

Abstract
I demonstrate the existence in protein domain sequences of sets of statistically significant periodic signals, characteristic of the architectures of those domains. It is shown that, although the frequencies of the signals characteristic of a particular architecture have definite wave numbers, they can occur in any physical property. The characteristic signals define sequence units, which may correspond to specific structural features. Study of signals in different architectures suggests that symmetric structures are characterized by sets of sequence signals, which are members of harmonic series and which therefore impose commensurate periodicities on the structure. Asymmetric structures are characterized by mutually incommensurate sets of sequence signals. It is pointed out that, although these periodic sequence units can be very short, their existence is a property of the entire sequence.
The rapidly growing database of experimentally determined protein structures has given new insight into the characteristics of the architectures into which protein domains can fold and the relationships between those architectures. It has become clear that there is a broad range of folds available, and it is accepted dogma that the choice of fold, as indeed the location of each atom in the protein, is dictated by amino acid sequence. In parallel with these developments, there has been intensive study of protein sequences. One of the most intriguing aspects of the latter work has been a series of papers by various authors (1–3) on the longitudinal correlation properties of sequences. These studies showed that, by various criteria, protein sequences behave in a manner indistinguishable from random.
Taken together, these results pose a riddle. Consider, for example, the fact that a significant number of protein architectures exhibit structural symmetry. (Two lovely examples of this phenomenon are the TIM barrel and neuraminidase folds.) If structure is determined by sequence, there must be signals in the sequences of these molecules that dictate their structural symmetry. Furthermore, because it is unreasonable to suppose that symmetrical proteins fold differently from their less decorative—but no less ordered—peers, one may justifiably presume that such architecture signals are a general feature of protein sequences. If, however, the sequences of proteins are truly longitudinally random, no such signal can exist.
One may also observe that a given architecture is, in general, assumed by proteins of widely differing molecular weights. It may then be asked what sequence characteristics are conserved in a length-independent manner.
In the present work, I investigate this problem. I demonstrate the existence of sequence signals characteristic of different folds. It is shown that the character of these signals is such that they are not seen by methods used previously to investigate sequence correlations. The nature of the signals gives essential information about the protein folding process.
METHODS
My strategy was as follows. I assembled groups of sequences associated with well-defined architectures, chosen so that they were not closely homologous. To isolate sequence signals, I carried out a Fourier analysis of the physical properties of the amino acids in these sequences. The concept of sequence Fourier analysis is not new. Eisenberg et al. (4), Hecht et al. (5, 6), and DeLisi et al. (7) have used Fourier analyses to demonstrate the effect of periodicity in determining certain types of local structure formation and in the detection of hydrophobicity. The novelty of the present work lies in the use of a complete, statistically uncorrelated representation of amino acid properties and in the application of Fourier techniques over a wide spectrum of wave numbers.
Sequence Representation.
I want to specify protein sequences in terms of the physical properties of the amino acids. There are, however, many available physical property data sets for the 20 amino acids. This leads to two problems. (i) If one selects a subset of those data to represent a sequence, one is not, in general, assured that enough properties have been used to completely describe the physics of the amino acids. (ii) Any two sets of properties containing data developed by different investigators using different methods are likely to exhibit undesirable correlations.
To obviate these problems, I made use of the results of Kidera et al. (8, 9), who carried out a factor analysis of essentially all of the data sets available for the amino acids. They were able to demonstrate that all of these data could be represented by a set of 10 factors, which together carry 86% of the variance of the entire data set. Four major factors each represent a single property. The remaining six factors, which carry less of the total variance, are linear combinations of several properties. (The definitions of these factors are shown in Table 1.) It follows that each amino acid can be represented by a 10-vector, which specifies almost completely its physical properties. I write
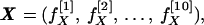 |
1 |
where X is the vector representing the amino acid X, and f[n]X is the nth property factor of X. A sequence of N amino acids can then be treated as a set of 10 N-strings, each specifying the course of a single property over the length of the chain. The value of the nth property factor as a function of chain position is thus given (in a convenient numbering scheme) by the set
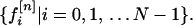 |
2 |
Table 1.
