

Abstract
Protein toxins from venomous animals exhibit remarkably specific and selective interactions with a wide variety of ion channels. Hanatoxin and grammotoxin are two related protein toxins found in the venom of the Chilean Rose Tarantula, Phrixotrichus spatulata. Hanatoxin inhibits voltage-gated K+ channels and grammotoxin inhibits voltage-gated Ca2+ channels. Both toxins inhibit their respective channels by interfering with normal operation of the voltage-dependent gating mechanism. The sequence homology of hanatoxin and grammotoxin, as well as their similar mechanism of action, raises the possibility that they interact with the same region of voltage-gated Ca2+ and K+ channels. Here, we show that each toxin can interact with both voltage-gated Ca2+ and K+ channels and modify channel gating. Moreover, mutagenesis of voltage-gated K+ channels suggests that hanatoxin and grammotoxin recognize the same structural motif. We propose that these toxins recognize a voltage-sensing domain or module present in voltage-gated ion channels and that this domain has a highly conserved three-dimensional structure.
The venom produced by many animals has been a rich source of ligands that interact with different types of ion channels. In some cases, these toxins bind to the outer vestibule of the pore and physically block ion conduction (1–6), whereas in other cases, the toxins modify channel gating. Toxins that act as modifiers of channel gating have been described for voltage-gated Na+ channels (7–11), voltage-gated K+ channels (12–14), and voltage-gated Ca2+ channels (15–17). The different effects of the pore-blocking toxins and the gating modifier toxins arise because they interact in very specific ways with different regions of ion channels (14).
Ion channel toxins exhibit highly selective interactions with their targets. For example, of the many K+ channel pore-blocking proteins discovered so far, none has been found to bind to any channel other than those containing K+ selective pores (18). It is interesting that some of these pore-blocking toxins, for example Lq2 from scorpion venom, will bind to Ca2+ activated K+ channels (19), voltage-gated K+ channels, (20) and inward rectifier K+ channels (21). These channels are very different in many ways but they all contain a K+ selective pore. The promiscuity of Lq2 suggests that the structure of K+ channel pores has been conserved throughout evolution, a notion supported by the conservation of pore-forming amino acids in K+ channels. If pore-blocking toxins underscore the conserved structure of the K+ channel pore, might gating modifier toxins reveal a voltage-sensing structure that may be conserved among channels with different ion selectivity?
We were led to ask this question when examining the sequences of two recently isolated toxins, hanatoxin and grammotoxin. Hanatoxin inhibits voltage-gated K+ channels (12–14) and grammotoxin inhibits voltage-gated Ca2+ channels (15, 16). Both toxins inhibit their respective channels by modifying voltage-dependent gating (13, 16). The conservation in the sequence of the two toxins and the similarity of their mechanisms of inhibition raise the possibility that they interact with very similar regions on voltage-gated K+ and Ca2+ channels. We examined this possibility and found that each of the two toxins can bind to and modify the gating of both voltage-gated Ca2+ and K+ channels. Several mutants of a voltage-gated K+ channel display altered binding of both hanatoxin and grammotoxin, suggesting that the two toxins recognize a structural motif that is conserved between voltage-gated Ca2+ and K+ channels.
MATERIALS AND METHODS
Channel Constructs.
A mutant construct (drk1Δ7) of the drk1 K+ channel was used because it is highly sensitive to pore-blocking proteins like agitoxin2 (Kd ≈ 70 pM), and thus allowing agitoxin2 to be used to subtract background conductances. This drk1Δ7 K+ channel contained the following mutations: Thr355Ser, Lys356Gly, Ala362Asp, Ser363Ala, Ile379Met, Tyr380Thr, and Lys382Val (22). The drk1Δ7 K+ channel cDNA, in a bluescript vector, was linearized with NotI and transcribed by using T7 RNA polymerase. cDNAs encoding rabbit brain α1A (BI-1) (23), rat brain β1b (24), and rabbit skeletal muscle α2δ (25) were kindly provided by Lutz Birnbaumer (Univ. of California, Los Angeles) in the pAGA2 vector (26). α1A and α2δ were linearized with XhoI, and β1b was linearized with HindIII. All of the subunits were transcribed by using T7 RNA polymerase. Approximately equal quantities of cRNA for the three subunits were injected into oocytes.
Electrophysiological Recordings.
Oocytes from Xenopus laevis frogs were removed surgically and incubated with agitation for 1.5 hrs in a solution containing: NaCl (82.5 mM), KCl (2.5 mM), MgCl2 (1 mM), Hepes (5 mM), and collagenase (2 mg/ml; Worthington), pH 7.6 with NaOH. Defolliculated oocytes were injected with cRNA and incubated at 17°C in a solution containing: NaCl (96 mM), KCl (2 mM), MgCl2 (1 mM), CaCl2 (1.8 mM), Hepes (5 mM), and gentamicin (50 μg/ml; GIBCO/BRL), pH 7.6 with NaOH for 16–24 hr before electrophysiological recording. Oocyte membrane voltage was controlled by using either an Axoclamp-2A two-electrode voltage clamp (Axon Instruments, Foster City, CA) or OC-725C oocyte clamp (Warner Instruments, Hamden, CT). Data were filtered at 1–2 kHz (8-pole Bessel) and digitized at 5–10 kHz. Microelectrode resistances were 0.1–0.8 MΩ when filled with 3 M KCl. Oocytes were studied in 100- or 160-μl recording chambers that were perfused with one of two different extracellular solutions. For recording of K+ channel currents, the extracellular solution contained (in mM): RbCl (50), NaCl (50), MgCl2 (1), CaCl2 (0.3), and Hepes (5), pH 7.6 with NaOH. For recording of Ca2+ channel currents, the extracellular solution contained (in mM): BaOH2 (10), NaCH3SO3 (100), and Hepes (10), pH 7.6 with NaOH. For recording K+ channel currents, chlorided silver ground wires were inserted directly into the recording chamber. For recording Ca2+ channel currents, agar salt bridges containing 1 M NaCl were used to connect the ground electrode pools and the recording chamber. All experiments were carried out at room temperature (≈22°C).
Toxins.
Synthetic grammotoxin was generously provided by Richard A. Keith and Richard A. Lampe, Department of Pharmacology, Zeneca Pharmaceuticals Group, Zeneca Inc. Hanatoxin was purified from Phrixotrichus spatulata (previously named Grammostola spatulata) venom as described (12). Hanatoxin used in these experiments is an approximately equal mixture of two isoforms that differ at position 13 where hanatoxin1 contains Ser and hanatoxin2 contains Ala. Venom was purchased from Spider Pharm (Feasterville, PA).
RESULTS AND DISCUSSION
Fig. 1 shows the sequence of hanatoxin and grammotoxin. The two toxins are highly related with >50% of the residues being either identical or highly conserved. This conservation seems to imply that voltage-gated Ca2+ and K+ channels share a similar binding surface for this type of inhibitor. To address this, we asked whether grammotoxin (the Ca2+ channel inhibitor) can interact with K+ channels and whether hanatoxin (the K+ channel inhibitor) can interact with Ca2+ channels. The drk1 voltage-gated K+ channel (27), a hanatoxin-sensitive K+ channel (12, 13), was expressed in Xenopus oocytes and studied by using a two-electrode voltage clamp amplifier. Fig. 2 shows that grammotoxin, an inhibitor of voltage-gated Ca2+ channels, also reversibly inhibits the drk1 voltage-gated K+ channel. Grammotoxin shifts the opening of channels to more depolarized voltages and speeds deactivation (Fig. 2 B and C), qualitatively similar to what has been observed previously for hanatoxin (13). These results suggest that grammotoxin-bound channels can open but do so with altered gating energetics. It is interesting that the effects of grammotoxin on deactivation kinetics are less pronounced when compared with the effects of hanatoxin (13). We examined the concentration dependence of K+ channel occupancy by grammotoxin using the same analysis previously described for hanatoxin (13). The concentration dependence for the fraction of uninhibited current at negative voltages (reflecting the fraction of unbound channels) could be well described by a model incorporating four independent and equivalent toxin-binding sites, each with a Kd for grammotoxin of 19 μM (Fig. 2 D and E).
Figure 1.

