

Abstract
Following facial nerve resection in the mouse, a substantial number of neurons reside in an atrophied state (characterized by cell shrinkage and decreased ability to uptake Nissl stain), which can be reversed by re-injury. The mechanisms mediating the reversal of neuronal atrophy remain unclear. Although T cells have been shown to prevent neuronal loss following peripheral nerve injury, it was unknown whether T cells play a role in mediating the reversal of axotomy-induced neuronal atrophy. Thus, we used a facial nerve re-injury model to test the hypothesis that the reversal of neuronal atrophy would be impaired in recombinase activating gene-2 knockout (RAG-2 KO) mice, which lack functional T and B cells. Measures of neuronal survival were compared in the injured facial motor nucleus (FMN) of RAG-2 KO and wild-type (WT) mice that received a resection of the right facial nerve followed by re-injury of the same nerve 10 weeks later (“chronic resection + re-injury”) or a resection of the right facial nerve followed by sham re-injury of the same nerve 10 weeks later (“chronic resection + sham”). We recently demonstrated that prior exposure to neuronal injury elicited a marked increase in T cell trafficking indicative of a T cell memory response when the contralateral FMN was injured later in adulthood. We examined if such a T cell memory response would also occur in the current re-injury model. RAG-2 KO mice showed no reversal of neuronal atrophy whereas WT mice showed a robust response. The reversal of atrophy in WT mice was not accompanied by a T cell memory response. Although the number of CD4+ and CD8+ T cells in the injured FMN did not differ from each other, double-negative T cells appear to be recruited in response to neuronal injury. Re-injury did not result in increased expression of MHC2 by microglia. Our findings suggest that T cells may be involved in reversing the axotomy-induced atrophy of injured neurons.
Keywords: motor neuron injury, facial nerve axotomy, T cells, microglia, neuronal atrophy
Introduction
Increasing evidence suggests that in some forms of neuronal injury, neurons may not actually die following nerve injury but reside in an atrophic state, characterized by extreme cell shrinkage and a decreased ability to take up Nissl stain (McPhail, et al., 2004). Recent studies by McPhail et al. demonstrated that a population of facial motor neurons undergo a protracted period of degeneration or atrophy following peripheral resection of the facial nerve in adult mice. Re-injuring the facial nerve stimulated a reversal in the atrophic status of the injured neurons, causing an increase in both their size and number. Reversal of neuronal atrophy has also been demonstrated in several different models of CNS nerve injury (Hagg, et al., 1989, Kwon, et al., 2002). The mechanisms that mediate this regenerative response after a prolonged survival period remain unknown.
Under normal conditions, the CNS is subject to continuous immune surveillance by low numbers of circulating peripheral T lymphocytes (Cose, et al., 2006, Hickey, et al., 1991). In pathogenic states such as experimental autoimmune encephalomyelitis (EAE) and infection, the presence of T cells in the brain can have detrimental effects (Martino and Hartung, 1999; Nau and Bruck, 2002), while in other contexts, T cells have been shown to act in concert with glial cells to promote neuroregeneration (Byram, et al., 2004, Martino and Hartung, 1999, Nau and Bruck, 2002, Raivich, et al., 1998, Schwartz, 2003). Studies have demonstrated the effectiveness of T cells in preventing neuronal loss following injury (Armstrong, et al., 2004, Jones, et al., 2005, Schwartz and Moalem, 2001). To date, research has focused on elucidating the role of T cells in preventing initial neuronal death or slowing the rate of neurodegeneration and gradual neuronal loss following facial nerve axotomy (Jones, et al., 2005, Serpe, et al., 1999, Serpe, et al., 2000). Given that a substantial number of facial motor neurons undergo atrophy following axotomy and that the atrophied state of these neurons is reversible by re-injury, T cells may also be involved in mediating the reversal of neuronal atrophy following re-injury. In this study, we therefore used the re-injury model described previously by McPhail et al. (2004, 2005) to test the hypothesis that the reversal of motor neuron atrophy (i.e., increase in cell number and size) elicited by nerve re-injury would be impaired in immunodeficient recombinase activating gene-2 knockout (RAG-2 KO) mice, which lack mature T and B cells. Neuronal cell counts and mean cell size were compared in mice that received an initial resection of the facial nerve followed by a re-injury of the same nerve 10 weeks later versus mice that received only a single resection followed by a sham re-injury 10 weeks later. In a landmark study, Raivich et al. (1998) demonstrated that T cells cross an intact blood-brain-barrier (BBB) and home to degenerating neuronal cell bodies following peripheral transection of the facial nerve, and established that the peak of this response occurs at 14 days post-axotomy. Measures of neuronal survival were therefore assessed at 14 days after the 2nd surgery in both groups, which also allowed us to assess the peak T cell response in the injured FMN. In a recent study, our lab showed that prior exposure to neuronal injury in the FMN early in adulthood induced a T cell memory response, characterized