The 10 property factors of Kidera et al. (8)
| Major factors |
| 1. α-helix or bend structure preference. |
| 2. Side chain bulk-related. |
| 3. β-structure preference. |
| 4. Hydrophobicity-related. |
| Lesser factors |
| 5. Double-bend frequency related. |
| 6. Average amino acid composition. |
| 7. Occurence in flat extended structure. |
| 8. Occurence in a region of Ramachandran map. |
| 9. pK-C |
| 10. Surrounding hydrophobicity. |
The Fourier Transform Approach.
I was interested in calculating sine and cosine transforms. I denote the sine and cosine Fourier coefficients of the nth property factor by
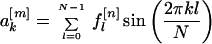 |
3 |
and
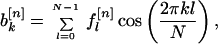 |
4 |
respectively. Note that each Fourier coefficient is a property of the entire sequence and that, because the sine and cosine functions form an orthogonal basis set, the coefficients are independent of one another.
I was interested primarily in the magnitude of each coefficient. It was therefore more convenient to work with the power spectrum, which is defined by the squares of the Fourier coefficients. My task was to identify those components of the power spectrum that occur in the members of a given group of sequences with magnitudes larger than one would expect in randomly selected sequences. I therefore needed to construct a scale on which to measure of the size of power spectral components.
I did this by simulation. For every sequence in a group of structurally similar, nonhomologous protein domains, I generated an ensemble of 10,000 sequences, each with a composition identical to that of the actual protein sequence but also with a randomly permuted ordering of amino acids. For each such random sequence, I calculated the values of the sine and cosine Fourier coefficients by using Eqs. 3 and 4. From these, I generated an average value of each of the Fourier coefficients over the ensemble and a standard deviation. These values, together with the values of the Fourier coefficients for the actual protein sequence, made it possible to generate reduced spectral power values as a function of k and n:
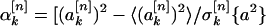 |
5 |
and
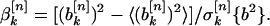 |
6 |
Here, the symbol 〈•〉 denotes an average over the ensemble of randomly permuted sequences, and σ[n]k{a2} is the corresponding standard deviation of the power spectral component inside the bracket for property factor n and wave number k. The quantities defined in Eqs. 5 and 6 give the deviation of the specified spectral power component of the actual protein sequence from its random ensemble average, measured in units of the SD. Thus, large positive values indicate power spectral components that are significantly larger than those that were measured for random sequences. These are the signals I sought.
I found it convenient to generate another quantity associated with the power spectrum. I refer to this as the “total power,” although it is actually a rms value of the spectral power, taken over all property factors, for a given value of k. I defined the functions
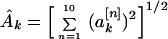 |
7 |
and
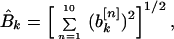 |
8 |
which are related to the sine and cosine power spectrum, respectively. I generated random ensemble averages of these quantities in the same way I did for the power spectra and defined the total power ratios
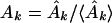 |
9 |
and
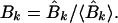 |
10 |
When the total sine (cosine) power for the actual protein sequence at specified k is greater than the ensemble average over random sequences, Ak (Bk) > 1. I exploited this property to construct a signal-averaging scheme that made it possible to find signals common to the sequences in a group, {P}, of nonhomologous, structurally similar domains. I denoted the values of the ratios in equations 9 and 10 for a particular protein, P, as Ak(P) and Bk(P). The group signal functions
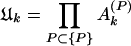 |
11 |
and
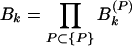 |
12 |
grow if many members of the group {P} have spectral power greater than average at wave number k and decrease if many members have less-than-average spectral power at k. Note an important feature of these group signal functions. Because they are multiplicative signal averages, the presence of even one very small (≈0) power ratio (Eqs. 9 or 10) can cause the corresponding group signal (Eqs. 11 or 12) to be very small. The group signal functions thus highlight signals at those wave numbers for which no power ratio is excessively small. I therefore could use them to extract signals common to many members of a group of sequences from the noise created by sequence features specific to individual members of the group. At the same time, special care needed to be taken not to lose signals that may be wiped out by the multiplicative effect I have mentioned. The final step in any analysis therefore was an examination of the occurrence of these signals in individual members of the group. A hierarchical approach eliminated the possibility of lost signals.
The Database.
