lewy-like inclusions,37 we performed most of the following experiments in two dopaminergic lines, ie, MES 23.5 and MN9D, and verified the generality of PUFAs effect in SK-N-SH neuronal cells overexpressing human αS. We chose clones with αS overexpression of onefold to twofold over the endogenous mouse brain expression level.
PUFA-Induced αS Oligomers Include Phosphorylated but Not Ubiquitinated Species
To better understand the nature of PUFA-inducible soluble αS oligomers, we asked whether they are phosphorylated at serine 129 or ubiquitinated. These two modifications have been observed in αS-positive inclusions in several human synucleinopathies and were shown to occur in high molecular weight insoluble αS forms.37,38,39,40,41,42,43 We supplemented the medium of human wt αS-expressing MES 23.5 dopaminergic cells with BSA only (at a concentration of 50 μmol/L) to assess basal oligomer levels or else BSA + 250 μmol/L 18:3 for 16 to 18 hours. We probed for the appearance of soluble oligomers by Western blotting with H3C (to total αS), anti-phosphoserine 129, and anti-ubiquitin antibodies.
In full accord with our previous work,17 PUFA treatment consistently induced the formation of soluble αS oligomers that were observed in the high-speed supernatant after heat delipidation (Figure 2). With the antibody H3C, we clearly detected αS dimers and trimers in the PUFA + BSA-treated cultures but in very low levels in the BSA-only cultures; the dimer was more abundant than the trimer (Figure 2a). Monomer levels in the cytosols were only slightly and insignificantly altered by the PUFA treatment, as reported.17 Anti-phosphoserine 129 reacted with the monomer, the abundant dimer, and the trimer (Figure 2b), suggesting that a fraction of the αS species is phosphorylated at serine 129. Western blotting the same samples with an anti-ubiquitin antibody (Stressgen) did not detect the soluble oligomers in these high-speed cytosols (Figure 2c). Further, no ubiquitin immunoreactivity was detected in αS oligomers that had been immunoprecipitated with an anti-αS Ab (using either H3C or Syn-1, data not shown). These results suggest that soluble oligomers initially induced by PUFAs in cultured dopaminergic cells are phosphorylated but not ubiquitinated. Next, we observed that PUFAs induce αS oligomer formation in a dose-dependent manner (Figure 2, d and e), and this begins at low physiological concentrations of 50 μmol/L of 18:3. At higher PUFA concentrations of 250 and 500 μmol/L that are still in the range found in normal serum, the level of soluble αS oligomers (specifically, dimers, trimers, tetramers, and heptamers) was further increased (Figure 2e) and the formation of insoluble, gel-excluded αS forms was detected (Figure 2d, arrow). Some variations in the basal and PUFA-induced levels of oligomers were observed. These variations were related to the αS expression level and the age of individual αS expressing lines [eg, compare the basal oligomers levels (without PUFA) in Figure 2, a and d (see below)].
Figure 2.

18:3 PUFA-induced αS oligomers are phosphorylated but not ubiquitinated. a: High-speed cytosols (15 μg protein) of human wt αS stably-transfected MES cells conditioned in serum-free medium with or without 18:3 (250 μmol/L) for 16 hours. Samples were treated at 65°C overnight before gel loading and blotting with H3C Ab. Samples are run in duplicate; each lane represents a separate dish (sister culture) treated and processed in parallel. b: The 18:3-induced αS oligomers are phosphorylated at serine 129. Immunoblot reacted with anti-phospho 129 Ab. c: The 18:3-induced αS soluble oligomers are not ubiquitinated. Immunoblot reacted with anti-ubiquitin Ab. d: 18:3-Induced αS oligomers rise in a dose-dependent manner. Cells were conditioned with 18:3 at the concentrations indicated, and protein samples were prepared and analyzed as in a. Immunoblot reacted with H3C Ab. e: Densitometric analysis of blot in d, showing the ratio of total oligomers (dimers, trimers, tetramers, and heptamers) to monomer.
PUFAs Induce the Formation of Lewy-Like Inclusions in αS-Overexpressing Neuronal Cell Lines
To search for a potential role of PUFA-dependent αS oligomerization in PD-type lesion formation, naïve and αS-overexpressing MES dopaminergic cells were conditioned overnight in serum-free medium supplemented with BSA (a well-characterized FA carrier protein) or with BSA-FA complexes. The cells were then processed for ICC to detect proteinaceous cytoplasmic inclusions and, in parallel, were subjected to Western blotting to analyze αS oligomerization. We systematically performed these analyses every 2 weeks to observe any gradual change in αS response to PUFA treatment throughout the lifetime of a specific human αS-expressing clone. This step is important in view of the reported tendency for apparently stable αS-expressing clonal lines to lose αS expression throughout time. Therefore, to improve consistency of results, stable clones were frozen in aliquots right after verifying αS overexpression at 65 ± 5 days after DNA transfection. A fresh aliquot was thawed every time a clone maintained in culture lost its αS expression (usually ∼17 to 19 weeks after DNA transfection or ∼8 to 10 weeks after thawing of a new aliquot). Overall, we followed two different wt αS MES clones thoroughly and then repeated specific time points with two additional wt αS MES clones. We then repeated this experiment with MN9D dopaminergic and SK-N-SH neuronal cells (at ∼12 to 14 weeks after DNA transfection) to ensure that our observations were not restricted to MES cells or dopaminergic cells.