by a robust increase in T cell trafficking to the FMN, when the contralateral nucleus was injured later in adulthood (Ha, et al., 2007). Interestingly, this enhanced T cell trafficking to the injured FMN was not correlated with improved neuronal survival. Interactions between T cells and microglia are important in the immune-mediated improvement of motor neuron survival (Byram, et al., 2004). That we did not see an improvement in neuronal survival in the presence of a T cell memory response could be due to the possibility that the memory T cells encountered microglia in the contralateral FMN which were naïve to prior injury and suggests that prior injury may be needed to sensitize microglia to encode a type of memory that permits them to interact with memory T cells to improve neuronal survival. Thus, secondary hypotheses we sought to test were whether prior exposure to neuronal injury could elicit a T cell memory response when the same FMN is injured later in adulthood, and if the postulated increase in T cell homing to the FMN is correlated with the improvement in neuronal outcome measures in the current re-injury model. Studies documenting the presence of CD4+ and CD8+ T cells in the injured FMN are limited (Ankeny and Popovich, 2007, Bohatschek, et al., 2004, Liu, et al., 2005), and none have been performed using this resection or re-injury model. To address the aforementioned T cell memory hypothesis and to assess the distribution of CD4+ and CD8+ T cells in the injured FMN, we compared the number of CD3+ T cells, CD4+ T cells, and CD8+ T cells in the FMN of 3 groups of WT mice. One group was assessed at 14 days post-resection but did not receive prior nerve injury and served as controls (acute resection). A second group received prior nerve injury and was assessed at 14 days after a re-injury of the same nerve (chronic resection + re-injury). The third group was assessed at 12 weeks after a single resection (chronic resection + sham). Finally, to determine whether prior injury alters the microglial response to re-injury, we also quantified the number of MHC2+ microglia in these subject groups.
Materials and methods
Animals
Wild-type (WT) and RAG-2 knockout (RAG-2 KO) were on a B6 background and were obtained from Taconic (Germantown, NY). RAG-2 KO mice originated on a 129 background and were backcrossed to the B6 background for 12 generations. Mice used in this study were cared for in compliance with the NIH Guide for the Care and Use of Laboratory Animals and were housed under specific pathogen-free conditions in individual microisolater cages.
Animal surgery
Mice were 8-weeks-old at the time of the first surgery. Animals were anesthetized with 4% isoflurane. The right facial nerve was exposed and resected to prevent reconnection of the nerve to its target, as described previously (McPhail, et al., 2004, McPhail, et al., 2005). At 10 weeks post-injury, the nerve was re-exposed and re-injured by removing the neuroma that formed near the proximal nerve stump (“chronic resection + re-injury”). In a second group of mice, we re-exposed the nerve but left the neuroma intact (“chronic resection + sham”). Raivich and colleagues (1998) demonstrated that the peak of T cell infiltration to the FMN and neuronal cell death occurs at 14 days after facial nerve transection. Thus, both groups of mice were sacrificed 14 days following the second surgery, or a total of 12 weeks after the initial surgery. In a third subset of WT and RAG-2 KO mice used as naïve controls (no prior nerve resection) to make comparisons of the T cell and microglial response, the right facial nerve was exposed but not resected. Ten weeks later, the right facial nerve was re-exposed and resected and mice were sacrificed at 14 days post-resection (“acute resection” group). For euthanasia, mice were anesthetized by intraperitoneal injection of a 0.5 mg/ml ketamine cocktail (ketamine/xylazine/acepromazine given in a 3:3:1 ratio) and were perfused with 1 X phosphate buffered saline (PBS). Brains were collected and stored at −80°C. Using the ambiguus nucleus and the facial nerve as the starting and ending points, respectively, 15 μm coronal sections were cut throughout the caudal-rostral extent of the facial motor nucleus. Sections were collected on Superfrost/Plus slides (Fisher Scientific) and stored at −80°C.
Light microscopic immunohistochemistry
Tissue sections were post-fixed in 4% paraformaldehyde (CD3 and MHC2 antibodies) or zinc fixative (Pharmingen; CD4 and CD8 antibodies) for 1 hour at room temperature. Tissue sections were incubated in normal goat serum (NGS) (Vector) for 1 hour at room temperature followed by overnight incubation with anti-mouse CD3 (17A2; PharMingen), CD4 (L3T4; Pharmingen), CD8 (H35-17.2; Pharmingen), or MHC2 I-A/I-E (M5/114.15.2; PharMingen) primary antibody at 4°C. Sections were washed in 1 × PBS after each incubation step. Visualization of the primary antibodies was performed by incubation of sections in goat anti-rat secondary antibody (1:2000, Vector Labs) for 1 h at room temperature followed by incubation in avidin-peroxidase conjugates (1:500, Sigma) for 1 h. No signal was obtained with each of the primary or secondary antibodies alone. The chromagen reaction was revealed by incubation in 3,3′-diaminobenzidine (DAB)-H2O2 solution (Sigma; 0.07% DAB/0.004% H2O2). Sections were counterstained with cresyl violet, dehydrated in ascending alcohol washes, cleared in xylenes, and coverslipped.