To identify sets of protein domains with common architecture and nonhomologous sequences, I used the CATH database of Orengo et al. (http://www.biochem.ucl.ac.uk/bsm/cath/index.html). In CATH, domains are classified by architectural type, and within architecture they are classified by sequence homology, an organizational scheme that is particularly useful for the present research. I required groups of proteins that share a common architecture but whose sequences fall into a large number of different homology classes. Two architectures satisfy this stringent requirement. The TIM barrels are represented in this work by 26 single-chain protein and domain sequences, with no pairwise identity >14%. The Ig fold is represented by 30 sequences, with no pairwise identity >23%. The feasibility of studying additional architectures is being investigated.
RESULTS
I first considered the TIM barrel (Table 2). In Fig. 1, I plot sine and cosine group signals (Eqs. 11 and 12). One is immediately struck by the enormous cosine peak at k = 21. Although this peak is overwhelmingly larger than any other group signal, there are other peaks that are clearly well above background. Those peaks that are statistically significantly larger than the background of observed signals have been identified in a objective fashion with the aid of a box plot. I found additional significant peaks in the cosine group signal at k = 10, 13, 18, 30, and 42. The statistically significant peaks in the sine group signal fall at k = 22, 30, and 47.
Table 2.
Property factors for which sequence signals are observed in TIM barrel proteins
| PDB code | cosine, k = 10 | cosine, k = 21 |
|---|---|---|
| 1TML | 6 | 2, 3, 7 |
| 1PII, domain 2 | 7 | |
| 1WSY, chain A | 7 | |
| 1NAL, chain 1 | 5, 8 | |
| 1NAR | 10 | |
| 1TPF, chain A | 2 | |
| 1LLO | 2 | 2 |
| 1PII, domain 1 | 5, 7 | |
| 1XYZ, chain A | 1, 9 | 6 |
| 2EBN | 1 | |
| 1ADS | 1 | |
| 1CEC | 9 | |
| 1FBA, chain A | 4 | |
| 1GHS, chain A | 1 | |
| 1RLC, chain L, domain 2 | 6, 7 | |
| 1EBH, chain A, domain 2 | 3* | |
| 2MNR | 4, 9 | |
| 1CHR, chain A | 10 | 10 |
| 1GOX | 4 | |
| 1ADD | 7 | |
| 2TMD, chain A, domain 1 | 4, 6 | 10 |
| 1PTA | 4 | 1, 5 |
| 1AMG, domain 1 | 8 | |
| 1XIS | 1 | |
| 1BTC | 5, 6 | |
| 1CBG | 2 |
β[n]21 = 1.98 in this case whereas in all other cases, β[n]21 ≥ 2.0 (see text).
Figure 1.

Sine and cosine signals for the set of 26 TIM-barrel proteins (Eqs. 11 and 12).
It was not sufficient to observe peaks in the group signal functions that are large. I wanted to examine the distribution of significant signals among the individual members of the group. I therefore examined the values of α[n]k and β[n]k (Eqs. 5 and 6) for 1 ≤ n ≤ 10 for each value of k at which a significant group signal occurs. I defined a significant signal as one for which α[n]k ≥ 2.0 or β[n]k ≥ 2.0. By definition, this means that the corresponding power spectral value is at least two SDs above the average observed for the ensemble of random sequences of the same composition.
I found that every member of the TIM barrel group has β[n]10 and/or β[n]21 ≥ 2.0 (with the exception of 1EBH, for which β[n]21 = 1.98, a statistically negligible difference). Within the limits of accuracy imposed by the discrete nature of the sequence, these two values of k constitute a fundamental frequency and its first harmonic. None of the other k values identified by the signal functions (Eqs. 11 and 12) give significant signals in as many members of the TIM barrel group. It should be noted, however, that the occurrence of cosine signals at k = 18, 30, and 42 and sine signals at k = 22 and 30 is a further indication of the importance of a harmonic series of architecture signals based on the fundamental k ≈ 10 in this set of molecules.
I further noted that the values of n—i.e., the property factors for which a signal is observed—are not the same for all of the proteins in the group. This is indicated in Table 2. It was seen that significant signals were observed in each of the property factors in one or another protein of the group.