Two weeks after thawing an aliquot of stable αS-expressing MES cells, we detected principally soluble αS that, by ICC, was diffusely distributed in the cytoplasm; this signal could be washed out by prepermeabilizing the cells with Triton X-100 (see Materials and Methods) (Figure 3). The remaining low signal that was consistently observed after this prepermeabilization step represents Triton-insoluble forms of αS and always appeared at higher levels in the 18:3-treated cultures (Figure 3b). Note that without Triton prepermeabilization, the differences between BSA and PUFA treatments could not be appreciated because of the overexpression of αS. At 2 to 4 weeks in culture (ie, after thawing an aliquot), the signal was still primarily diffuse but it was washed out less completely by the prepermeabilization step (Figure 4a). No discrete inclusions were detected at this time point, either with or without 16 to 18 hours of PUFA treatment (quantified in Table 1). However, spherical inclusions were clearly and consistently observed in clones maintained in culture for 5 to 6 weeks after thawing and then treated with 18:3 for 16 to 18 hours (Figures 4b and 5b; Table 1). In the longest-lived clones (cultured for 6 to 8 weeks after thawing), spherical inclusions were frequently detected in the BSA-treated culture, and 16 hours of treatment with BSA-18:3 increased both the size and the number of the inclusions (Figures 4c and 5c; Table 1).
Figure 3.

The detection of insoluble αS forms by ICC requires the removal of soluble αS forms. a: Stably αS-transfected MES cells were conditioned in serum-free medium supplemented with the PUFA 18:3 (right) or without (left) for 16 hours and processed for ICC with anti-αS Ab (LB509, Zymed) followed by Alexa 488 (green). b: Cells were treated and processed for ICC as in a, but the soluble αS forms were washed out by prepermeabilization of the cells with 0.002 Triton X-100 before fixation. Note that the pictures in a and b were taken under identical conditions of laser intensity and exposure.
Figure 4.

PUFAs induce the formation of Lewy-like inclusions in dopaminergic MES 23.5 cells. αS-Overexpressing MES 23.5 lines were maintained in culture under standard serum conditions for the times indicated and then transferred to serum-free medium supplemented with BSA + PUFA (250 μmol/L of 18:3) for 16 to 18 hours. Cells were then processed for ICC using antibodies against αS (LB509, Zymed) and ubiquitin (DAKO), followed by Alexa 488 and Cy5, respectively. Note that differences in morphology of αS-overexpressing MES23.5 cells are routinely observed with time in culture. Scale bars = 10 μm.
Table 1.
The Number and Size of Proteinaceous Cytosolic Inclusions in MES Cells
| Time in culture | BSA alone
|
BSA + ALA ( 250 μM, 18:3)
|
||
|---|---|---|---|---|
| Inclusions/cell (cells counted) | Inclusion diameter (μM) ± SEM | Inclusions/cell (cells counted) | Inclusion diameter (μM) ± SEM | |
| 2–4 weeks | 0.07 (55) | 0.19* (54) | ||
| 4–6 weeks | 0.41 (53) | 2.65 ± 1.47 | 6.84* (49) | 10.99 ± 5.69 |
| 6–8 weeks | 1.33 (42) | 3.96 ± 1.26 | 14.16* (45) | 11.50 ± 4.69 |
P < 0.05 (t-test).
Figure 5.

PUFAs induce the formation of Lewy-like inclusions in dopaminergic MES cells. ICC using antibodies against αS and HSP70 followed by Alexa 488 and Cy5, respectively. Conducted just as in legend of Figure 4.
To characterize further these PUFA-induced inclusions, we stained the inclusions for specific proteins that have been reported to be constituents of human LBs. We double-stained for αS plus ubiquitin43,44(Figure 4), αS plus HSP-7045(Figure 5), and phosphoSer129 plus ubiquitin46(Figure 6a). We found that the PUFA-induced inclusions in these dopaminergic cells contain proteins that are components of the human LB. We therefore designate the inclusions as Lewy-like inclusions. Quantification revealed that the average number of Lewy-like inclusions per cell rose ∼9- to 16-fold after 18:3 treatment, and their mean size increased approximately threefold to fourfold (Table 1). A longer chain PUFA, ie, 20:4, was more potent in inducing Lewy-like inclusions than was 18:3 (data not shown). Very importantly, no inclusions were detected in cells treated at the same concentrations with an SFA of identical carbon chain length (18:0 or 20:0; n = 37 and n = 32 cells counted, respectively). We conclude that there is a gradual appearance of spherical cytoplasmic inclusions immunoreactive for αS that rise both in size and number during the time an MES dopaminergic culture is expressing human αS, and this process is markedly and significantly enhanced by exposing the cells overnight to 18:3 PUFA but not to 18:0 SFA. To ensure that the formation of PUFA-induced inclusions was not restricted to the MES 23.5 or to the dopaminergic cell line, we treated MN9D, a second dopaminergic cell line, and also SK-N-SH neuronal cells with PUFA and found similar inclusions in the αS-overexpressing cells (Figure 6, b and c). These inclusions were indistinguishable from the inclusions detected in the αS-overexpressing MES cells (Figures 4, 5, and 6a). Importantly, we tested the specificity of PUFA effect on αS deposition in Lewy-like inclusions. For this we have treated βS-, in parallel to αS-overexpressing MES cells, with 18:3 (at 250 μmol/L) and analyzed them for inclusion formation by ICC. Inclusions were not detected in the βS-overexpressing cells, which were clearly and abundantly detected in the αS-overexpressing cells treated and analyzed in parallel. Therefore, PUFAs specifically induce αS cytopathology.