Quantification and statistical analysis
Sections throughout the FMN were assessed by an experimenter under blind conditions. Four sections (approximately 1/10 of the entire facial motor nucleus) were used to assess the number of CD3+ T cells, CD4+ T cells, CD8+ T cells, or MHC2+ microglia per mouse. The number of Nissl stained motor neurons was quantified by counting neuronal profiles that contained a visible nucleolus in 8 representative sections throughout the extent of the FMN, as described previously (Deboy, et al., 2006). Neuronal cell sizes were measured in 3 representative sections of the FMN each spaced 90μm apart using ImageJ software (National Institutes of Health). Based on previous studies by McPhail et al. (2004), the medial nucleus, which is innervated by the auricular branch of the facial nerve, remained uninjured and was thus excluded from our analyses. The number of neurons in the injured FMN was expressed as a percentage of the number of neurons in the uninjured, contralateral FMN. Analysis of variance (ANOVA) was used to make statistical comparisons.
Results
Effect of immunodeficiency on the reversal of neuronal atrophy
We compared the effect of treatment (chronic resection + re-injury vs. chronic resection + sham re-injury) on motor neuron survival in RAG-2 KO and WT mice. As seen in Figure 1A, as expected from the literature, motor neuron survival was significantly increased in chronically resected WT mice that received nerve re-injury compared to those that received sham re-injury (F(1,10)=6.083, p<0.03, 6 mice/treatment). By contrast, the level of motor neuron survival did not differ between chronically resected RAG-2 KO mice that received re-injury (n=5) or sham re-injury (n=6). We also compared the effect of treatment on the cross-sectional area of neurons measured in 3 representative sections throughout the FMN in RAG-2 KO and WT mice. As seen in Figure 1B, average cell size was significantly increased in chronically resected WT mice that received re-injury compared to those that received sham re-injury (F(1,6)=18.981, p<0.01, n=4 mice/treatment). By contrast, average cell size did not differ between chronically resected RAG-2 KO mice that received re-injury or sham re-injury (n=3 mice/treatment). As seen in Figures 2A–2B, binning of the neurons by cell size revealed a noticeable shift from smaller to larger cell sizes following nerve re-injury in WT mice. This shift in cell size following nerve re-injury was not apparent in the RAG-2 KO mice (Figures 2C–2D). Figure 3 shows representative sections of the FMN from chronically resected WT and RAG-2 KO mice that received re-injury or sham re-injury. In Figures 3B–3C, note the increase in the number and size of motor neurons in re-injured WT compared to sham re-injured WT mice. In Figure 3F, motor neurons remained shrunken or appeared to be lost to axotomy in RAG-2 KO mice that received re-injury.
Figure 1.

Quantification of mean cell counts (A) and mean cell size (B) in chronically resected WT and RAG-2 KO mice that received nerve re-injury or sham re-injury. Each bar in A represents the S.E.M. of 6 WT mice/treatment and the S.E.M. of 6 (chronic resection + sham) and 5 (chronic resection + re-injury) RAG-2 KO mice. Each bar in B represents the S.E.M. of 4 WT mice/treatment and the S.E.M. of 3 RAG-2 KO mice/treatment. *p<0.05, **p<0.01
Figure 2.

Facial motor neurons binned according to cell size following sham re-injury and re-injury. Note that the distribution of neurons in the injured FMN shifts from small to large cell sizes and is normalized to the contralateral uninjured side following re-injury in B6 but not in RAG-2 KO mice.
Figure 3.

Photomicrographs of Nissl stained facial motor neurons in WT and RAG-2 KO mice. The facial motor nucleus is oriented so that the medial sub-nucleus is located on the right. There was a significant loss and shrinkage of neurons in WT and RAG-2 KO mice that received a chronic resection + sham injury (12 weeks after initial resection; B & E) compared to their respective contralateral controls (A & D). Following nerve re-injury, the number and size of injured motor neurons were markedly increased in WT (C) but not in RAG-2 KO mice (F).