I now turn to the Ig fold (10) (Table 3). Applying the analytic procedure set forth above, I found significant sine group signals at k = 3, 7, 20, 37, 38, and 41, and cosine group signals at k = 16, 39, 48, 49, 51, 54, and 59 (Fig. 2). Examination of α[n]k and β[n]k values, however, revealed a different situation than that encountered in the TIM barrel proteins. Rather than detecting two harmonically related wave numbers, at one or both of which significant signals occur in every protein of the group, I found no apparent harmonic relationships. I also found no single wave number that gave rise to significant signals in more than half of the proteins. These observations suggest that there may be significant signals that do not show up in the initial analysis—a possibility that was noted above. To clarify this point, I selected one prominent group signal and considered the subset of proteins that did not show significant signals at that wave number. I chose the subset of proteins that showed no cosine signal at k = 16 for this purpose. I repeated the analysis on this subset. It was found that the set of Ig proteins in which there was no cosine signal at k = 16, containing 15 proteins, exhibited a cosine group signal at k = 15. Analysis of this second group showed that nine proteins in this set showed significant cosine signals at k = 15. I thus found that signals at one or both of these wave numbers occurred in 24 of the 30 proteins in this group. Given the discrete nature of the Fourier transforms, signals at k = 15 and k = 16 can be regarded as essentially equivalent. The remaining six proteins in this group showed significant signals at values of k corresponding to other significant group signals. As in the case of the TIM barrels, the significant signals in the Ig group occurred for different values of n—i.e., in different physical properties—in different proteins (Table 3).
Table 3.
Property factors for which sequence signals are observed in Ig-like proteins
| PDB code | cosine, k = 16 | cosine, k = 15 |
|---|---|---|
| 1HOE | 3, 8 | 1 |
| 3HHR, chain B, domain 1 | 1, 3 | |
| 3HHR, chain B, domain 2 | 3 | |
| 1WAP, chain A | 3, 4, 7, 8, 10 | |
| 1NCG | ||
| 1GGT, chain A, domain 3 | ||
| 1GGT, chain A, domain 4 | ||
| 1HNF, domain 2 | 2 | |
| 1HFT, domain 1 | 4 | |
| 1CGT, domain 4 | 10 | |
| 1MFA, chain H | * | 5 |
| 1TTF | 8 | |
| 1CFB, domain 1 | 2, 10 | |
| 1RSY | 8 | |
| 1CFB, domain 2 | 1, 5, 10 | |
| 1PLC | 10 | |
| 1ETA, chain 1 | 3 | |
| 1VCA, chain A, domain 2 | 1 | |
| 1SXA chain A | 1 | 3 |
| 1TNM | 10 | |
| 1AOZ, chain A, domain 1 | 2 | 7 |
| 1AKP | 1 | |
| 1CLC, domain 1 | 3 | 7 |
| 1CGT, domain 3 | ||
| 1GOF, domain 3 | ||
| 1NIF, domain 1 | 3, 4 | |
| 1CTN, domain 1 | 5 | |
| 1EXG | 4 | |
| 1CID | § | |
| 3CD4 |
β[n]16 = 1.44
β[n]16 = 1.56
Figure 2.

Sine and cosine signal for the set of 30 Ig-like proteins.
One may ask what relationship these sequence signals have to time-averaged structural features of the protein. The connection between sequence signals and structure can be made by noting that a signal of wave number k divides the sequence into subsequences of length N/k. Such a signal therefore implicates units of this size (which I refer to as “sequence units”) in the determination of the architecture of the molecule. In a sequence in which several signals are important, the sizes and positions of the sequence units will be determined by interference between those signals. With that in mind, I asked to what extent the principal sequence signal alone relates to recognizable structural features in the two architectures I considered. In Fig. 3, I examine the positions of such sequence units in a TIM barrel protein for k = 21. It was found that each strand of the central barrel is composed of one sequence unit. In contrast, several of the helices that surround the central barrel contain parts of more than one sequence unit. Similar behavior was observed in other TIM barrels. Examination of TIM barrels for which only k = 10 is a significant signal revealed similar behavior in which essentially every sequence unit contains one strand. These observations provide a direct connection between the architecture of these molecules and the principal sequence signal and suggest that the formation of the central barrel, or at least of the strands that comprise it, is a critical event in the folding of these proteins.