Figure 6.

PUFAs induce the formation of Lewy-like inclusions in MN9D dopaminergic and SK-N-SH cells. a: The inclusions in MES 23.5 dopaminergic cells are immunoreactive for phosphoSer-129 αS. ICC performed as in Figure 4 using antibodies against phosphoSer129 αS and ubiquitin, followed by Alexa 488 and Cy5, respectively. b: Similar inclusions arise in MN9D dopaminergic cells with antibodies against αS (H3C) and ubiquitin (DAKO). c: Similar inclusions arise in SK-S-SH undifferentiated cells.
Soluble αS Oligomers Precede the Formation of Lewy-Like Cytoplasmic Inclusions
To determine whether PUFA-induced soluble oligomers precede the formation of Lewy-like inclusions, we followed the time course of oligomer accumulation using Western blotting in parallel to detection of inclusion formation by ICC and using two different 18:3 concentrations (ie, 125 and 250 μmol/L). We first treated αS-expressing MES cells with 250 μmol/L 18:3 and asked how soon the cell responds to PUFA by αS oligomerization and by inclusion formation. Cells were conditioned in the specific medium for the number of hours indicated in Figure 6. Cells were then harvested, and equal protein amounts of the high-speed cytosols were incubated at 65°C and Western blotted. In accord with our previous results,17 the levels of αS oligomers, particularly the dimer, rose beginning 2 hours after addition of PUFA to the culture medium (Figure 7b). A further increase in the amount of αS oligomers occurred with time up to 8 hours. Some reduction in the amount of soluble αS oligomers was observed after 16 hours of incubation with 18:3 at 250 μmol/L. This reduction occurs in parallel with an accumulation of insoluble αS forms, as reported previously.17 In sister cultures treated in parallel and processed for ICC, we detected multiple cytoplasmic inclusions starting at 6 hours. A further increase in the number of inclusions per cell occurred with time up to 16 hours (Figure 7a).
Figure 7.
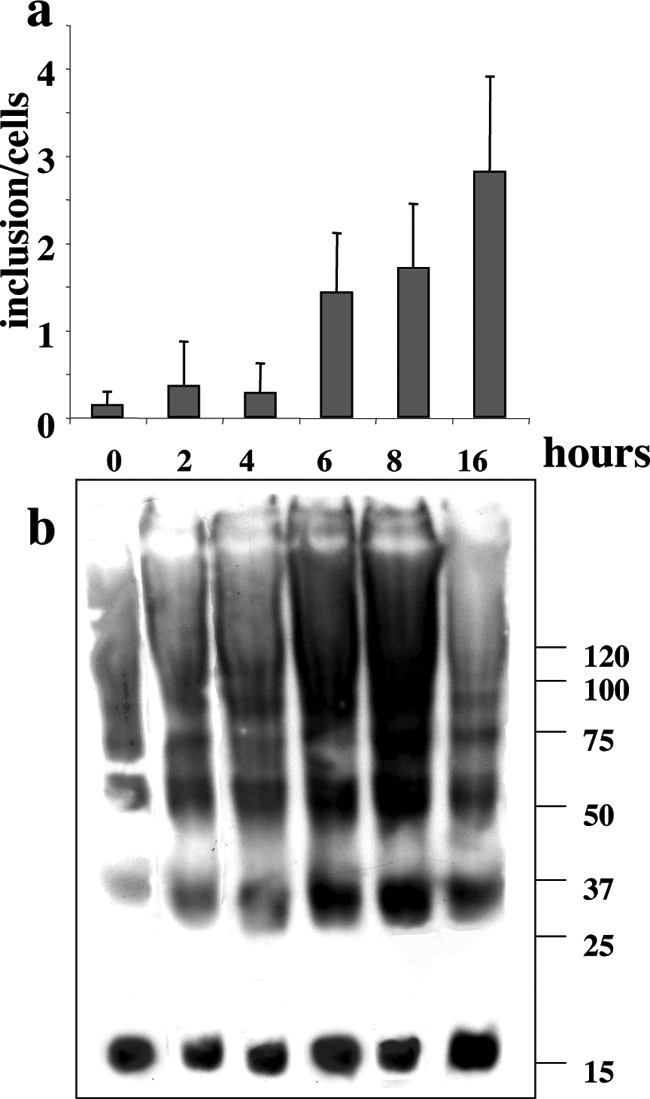
PUFA-induced soluble oligomers precede the formation of Lewy-like inclusions. Cells were conditioned in serum-free medium supplemented with BSA alone or BSA + PUFA (18:3, 250 μmol/L) for the times indicated. a: Samples (15 μg) of high-speed supernatant (after 280,000 × g cytosol) were treated at 65°C for 16 hours before gel loading and blotting with H3C Ab. b: Sister cultures were processed for ICC, and Lewy-like inclusions were detected using anti-αS and anti-ubiquitin Abs. Mean number of inclusions per cell (n = 32 to 50 cells counted at each time point) ± SD. Inclusions were counted independently by two investigators blinded to the experimental conditions. Pictures were captured with Image Pro and processed with Image J software (Media Cybernetics Inc., Silver Spring, MD).