Effect of nerve re-injury on the T cell response in the FMN
To determine whether a T cell memory response is elicited following nerve re-injury, we compared the effect of treatment on the number of CD3+ T cells in the FMN of WT mice. Mice that did not receive prior nerve injury were assessed for their T cell response in the FMN at 14 days post-resection (acute resection) and were used as controls. As depicted in Figure 4A, there was a significant decrease in the number of CD3+ T cells in chronically resected mice that received re-injury (F(1,9)=7.943, p<0.05, n=6) compared to acute resection (n=5). There was also a significant decrease in the number of CD3+ T cells in chronically resected mice that received sham re-injury (F(1,9)=11.577, p<0.01, n=6) compared to acute resection. The number of T cells in the injured FMN was comparable between chronically resected mice that received re-injury or sham re-injury. By 12 weeks post-injury, there was an occasional T cell (<1 T cell/section) in the injured FMN of chronically resected mice that received sham re-injury.
Figure 4.

Quantification of CD3+, CD4+, and CD8+ T cells in the FMN of WT mice. For A and C, each bar represents the S.E.M. of 5 (acute resection) and 6 (chronic resection + re-injury, chronic resection + sham) mice. For B, each bar represents that S.E.M. of 5 (acute resection, chronic resection + re-injury) and 6 (chronic resection + sham) mice. *p<0.05, **p<0.01 (compared to acute resection)
In Figures 4B and 4C, we quantified the number of CD4+ and CD8+ T cells, respectively, that were recruited to the FMN in acutely resected WT mice to determine whether there is a predominance of either T cell subtype in the FMN. The number of CD4+ and CD8+ T cells was comparable in the FMN of acutely resected mice. We also compared the number of CD4+ and CD8+ T cells in the FMN of chronically resected mice that received re-injury or sham re-injury versus the acute resection group to determine whether re-injury alters their distribution in the FMN. ANOVA revealed a significant decrease in the number of CD4+ T cells in chronically resected WT mice that received re-injury (F(1,8)=5.366, p<0.05, n=5) or sham re-injury (F(1,9)=15.771, p<0.01, n=6) compared to acute resection (n=5). The number of CD4+ cells did not differ between the chronic axotomy groups that received re-injury or sham re-injury. Similarly, there was a significant decrease in the number of CD8+ T cells in chronically resected mice that received re-injury (F(1,9)=6.376, p<0.05, n=6) or sham re-injury (F(1,9)=7.621, p<0.05, n=6) compared to acute resection (n=5). The number of CD8+ T cells did not differ between the chronic axotomy groups that received re-injury or sham re-injury.
Effect of nerve re-injury on microglial response
To determine whether nerve re-injury alters microglial reactivity in the FMN, we compared the number of MHC2+ microglia between chronically resected WT mice that received re-injury and acutely resected WT mice. As shown in Figure 5, although there were fewer MHC2+ microglia following re-injury in chronically resected mice that received re-injury compared to acute resection mice, the groups were not statistically significant. ANOVA revealed a significant decrease in the number of MHC2+ microglia in chronically resected WT mice that received sham re-injury (n=5) when compared to those that received re-injury (F(1,9)=16.771, p<0.01, n=6) or acute resection (F(1,8)=13.236, p<0.01, n=5).
Figure 5.
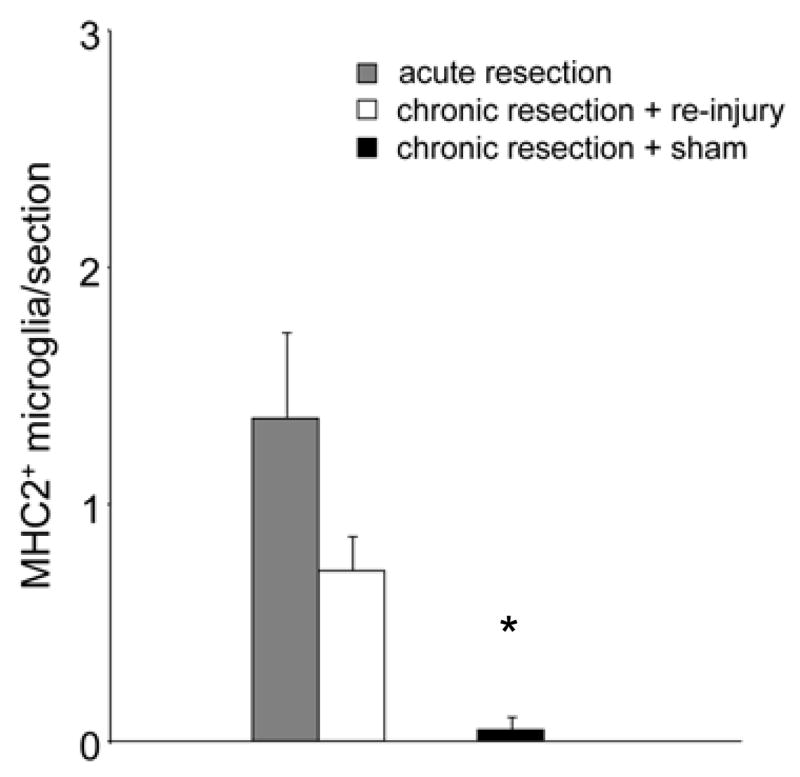
Quantification of MHC2+ microglia in the FMN of WT mice. Each bar represents the S.E.M. of 5 (acute resection, chronic resection + sham) and 6 (chronic resection + re-injury) mice. *p<0.01 (compared to acute resection and chronic resection resection + re-injury)
Discussion