Figure 3.

Structure of the TIM-barrel protein Endo-1,4-β-d-glucanase (1TML), indicating the locations of the sequence units determined by the peak in the cosine power spectrum at k = 21. Units are alternately colored red and white, with the exception of the N-terminal unit, which is colored yellow, and the C-terminal unit, which is colored blue.
I did not observe such a neat coincidence in the Ig proteins between structural elements and sequence units determined by a single wave number. This fact is demonstrated in Fig. 4, in which I show an Ig structure in which the periodic units determined by the cosine group signal with k = 16 are indicated in color. I suggest that this observation reflects the fact that, in these proteins, a number of important group signals occur at wave numbers that are not members of a harmonic series and which therefore give rise to mutually incommensurate periodicities. The sizes of the structural units are defined by interference between these incommensurate periodicities and are therefore substantially less regular in distribution than those observed in the TIM barrels. This may also result in a less symmetric structure than shown by the TIM barrels, in which most of the group signals are members of a single harmonic series and constructively reinforce a limited set of periodicities.
Figure 4.

Structure of the Ig-like protein superoxide dismutase (1SXA), indicating the locations of the periodic units determined by the peak in the cosine power spectrum at k = 16. The color scheme is the same as in Fig. 3. Two equivalent molecules are shown, oppositely oriented, to exhibit both sides of the structure.
These results have a number of implications: (i) Protein sequences are not random. They exhibit periodic signals in various physical properties that are statistically significantly different from what one would expect from random sequences. Nonrandomness was not observed in previous studies in which sequences have not been represented by a complete, uncorrelated set of properties. (ii) Distinct sets of sequence signals characterize individual protein architectures. I speculate that, when these sequence signals are members of a harmonic series, the resulting limited set of periodicities gives rise to a relatively symmetric structure. When the sequence signals are incommensurate, less symmetric structures may be produced. (iii) These sequence signals can occur in any amino acid property. It is the frequency of the signals, rather than the physical property in which they occur, that determines the architecture. Because of this fact, sequences that apparently are unrelated can give rise to similar architectures. (iv) The size of the sequence fragments responsible for architecture choice are related inversely to the wave numbers of the sequence signals. However, no matter how small these sequence fragments are, the signal is a property of the entire sequence.
Acknowledgments
I thank Dr. Christine Orengo for much useful information and practical help in connection with the CATH database. I also thank Prof. Craig Benham for commenting on the manuscript and Sidney B. Klein, of Analytic Decisions Corporation, for many helpful and interesting discussions.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviation: TIM, Triose phosphate isomerase.
References
- 1.White S H, Jacobs R E. Biophys J. 1990;57:911–921. doi: 10.1016/S0006-3495(90)82611-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.White S H, Jacobs R E. J Mol Evol. 1993;36:79–95. doi: 10.1007/BF02407307. [DOI] [PubMed] [Google Scholar]
- 3.Rahman R S, Rackovsky S. Biophys J. 1995;68:1531–1539. doi: 10.1016/S0006-3495(95)80325-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eisenberg D, Weiss R M, Terwilliger T C. Proc Natl Acad Sci USA. 1984;81:140–144. doi: 10.1073/pnas.81.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiong H, Buckwalter B L, Shieh H-M, Hecht M H. Proc Natl Acad Sci USA. 1995;92:6349–6353. doi: 10.1073/pnas.92.14.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.West M W, Hecht M H. Protein Sci. 1995;4:2032–2039. doi: 10.1002/pro.5560041008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cornette J L, Cease K B, Margalit H, Spouge J L, Berzofsky J A, DeLisi C. J Mol Biol. 1987;195:659–685. doi: 10.1016/0022-2836(87)90189-6. [DOI] [PubMed] [Google Scholar]
- 8.Kidera A, Konishi Y, Oka M, Ooi T, Scheraga H A. J Protein Chem. 1985;4:23–54. [Google Scholar]
- 9.Kidera A, Konishi Y, Ooi T, Scheraga H A. J Protein Chem. 1985;4:265–297. [Google Scholar]
- 10.Chothia C, Gerstein M. Nature (London) 1997;385:579–580. doi: 10.1038/385579a0. [DOI] [PubMed] [Google Scholar]