The above experiments involved exposing the cells to concentrations of 250 μmol/L FA for 16 hours. This FA concentration is lower than plasma FA levels (∼500 μmol/L), but it represents a high concentration for a single FA. We therefore conducted similar experiments but with a concentration of 125 μmol/L and obtained very similar results, although with an expanded time scale. To observe PUFA-induced oligomerization and inclusions with the lower concentration, we conditioned αS-overexpressing cells in 18:3-containing medium for 8, 16, 24, and 48 hours. Sister cultures treated with BSA alone were maintained in parallel for the same intervals. Cells were then processed for ICC, or else they were harvested and equal protein amounts of the high-speed cytosols were Western blotted. An increase in the levels of sodium dodecyl sulfate-stable, soluble αS oligomers in the cytosols occurred in response to 125 μmol/L 18:3 with time up to 16 hours. Longer incubation times of 24 and 48 hours resulted in a decline in total soluble oligomer levels by Western blot (Figure 8, a and b). Cytoplasmic inclusions were detected after 24 hours, and a higher number of inclusions was detected after 48 hours in αS-overexpressing cells conditioned in medium supplemented with 125 μmol/L 18:3 but not in parallel cultures treated with BSA alone or 18:1. A gradual shift in the ratio of oligomers to inclusion number was observed. Maximal oligomer levels with very few inclusions are detected at 16 hours; however, at 48 hours the inverse situation is observed, with reduced oligomer levels and a dramatic increase in the number of inclusions. This result suggests that oligomers are consumed to form inclusions. The inclusions detected on 125 μmol/L PUFA treatment did not differ significantly from those detected with our 250 μmol/L PUFA protocol in terms of size and immunoreactivity to αS, HSP 70, and ubiquitin (data not shown). Therefore, the results with lower PUFA levels further support the conclusion that αS dynamically responds to PUFA (but not equimolar SFA or MUFA) by oligomerization and aggregation, and αS oligomers precede the formation of Lewy-like inclusions in this system.
Figure 8.

PUFA-induced αS oligomers induce cytotoxicity. αS-Overexpressing MES dopaminergic cells were conditioned with BSA alone or BSA plus FA (125 μmol/L) for the indicated time. a: Samples (15 μg) of high-speed supernatant (after 280,000 × g) were incubated at 65°C overnight before gel loading and blotting with H3C Ab. B, BSA-treated; 2, 8, 16, 24, 48 = hours of conditioning in the presence of 18:3. b: Densitometric analysis of the blot in a, presented as the ratio of total oligomers to monomer shown in comparison to the number of inclusions in sister cultures treated in parallel for each time point. Inclusions were counted as described in legend of Figure 6b. nd, not done. c: Values of XTT assay representing cellular metabolic activity at the indicated time points in sister cultures (conducted in parallel to a and b), presented as the relative activity in 18:3-treated to 18:1 (control) treated cultures. Mean of five to six replicates at each time point ± SD; experiment repeated three times.
The Kinetics of Cell Viability Suggest that Oligomers Are Toxic and Inclusions Are Protective
To search for cell toxicity effects of the PUFA-induced αS oligomers and higher molecular weight aggregates, we compared cell viability using two cytotoxicity assays, both relying on the cleavage of tetrazolium salts and producing soluble formazan salts (XTT or WST1; Roche Diagnostics). The assays determine the metabolic activity of the cells through the specific enzymatic activity tested.
The above time course with the lower 18:3 concentration (125 μmol/L) indicated that αS-immunoreactive inclusions appeared only after ∼24 hours of PUFA treatment. Before 24 hours, PUFA treatment was principally associated with αS oligomer formation. These experimental conditions enabled us to discriminate between the accumulation of oligomers and the development of microscopically visible inclusions. Naïve (untransfected) and αS-overexpressing MES cell lines were conditioned in parallel with and without 125 μmol/L 18:3 or 18:1 for the times indicated in Figure 8 and then tested with both cell viability assays. The MUFA 18:1 was used as a control for the effects of a FA of identical carbon chain length that does not induce αS oligomerization.17
Whereas the FAs did not affect cell viability values of naïve MES cells, they did affect the viability of αS-overexpressing lines. The maximal effect on cell viability was observed for αS-overexpressing cells after 16 hours of exposure to 18:3 (125 μmol/L), with a statistically significantly reduced viability of ∼12% (t-test, P = 0.0402) relative to cells conditioned in parallel with 18:1 (125 μmol/L) (Figure 8c). After 24 hours of exposure to 18:3, viability was restored for the αS-overexpressing cells and was very similar to the value observed for the naïve MES cells. Only slight changes in viability were observed between 24 and 48 hours of FAs treatment. Therefore, cell toxicity was associated with the occurrence of oligomers and not with the advent of inclusion formation.
Discussion
We studied the role of PUFAs and their interactions with αS in relation to the development of cytopathogenic features resembling those of PD. We found that exposing cultured dopaminergic cells to PUFA at physiological FA levels resulted in the accumulation of αS-soluble oligomers that included phosphorylated but not ubiquitinated αS species. Using kinetic experiments, we found that PUFA-induced soluble oligomers invariably precede the formation of Lewy-like cytoplasmic proteinaceous inclusions in our cultures and therefore may be intermediates in the time-dependent process leading to deposition of LBs. Importantly, the effects observed in response to exposure to PUFAs were not detected with SFAs or MUFAs of identical carbon chain length. Based on cell viability measurements, we found that PUFA-induced αS oligomers are associated with cytotoxicity, ie, reduced cellular metabolic activity, when compared with PUFA-treated naïve cells that do not overexpress αS. Further, we found that although PUFA-induced αS oligomers are associated with cytotoxicity, PUFA-induced inclusions appear to be protective, ie, they are associated with cellular metabolic activity comparable to those of naïve cells. The peak in cytotoxicity observed under our experimental conditions, ie, at 16 hours of exposure to 125 μmol/L 18:3, correlates with the accumulation of αS oligomers, whereas the restoration of viability at 24 and 48 hours correlates with the appearance of Lewy-like inclusions.