In the current study, we demonstrated that immunodeficiency prevents the reversal of neuronal atrophy following nerve re-injury in the mouse. Although the number and size of injured motor neurons was not increased in response to nerve re-injury in RAG-2 KO mice, there was a significant increase in motor neuron survival from 38 ± 3.2% to 49 ± 3.2% in chronically resected WT mice that received sham or re-injury, respectively. Furthermore, there was a marked increase in the average cell size of motor neurons from 56 ± 5.2% to 99 ± 8.4% in chronically resected WT mice that received sham or re-injury, respectively. The degree of improvement in neuronal survival observed in our study, however, was not as robust as the improvement seen by McPhail et al. (2004, 2005) likely because we made our assessments at 2 weeks post re-injury rather than at 1 week post-re-injury. Altogether, our data indicate that T cells may be involved in reversing the axotomy-induced atrophy of injured neurons. In mice given a single resection and assessed at 12 weeks post-injury, we did not detect a significant difference in the level of motor neuron survival between WT and RAG-2 KO mice. In a study comparing the levels of motor neuron survival in severe combined immunodeficient (scid) mice, immune reconstituted scid mice, and WT controls, the effect of T cells in slowing motor neuron degeneration and loss was found to be time-dependent, with a substantial reduction in neuroprotection seen between 4 and 10 weeks post-axotomy (Serpe, et al., 2000). This time-dependent reduction in neuroprotection coincides with the substantial reduction in T cells in the axotomized FMN between 4 and 10 weeks (where few T cells are present by 10 weeks post-axotomy) found by Ravich and colleagues (1998).
Contrary to our hypothesis, prior exposure to neuronal injury early in adulthood did not elicit a T cell memory response (as assessed by the number of T cells in the FMN) following re-injury of the same FMN later in adulthood. Despite the absence of a T cell memory response, there was still a marked improvement in neuronal survival in WT mice. Although a T cell memory response is typically characterized by an increased number of T cells responding to re-exposure to a specific antigen (Rogers, et al., 2000), it is plausible that re-injury re-activated the few T cells that we did find in the FMN. Prior exposure to neuronal injury may have resulted in a functional enhancement of these cells, allowing them to mediate the improvement in neuronal survival that we observed in WT mice following re-injury. Alternatively, it is possible that the neuroprotective benefit of T cells could be attributable to their actions following the initial injury where they may sustain the viability of injured neurons by maintaining them in an atrophied state (and the atrophied neurons subsequently become detectable following re-injury). Future studies will be needed to clarify these intriguing possibilities. In a recent study, we demonstrated that prior exposure to neuronal injury elicited a two-fold increase in T cell homing to the FMN when the contralateral FMN was injured later in adulthood (Ha, et al., 2007). In that double injury paradigm, the second transection was performed on the contralateral nerve, thus exposing sensitized T cells to neurons that were naïve to injury and were presumably undergoing degeneration. Given that additional T cells did not infiltrate the FMN following re-injury in the current model, it is plausible that T cells are not recruited in response to neurons undergoing a reversal of atrophy (where the size and number of neurons were increased following re-injury) but are recruited under conditions of neurodegeneration. In keeping with this possibility, we found that B6 mice, which showed greater neuronal death following axotomy than the 129 strain of mice, exhibited more T cells in the FMN following nerve transection than the 129 mice, which did not display a notable T cell response (Ha, et al., 2006). Furthermore, the survival of the majority of neurons following facial nerve axotomy in the rat is generally associated with the lack of a prominent T cell response. By contrast, facial nerve axotomy in the mouse results in more substantial neuronal loss that is generally accompanied by a more prominent T cell response (Moran and Graeber, 2004). Because we assessed the T cell response following nerve re-injury at a single time point, it is also plausible that the T cell response occurred earlier than 2 weeks post-re-injury and provided the cytokines, chemokines, and/or neurotrophic factors (i.e., brain derived neurotrophic factor) necessary to mediate the subsequent reversal of neuronal atrophy.