The current study is an extension of a series of recent studies in which we have documented different aspects of the interaction of neuronal αS with FA, in particular with PUFAs, under normal and pathological conditions.9,17,20 In relation to pathogenesis, we found that the accumulation of αS in high molecular weight assemblies, including certain soluble oligomers and insoluble aggregates, is associated with alterations in brain PUFA composition but not MUFA or SFA. In an attempt to correlate the altered PUFA composition in the brains of patients with PD or dementia with Lewy bodies with αS expression and accumulation as high molecular weight assemblies, we examined FA profiles in mesencephalic dopaminergic cells that stably express αS and found accumulations of PUFAs in the high-speed cytosol as well as in membrane fractions on αS overexpression.20 Conversely, declines in certain PUFAs were detected in the cytosolic and membrane fractions of brains of αS−/− compared to wt mice. We also found that the αS-dependent changes in membrane PUFA levels were reflected in altered biophysical properties such as membrane fluidity.20
A common pathogenic finding among clinically, pathologically, and biochemically diverse neurodegenerative diseases such as PD, Alzheimer’s disease, amyotrophic lateral sclerosis, Huntington’s disease, and prion disorders is the deposition of inclusion bodies that contain abnormally aggregated proteins. Although inclusion bodies are a frequent, often invariant, cytopathological feature of human neurodegeneration, there is an ongoing debate as to the role of the aggregation process in the pathogenesis of each disease. Indirect lines of evidence have linked protein oligomerization and neurotoxicity and have, on the other hand, associated inclusion body formation with a cellular protective response. For example, impaired ubiquitination and degradation of mutant ataxin-1 with a polyglutamine expansion resulted in accumulation of aggregated ataxin-1 with reduced nuclear inclusion formation and an accelerated polyglutamine-induced cytotoxic phenotype in vivo.47,48,49 As another example, the survival of neurons expressing mutant Huntingtin (Htt) was measured by an advanced microscopy technique that enables the following of specific neurons as a function of time. Using this technique, it was found that improved neuronal survival is correlated with inclusion formation and reduced free Htt protein levels.50
Here, we show that PUFA-induced αS high molecular weight assemblies are associated with decreased cell viability and that inclusion formation is associated with restored cell viability. Using a low concentration of 18:3 (125 μmol/L) for long incubation times of up to 48 hours, we were able to find a narrow window of time in which oligomeric αS assemblies were present but not yet deposited into microscopically visible inclusions, and this period was associated with the appearance of cytotoxicity. However, viability was normal once inclusions were present. We used a cell culture model to test the effects of PUFAs on αS oligomerization and inclusion formation and the resultant effect on cellular metabolic activity. This cellular model raises questions regarding the role of cellular metabolism in neurodegeneration. Specifically, why in some neurons are inclusions formed and the neurons survive, whereas other neurons fail to produce inclusions and degenerate? Partial explanation may be attributed to the neuron metabolic state. It is suggested that neuronal metabolic activity plays a major role in neurodegeneration in general51,52 and in PD in particular.53,54,55
The cytoplasmic inclusions in the dopaminergic and nondopaminergic cell models described here share some characteristics with LB. They contain specific proteins that have been shown to be major constituents of LB, such as αS, phosphorylated (ser129) αS, ubiquitin, and HSP 70. They appear in an age-dependent manner in culture and are usually round. Lewy-like inclusions were previously described in several other experimental cellular systems. The appearance of inclusion bodies in cultured cells was first described on overexpression of αS and synphilin,56 and ubiquitinated inclusions were detected when parkin was overexpressed together with αS and synphilin.57 Recently, the inclusions observed with αS, synphilin, and parkin were shown to be eosinophilic and to contain filamentous αS structures.38 Nevertheless, it is questionable whether parkin expression is crucial for Lewy-like inclusion formation.58 Additional experimental approaches to induce inclusion formation in αS-overexpressing cultured mammalian cells include oxidative stress37,59 and proteosomal inhibition,59,60,61,62 and in yeast, αS overexpression also resulted in inclusion formation.63
A short exposure of 6 to 8 hours to physiological FA concentrations (250 μmol/L) resulted in induced Lewy-like inclusion formation in our experimental models. An interesting feature of the Lewy-like inclusions we describe is that their formation is dependent on the gradual changes that a specific αS-overexpressing clone undergoes with time in culture. A clone will transition from having soluble, immunocytochemically diffuse protein to an accumulation of αS in insoluble particles as the clone is aged in culture. Inclusions can appear in the older stable clones even without PUFA treatment. This result emphasizes the relevance of the αS culture model, as lesion formation in PD is a time-dependent process.
An unresolved question that is relevant to the observations presented here is whether αS interacts with free FA or FA assembled in more complex lipids, such as phospholipids. In cells, FAs are bound mainly to phospholipid membranes and to FA-binding proteins (FABPs), when the latter are present.64 Free cytosolic FA concentrations are generally low, ie, in the nmol/L range. A principal force keeping them low is the formation of a thioester linkage between the FA carboxyl group and the thiol group of coenzyme A (yielding fatty acyl-CoA) within minutes after an FA enters a cell.65 The esterified FAs are then consumed for energy production by mitochondria and peroxisomes or else used for synthesis of lipids. FAs are assumed to cross cell membranes continuously, either actively by specific protein transporters or via flip-flop of the FA through the membrane.66 We have previously reported that αS shares some regional sequence homologies with a FABP signature motif and that αS can bind free radiolabeled 18:1 with low affinity, in a manner reminiscent of FABPs.9 However, this in vitro result has not been matched by additional in vitro studies. Unlike classical FABPs, no specific FA-binding sites were detected for αS using NMR spectroscopy.18 Further, no conformational similarities of αS to the characteristic FABP tertiary structure was observed using this method.18 Instead, the formation of high molecular weight complexes of purified αS and specific FAs were detected in this in vitro study using purified components.18 Similar to the FABPs, αS has been suggested to be involved in cellular FA uptake and metabolism. Specifically, altered FA uptake and metabolism were observed in αS-null mice infused with 14C 16:0,28 and altered acyl side chain composition characterized by decreased n-6 PUFAs and increased SFAs composition were observed in certain brain phospholipids of αS−/− mice.29,67 However, no stable binding of αS to 16:0 or 18:1 was detected using titration microcalorimetry. It appears at this juncture, that αS is involved with FAs in ways that are similar but not identical to classical FABPs, but much further biochemical work is needed to understand the nature of αS interactions with lipids in general and with FA in particular.
In conclusion, our findings demonstrate a role for PUFAs in helping to induce the oligomerization of αS monomers into stable but still soluble higher molecular weight aggregates and ultimately into larger cytoplasmic inclusions that may modulate neurodegeneration in human α-synucleinopathies such as PD.
Footnotes
Address reprint requests to Ronit Sharon, Department of Cellular Biochemistry and Human Genetics, Hebrew University, Hadassah Medical School, Jerusalem, Israel. E-mail: rsharon@md.huji.ac.il.
Supported by the National Institutes of Health (grant R01 NS051318).
References
- Lee VM, Trojanowski JQ. Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52:33–38. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Hardy J, Cai H, Cookson MR, Gwinn-Hardy K, Singleton A. Genetics of Parkinson’s disease and parkinsonism. Ann Neurol. 2006;60:389–398. doi: 10.1002/ana.21022. [DOI] [PubMed] [Google Scholar]
- Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- Maroteaux L, Scheller RH. The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res Mol Brain Res. 1991;11:335–343. doi: 10.1016/0169-328x(91)90043-w. [DOI] [PubMed] [Google Scholar]
- Chen X, de Silva HA, Pettenati MJ, Rao PN, St. George-Hyslop P, Roses AD, Xia Y, Horsburgh K, Ueda K, Saitoh T. The human NACP/alpha-synuclein gene: chromosome assignment to 4q21.3-q22 and TaqI RFLP analysis. Genomics. 1995;26:425–427. doi: 10.1016/0888-7543(95)80237-g. [DOI] [PubMed] [Google Scholar]
- Sharon R, Goldberg MS, Bar-Josef I, Betensky RA, Shen J, Selkoe DJ. Alpha-synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc Natl Acad Sci USA. 2001;98:9110–9115. doi: 10.1073/pnas.171300598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YS, Laurine E, Woods W, Lee SJ. A novel mechanism of interaction between alpha-synuclein and biological membranes. J Mol Biol. 2006;360:386–397. doi: 10.1016/j.jmb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH. Lipid rafts mediate the synaptic localization of alpha-synuclein. J Neurosci. 2004;24:6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Li J, Fink AL. The association of alpha-synuclein with membranes affects bilayer structure, stability, and fibril formation. J Biol Chem. 2003;278:40186–40197. doi: 10.1074/jbc.M305326200. [DOI] [PubMed] [Google Scholar]
- Zhu M, Fink AL. Lipid binding inhibits alpha-synuclein fibril formation. J Biol Chem. 2003;278:16873–16877. doi: 10.1074/jbc.M210136200. [DOI] [PubMed] [Google Scholar]
- Bussell R, Jr, Ramlall TF, Eliezer D. Helix periodicity, topology, and dynamics of membrane-associated alpha-synuclein. Protein Sci. 2005;14:862–872. doi: 10.1110/ps.041255905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisaglia M, Tessari I, Pinato L, Bellanda M, Giraudo S, Fasano M, Bergantino E, Bubacco L, Mammi S. A topological model of the interaction between alpha-synuclein and sodium dodecyl sulfate micelles. Biochemistry. 2005;44:329–339. doi: 10.1021/bi048448q. [DOI] [PubMed] [Google Scholar]