That few T cells were observed in the FMN following re-injury does not preclude the possibility that T cells might exert their effects peripherally at the site of nerve injury, as T cells have been shown to accumulate at the nerve stump following peripheral nerve injury (Kleinschnitz, et al., 2006, Moalem, et al., 2004). Moreover, facial motor neurons showed an upregulated expression of the anti-apoptotic gene bcl-2 following induction of an inflammatory response in the facial muscles of the rat, indicating that facial motor neurons are able to respond to peripheral immune signals (Mariotti, et al., 2001). The effectiveness of T cells in conferring neuroprotection peripherally (at the site of nerve injury by releasing cytokines or other factors) versus centrally (at site of injured neuronal cell bodies) is currently unknown. An alternative, albeit unlikely, explanation for the lack of a prominent T cell response following nerve re-injury could be that exposure to previously injured neurons produced T cell tolerance that resulted in a decreased number of T cells in the FMN following re-injury.
Facial nerve resection in immunologically intact mice resulted in the recruitment of CD4+ and CD8+ T cells to the FMN, although there did not appear to be a predominance of either T cell subtype. Nerve re-injury resulted in a significant decrease in the number of both CD4+ and CD8+ T cells in the FMN, but the number of either sub-type did not differ from each other. The combined number of CD4+ and CD8+ T cells could not account for the total number of CD3+ T cells in the injured FMN, suggesting the possibility that double-negative T cells (DN T cells, CD3+/CD4−/CD8− T cells) were also recruited in response to nerve injury. Characterizing DN T cells by immunohistochemistry has proven to be difficult, as the B220 marker that is typically used to label DN T cells is also shared by B cells. Thus, isolating cells from the injured FMN and characterizing them simultaneously for CD3, CD4, and CD8 using multi-color flow cytometry will be required to definitively determine the presence of DN T cells in the injured FMN (James, et al., 2006). DN T cells have been seen in pathological CNS states such as lupus autoimmune encephalomyelitis, and have been suggested to be involved in phenotype switching and inhibiting autoimmune processes (James, et al., 2006, Lider, et al., 1991, Matsumoto, et al., 1996). The presence of DN T cells in the injured FMN may serve to downregulate the actions of CD4+ and CD8+ T cells and prevent autoimmune processes in the brain as a result of injury. Since we were only able to visualize CD4+ and CD8+ T cells by post-fixing sections with zinc fixative, and not with paraformaldehyde as was done with CD3 staining, it is also possible that differences in our staining methodology prevented us from accounting for all of the T cells in a particular section. Although Serpe et al. (2003) showed by adoptive transfer that the CD4+ T cell sub-type was sufficient to prevent neuronal loss in immunodeficient mice, interactions between different T cell sub-types may be important to mediate neuroprotection following neuronal injury in immunologically intact mice.
Although the astrocytic response to re-injury was examined by McPhail et al. (2004), the response of microglia, the resident immune cells of the CNS, had not been addressed. Prior exposure to neuronal injury did not result in enhanced MHC2+ expression by microglia following re-injury. Comparisons between acutely resected and re-injured mice did not reveal an increase in the number of MHC2+ microglia, but we did observe a significant increase in re-injured mice, compared to mice that were assessed at 12 weeks post-resection, suggesting that microglia were re-activated in response to nerve re-injury. By contrast, McPhail et al. showed that the astrocytic response, as assessed by glial fibrillary atrocytic protein (GFAP) expression, remained high following injury and did not increase following re-injury. Microglia possess the ability to phagocytose neuronal debris and can present neural antigen to infiltrating CD4+ T cells via the MHC2 complex (Olson and Miller, 2004, Streit, et al., 1999). Moreover, microglia produce brain derived neurotrophic factor (BDNF), a potent growth factor of motor neurons, in response to neuronal injury (Batchelor, et al., 1999, Yan, et al., 1993, Yan, et al., 1994). That microglia were reactivated in response to nerve re-injury suggests their involvement in reversing neuronal atrophy.