- Bussell R, Jr, Eliezer D. A structural and functional role for 11-mer repeats in alpha-synuclein and other exchangeable lipid binding proteins. J Mol Biol. 2003;329:763–778. doi: 10.1016/s0022-2836(03)00520-5. [DOI] [PubMed] [Google Scholar]
- Sharon R, Bar-Joseph I, Frosch MP, Walsh DM, Hamilton JA, Selkoe DJ. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron. 2003;37:583–595. doi: 10.1016/s0896-6273(03)00024-2. [DOI] [PubMed] [Google Scholar]
- Lücke C, Gantz DL, Klimtchuk E, Hamilton JA. Interactions between fatty acids and alpha-synuclein. J Lipid Res. 2006;47:1714–1724. doi: 10.1194/jlr.M600003-JLR200. [DOI] [PubMed] [Google Scholar]
- Perrin RJ, Woods WS, Clayton DF, George JM. Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J Biol Chem. 2001;276:41958–41962. doi: 10.1074/jbc.M105022200. [DOI] [PubMed] [Google Scholar]
- Sharon R, Bar-Joseph I, Mirick GE, Serhan CN, Selkoe DJ. Altered fatty acid composition of dopaminergic neurons expressing alpha-synuclein and human brains with alpha-synucleinopathies. J Biol Chem. 2003;278:49874–49881. doi: 10.1074/jbc.M309127200. [DOI] [PubMed] [Google Scholar]
- Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, Nussbaum RL. Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J Biol Chem. 2002;277:6344–6352. doi: 10.1074/jbc.M108414200. [DOI] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Chandra S, Fornai F, Kwon HB, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, Castillo PE, Sudhof TC. Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc Natl Acad Sci USA. 2004;101:14966–14971. doi: 10.1073/pnas.0406283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherzer CR, Jensen RV, Gullans SR, Feany MB. Gene expression changes presage neurodegeneration in a Drosophila model of Parkinson’s disease. Hum Mol Genet. 2003;12:2457–2466. doi: 10.1093/hmg/ddg265. [DOI] [PubMed] [Google Scholar]
- Willingham S, Outeiro TF, DeVit MJ, Lindquist SL, Muchowski PJ. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science. 2003;302:1769–1772. doi: 10.1126/science.1090389. [DOI] [PubMed] [Google Scholar]
- Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagnet PI, Golovko MY, Barcelo-Coblijn GC, Nussbaum RL, Murphy EJ. Fatty acid incorporation is decreased in astrocytes cultured from alpha-synuclein gene-ablated mice. J Neurochem. 2005;94:839–849. doi: 10.1111/j.1471-4159.2005.03247.x. [DOI] [PubMed] [Google Scholar]
- Golovko MY, Faergeman NJ, Cole NB, Castagnet PI, Nussbaum RL, Murphy EJ. Alpha-synuclein gene deletion decreases brain palmitate uptake and alters the palmitate metabolism in the absence of alpha-synuclein palmitate binding. Biochemistry. 2005;44:8251–8259. doi: 10.1021/bi0502137. [DOI] [PubMed] [Google Scholar]
- Barceló-Coblijn G, Golovko MY, Weinhofer I, Berger J, Murphy EJ. Brain neutral lipids mass is increased in alpha-synuclein gene-ablated mice. J Neurochem. 101:134–142. doi: 10.1111/j.1471-4159.2006.04348.x. [DOI] [PubMed] [Google Scholar]
- Barceló-Coblijn G, Kitajka K, Puskas LG, Hogyes E, Zvara A, Hackler L, Jr, Farkas T. Gene expression and molecular composition of phospholipids in rat brain in relation to dietary n-6 to n-3 fatty acid ratio. Biochim Biophys Acta. 2003;1632:72–79. doi: 10.1016/s1388-1981(03)00064-7. [DOI] [PubMed] [Google Scholar]
- Kitajka K, Sinclair AJ, Weisinger RS, Weisinger HS, Mathai M, Jayasooriya AP, Halver JE, Puskas LG. Effects of dietary omega-3 polyunsaturated fatty acids on brain gene expression. Proc Natl Acad Sci USA. 2004;101:10931–10936. doi: 10.1073/pnas.0402342101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. Alpha-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Crawford GD, Jr, Le WD, Smith RG, Xie WJ, Stefani E, Appel SH. A novel N18TG2 x mesencephalon cell hybrid expresses properties that suggest a dopaminergic cell line of substantia nigra origin. J Neurosci. 1992;12:3392–3398. doi: 10.1523/JNEUROSCI.12-09-03392.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HK, Won LA, Kontur PJ, Hammond DN, Fox AP, Wainer BH, Hoffmann PC, Heller A. Immortalization of embryonic mesencephalic dopaminergic neurons by somatic cell fusion. Brain Res. 1991;552:67–76. doi: 10.1016/0006-8993(91)90661-e. [DOI] [PubMed] [Google Scholar]
- Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, Selkoe DJ. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. doi: 10.1016/s0896-6273(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Odoy S, Okamoto N, Jacobsen H, Iwatsubo T, Trojanowski JQ, Takahashi H, Wakabayashi K, Bogdanovic N, Riederer P, Kretzschmar HA, Haass C. Selective insolubility of alpha-synuclein in human Lewy body diseases is recapitulated in a transgenic mouse model. Am J Pathol. 2001;159:2215–2225. doi: 10.1016/s0002-9440(10)63072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Margolis RL, Li X, Troncoso JC, Lee MK, Dawson VL, Dawson TM, Iwatsubo T, Ross CA. Alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J Neurosci. 2005;25:5544–5552. doi: 10.1523/JNEUROSCI.0482-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Feany MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ. Phosphorylation of Ser 129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 281:29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Fujiwara H, Nonaka T, Wakabayashi K, Takahashi H, Lee VM, Trojanowski JQ, Mann D, Iwatsubo T. Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J Biol Chem. 2002;277:49071–49076. doi: 10.1074/jbc.M208046200. [DOI] [PubMed] [Google Scholar]
- Sampathu DM, Giasson BI, Pawlyk AC, Trojanowski JQ, Lee VM. Ubiquitination of alpha-synuclein is not required for formation of pathological inclusions in alpha-synucleinopathies. Am J Pathol. 2003;163:91–100. doi: 10.1016/s0002-9440(10)63633-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzuhara S, Mori H, Izumiyama N, Yoshimura M, Ihara Y. Lewy bodies are ubiquitinated. A light and electron microscopic immunocytochemical study. Acta Neuropathol (Berl) 1988;75:345–353. doi: 10.1007/BF00687787. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Auluck PK, Chan E, Trojanowski JQ, Lee VM-Y, Bonini NM. Chaperon suppression of a-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science. 2002;295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. Alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Al-Ramahi I, Lam YC, Chen HK, de Gouyon B, Zhang M, Perez AM, Branco J, de Haro M, Patterson C, Zoghbi HY, Botas J. CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J Biol Chem. 2006;281:26714–26724. doi: 10.1074/jbc.M601603200. [DOI] [PubMed] [Google Scholar]
- Bowman AB, Yoo SY, Dantuma NP, Zoghbi HY. Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet. 2005;14:679–691. doi: 10.1093/hmg/ddi064. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Reinstein E, Sun Y, Antalffy B, Jiang Y, Ciechanover A, Orr HT, Beaudet AL, Zoghbi HY. Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron. 1999;24:879–892. doi: 10.1016/s0896-6273(00)81035-1. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ, Pappolla M, Pelaez RP, Bazan NG. Alzheimer’s disease—a dysfunction in cholesterol and lipid metabolism. Cell Mol Neurobiol. 2005;25:475–483. doi: 10.1007/s10571-005-4010-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D. Glucose metabolism and Alzheimer’s disease. Ageing Res Rev. 2005;4:240–257. doi: 10.1016/j.arr.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Kawahara K, Bar-On P, Rockenstein E, Crews L, Masliah E. The role of alpha-synuclein assembly and metabolism in the pathogenesis of Lewy body disease. J Mol Neurosci. 2004;24:343–352. doi: 10.1385/JMN:24:3:343. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A, Hoffer B, Cullheim S, Mohammed AH, Olson L, Larsson NG. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci USA. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofaris GK, Layfield R, Spillantini MG. Alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001;509:22–26. doi: 10.1016/s0014-5793(01)03115-5. [DOI] [PubMed] [Google Scholar]
- Engelender S, Kaminsky Z, Guo X, Sharp AH, Amaravi RK, Kleiderlein JJ, Margolis RL, Troncoso JC, Lanahan AA, Worley PF, Dawson VL, Dawson TM, Ross CA. Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nat Genet. 1999;22:110–114. doi: 10.1038/8820. [DOI] [PubMed] [Google Scholar]
- Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med. 2001;7:1144–1150. doi: 10.1038/nm1001-1144. [DOI] [PubMed] [Google Scholar]
- von Coelln R, Thomas B, Andrabi SA, Lim KL, Savitt JM, Saffary R, Stirling W, Bruno K, Hess EJ, Lee MK, Dawson VL, Dawson TM. Inclusion body formation and neurodegeneration are parkin independent in a mouse model of alpha-synucleinopathy. J Neurosci. 2006;26:3685–3696. doi: 10.1523/JNEUROSCI.0414-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev N, Melamed E, Offen D. Proteasomal inhibition hypersensitizes differentiated neuroblastoma cells to oxidative damage. Neurosci Lett. 2006;399:27–32. doi: 10.1016/j.neulet.2005.09.086. [DOI] [PubMed] [Google Scholar]
- Sawada H, Kohno R, Kihara T, Izumi Y, Sakka N, Ibi M, Nakanishi M, Nakamizo T, Yamakawa K, Shibasaki H, Yamamoto N, Akaike A, Inden M, Kitamura Y, Taniguchi T, Shimohama S. Proteasome mediates dopaminergic neuronal degeneration, and its inhibition causes alpha-synuclein inclusions. J Biol Chem. 2004;279:10710–10719. doi: 10.1074/jbc.M308434200. [DOI] [PubMed] [Google Scholar]
- Rideout HJ, Larsen KE, Sulzer D, Stefanis L. Proteasomal inhibition leads to formation of ubiquitin/alpha-synuclein-immunoreactive inclusions in PC12 cells. J Neurochem. 2001;78:899–908. doi: 10.1046/j.1471-4159.2001.00474.x. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Olanow CW, Halliwell B, Isacson O, Jenner P. Failure of the ubiquitin-proteasome system in Parkinson’s disease. Nat Rev Neurosci. 2001;2:589–594. doi: 10.1038/35086067. [DOI] [PubMed] [Google Scholar]
- Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science. 2003;302:1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JA, Kamp F. How are free fatty acids transported in membranes? Is it by proteins or by free diffusion through the lipids? Diabetes. 1999;48:2255–2269. doi: 10.2337/diabetes.48.12.2255. [DOI] [PubMed] [Google Scholar]
- Nelson D, Cox M. New York: Worth Publishers,; Lehninger Principles of Biochemistry. 2000:599–622. Chapter 17. [Google Scholar]
- Hamilton JA. Transport of fatty acids across membranes by the diffusion mechanism. Prostaglandins Leukot Essent Fatty Acids. 1999;60:291–297. doi: 10.1016/s0952-3278(99)80002-7. [DOI] [PubMed] [Google Scholar]
- Ellis CE, Murphy EJ, Mitchell DC, Golovko MY, Scaglia F, Barcelo-Coblijn GC, Nussbaum RL. Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol Cell Biol. 2005;25:10190–10201. doi: 10.1128/MCB.25.22.10190-10201.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]