In conclusion, we demonstrated that the reversal of neuronal atrophy following nerve re-injury is impaired by immunodeficiency. The reversal of neuronal atrophy was not associated with increased T cell homing to the FMN indicative of a T cell memory response in the brain. Additional study is needed to determine possible peripheral actions of T cells on injured neurons. Our findings suggest a potentially important and novel role of T cells in reversing the process of neuronal atrophy. Further characterization of the effect of T cells on atrophied neurons may have important implications for understanding the role of T cells in influencing neuronal integrity and function and could help guide future intervention strategies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ankeny DP, Popovich PG. Central nervous system and non-central nervous system antigen vaccines exacerbate neuropathology caused by nerve injury. Eur J Neurosci. 2007;25:2053–2064. doi: 10.1111/j.1460-9568.2007.05458.x. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong BD, Abad C, Chhith S, Rodriguez W, Cheung-Lau G, Trinh V, Waschek JA. Restoration of axotomy-induced PACAP gene induction in SCID mice with CD4+ T-lymphocytes. Neuroreport. 2004;15:2647–2650. doi: 10.1097/00001756-200412030-00018. [DOI] [PubMed] [Google Scholar]
- 3.Batchelor PE, Liberatore GT, Wong JY, Porritt MJ, Frerichs F, Donnan GA, Howells DW. Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J Neurosci. 1999;19:1708–1716. doi: 10.1523/JNEUROSCI.19-05-01708.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bohatschek M, Kloss CU, Hristova M, Pfeffer K, Raivich G. Microglial major histocompatibility complex glycoprotein-1 in the axotomized facial motor nucleus: regulation and role of tumor necrosis factor receptors 1 and 2. J Comp Neurol. 2004;470:382–399. doi: 10.1002/cne.20017. [DOI] [PubMed] [Google Scholar]
- 5.Byram SC, Carson MJ, DeBoy CA, Serpe CJ, Sanders VM, Jones KJ. CD4-positive T cell-mediated neuroprotection requires dual compartment antigen presentation. J Neurosci. 2004;24:4333–4339. doi: 10.1523/JNEUROSCI.5276-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cose S, Brammer C, Khanna KM, Masopust D, Lefrancois L. Evidence that a significant number of naive T cells enter non-lymphoid organs as part of a normal migratory pathway. Eur J Immunol. 2006;36:1423–1433. doi: 10.1002/eji.200535539. [DOI] [PubMed] [Google Scholar]
- 7.Deboy CA, Xin J, Byram SC, Serpe CJ, Sanders VM, Jones KJ. Immune-mediated neuroprotection of axotomized mouse facial motoneurons is dependent on the IL-4/STAT6 signaling pathway in CD4(+) T cells. Exp Neurol. 2006;201:212–224. doi: 10.1016/j.expneurol.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 8.Ha GK, Huang Z, Petitto JM. Prior facial motor neuron injury elicits endogenous T cell memory: relation to neuroregeneration. J Neuroimmunol. 2007;183:111–117. doi: 10.1016/j.jneuroim.2006.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ha GK, Huang Z, Streit WJ, Petitto JM. Endogenous T lymphocytes and microglial reactivity in the axotomized facial motor nucleus of mice: effect of genetic background and the RAG2 gene. J Neuroimmunol. 2006;172:1–8. doi: 10.1016/j.jneuroim.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 10.Hagg T, Fass-Holmes B, Vahlsing HL, Manthorpe M, Conner JM, Varon S. Nerve growth factor (NGF) reverses axotomy-induced decreases in choline acetyltransferase, NGF receptor and size of medial septum cholinergic neurons. Brain Res. 1989;505:29–38. doi: 10.1016/0006-8993(89)90112-1. [DOI] [PubMed] [Google Scholar]
- 11.Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J Neurosci Res. 1991;28:254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- 12.James WG, Hutchinson P, Bullard DC, Hickey MJ. Cerebral leucocyte infiltration in lupus-prone MRL/MpJ-fas lpr mice--roles of intercellular adhesion molecule-1 and P-selectin. Clin Exp Immunol. 2006;144:299–308. doi: 10.1111/j.1365-2249.2006.03056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones KJ, Serpe CJ, Byram SC, Deboy CA, Sanders VM. Role of the immune system in the maintenance of mouse facial motoneuron viability after nerve injury. Brain Behav Immun. 2005;19:12–19. doi: 10.1016/j.bbi.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Kleinschnitz C, Hofstetter HH, Meuth SG, Braeuninger S, Sommer C, Stoll G. T cell infiltration after chronic constriction injury of mouse sciatic nerve is associated with interleukin-17 expression. Exp Neurol. 2006;200:480–485. doi: 10.1016/j.expneurol.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 15.Kwon BK, Liu J, Messerer C, Kobayashi NR, McGraw J, Oschipok L, Tetzlaff W. Survival and regeneration of rubrospinal neurons 1 year after spinal cord injury. Proc Natl Acad Sci U S A. 2002;99:3246–3251. doi: 10.1073/pnas.052308899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lider O, Miller A, Miron S, Hershkoviz R, Weiner HL, Zhang XM, Heber-Katz E. Nonencephalitogenic CD4-CD8- V alpha 2V beta 8.2+ anti-myelin basic protein rat T lymphocytes inhibit disease induction. J Immunol. 1991;147:1208–1213. [PubMed] [Google Scholar]
- 17.Liu ZQ, Bohatschek M, Pfeffer K, Bluethmann H, Raivich G. Major histocompatibility complex (MHC2+) perivascular macrophages in the axotomized facial motor nucleus are regulated by receptors for interferon-gamma (IFNgamma) and tumor necrosis factor (TNF) Neuroscience. 2005;131:283–292. doi: 10.1016/j.neuroscience.2004.10.043. [DOI] [PubMed] [Google Scholar]
- 18.Mariotti R, Tongiorgi E, Bressan C, Armellin M, Kristensson K, Bentivoglio M. Retrograde response of the rat facial motor nucleus to muscle inflammation elicited by phytohaemagglutinin. Eur J Neurosci. 2001;13:1329–1338. doi: 10.1046/j.0953-816x.2001.01507.x. [DOI] [PubMed] [Google Scholar]
- 19.Martino G, Hartung HP. Immunopathogenesis of multiple sclerosis: the role of T cells. Curr Opin Neurol. 1999;12:309–321. doi: 10.1097/00019052-199906000-00010. [DOI] [PubMed] [Google Scholar]
- 20.Matsumoto Y, Abe S, Tsuchida M, Hirahara H, Abo T, Shin T, Tanuma N, Kojima T, Ishihara Y. Characterization of CD4-CD8- T cell receptor alpha beta + T cells appearing in the subarachnoid space of rats with autoimmune encephalomyelitis. Eur J Immunol. 1996;26:1328–1334. doi: 10.1002/eji.1830260623. [DOI] [PubMed] [Google Scholar]
- 21.McPhail LT, Fernandes KJ, Chan CC, Vanderluit JL, Tetzlaff W. Axonal reinjury reveals the survival and re-expression of regeneration-associated genes in chronically axotomized adult mouse motoneurons. Exp Neurol. 2004;188:331–340. doi: 10.1016/j.expneurol.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 22.McPhail LT, Oschipok LW, Liu J, Tetzlaff W. Both positive and negative factors regulate gene expression following chronic facial nerve resection. Exp Neurol. 2005;195:199–207. doi: 10.1016/j.expneurol.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 23.Moalem G, Xu K, Yu L. T lymphocytes play a role in neuropathic pain following peripheral nerve injury in rats. Neuroscience. 2004;129:767–777. doi: 10.1016/j.neuroscience.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 24.Moran LB, Graeber MB. The facial nerve axotomy model. Brain Res Brain Res Rev. 2004;44:154–178. doi: 10.1016/j.brainresrev.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 25.Nau R, Bruck W. Neuronal injury in bacterial meningitis: mechanisms and implications for therapy. Trends Neurosci. 2002;25:38–45. doi: 10.1016/s0166-2236(00)02024-5. [DOI] [PubMed] [Google Scholar]
- 26.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 27.Raivich G, Jones LL, Kloss CU, Werner A, Neumann H, Kreutzberg GW. Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J Neurosci. 1998;18:5804–5816. doi: 10.1523/JNEUROSCI.18-15-05804.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rogers PR, Dubey C, Swain SL. Qualitative changes accompany memory T cell generation: faster, more effective responses at lower doses of antigen. J Immunol. 2000;164:2338–2346. doi: 10.4049/jimmunol.164.5.2338. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz M. Macrophages and microglia in central nervous system injury: are they helpful or harmful? J Cereb Blood Flow Metab. 2003;23:385–394. doi: 10.1097/01.WCB.0000061881.75234.5E. [DOI] [PubMed] [Google Scholar]
- 30.Schwartz M, Moalem G. Beneficial immune activity after CNS injury: prospects for vaccination. J Neuroimmunol. 2001;113:185–192. doi: 10.1016/s0165-5728(00)00447-1. [DOI] [PubMed] [Google Scholar]
- 31.Serpe CJ, Coers S, Sanders VM, Jones KJ. CD4+ T, but not CD8+ or B, lymphocytes mediate facial motoneuron survival after facial nerve transection. Brain Behav Immun. 2003;17:393–402. doi: 10.1016/s0889-1591(03)00028-x. [DOI] [PubMed] [Google Scholar]
- 32.Serpe CJ, Kohm AP, Huppenbauer CB, Sanders VM, Jones KJ. Exacerbation of facial motoneuron loss after facial nerve transection in severe combined immunodeficient (scid) mice. J Neurosci. 1999;19:RC7. doi: 10.1523/JNEUROSCI.19-11-j0004.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Serpe CJ, Sanders VM, Jones KJ. Kinetics of facial motoneuron loss following facial nerve transection in severe combined immunodeficient mice. J Neurosci Res. 2000;62:273–278. doi: 10.1002/1097-4547(20001015)62:2<273::AID-JNR11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 34.Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog Neurobiol. 1999;57:563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- 35.Yan Q, Elliott JL, Matheson C, Sun J, Zhang L, Mu X, Rex KL, Snider WD. Influences of neurotrophins on mammalian motoneurons in vivo. J Neurobiol. 1993;24:1555–1577. doi: 10.1002/neu.480241202. [DOI] [PubMed] [Google Scholar]
- 36.Yan Q, Matheson C, Lopez OT, Miller JA. The biological responses of axotomized adult motoneurons to brain-derived neurotrophic factor. J Neurosci. 1994;14:5281–5291. doi: 10.1523/JNEUROSCI.14-09-05281.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]