Amino acid sequence of hanatoxin1 (12) and grammotoxin (15). Dashes indicate identity with hanatoxin1. Asterisks above the sequence indicate conservation among the toxins.
Figure 2.

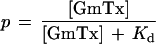 |
We next asked whether hanatoxin, the K+ channel inhibitor, interacts with a voltage-gated Ca2+ channel. The α1A voltage-gated Ca2+ channel (23) was examined because it is known to contain at least one high affinity receptor (Kd in low nanomolar range) for grammotoxin (16). Fig. 3 shows that hanatoxin inhibits the Ca2+ channel. Ba2+ currents during weak depolarization were inhibited to a larger extent than those elicited by strong depolarization, consistent with observations of hanatoxin inhibition of K+ channels (13) and grammotoxin inhibition of Ca2+ channels (16). The voltage-dependence of the fraction of uninhibited current at 10 μM hanatoxin is shown in Fig. 3D. The fraction of uninhibited current increased with larger depolarization. This voltage-dependence may result from the opening of toxin bound channels (13, 16) or the unbinding of the toxin at depolarized voltages. Without high resolution tail current measurements, we cannot distinguish between these possibilities. The fraction of uninhibited current reached a plateau at negative voltages similar to what has been previously observed for hanatoxin inhibition of the drk1 K+ channel. The plateau value for the fraction of uninhibited Ba2+ current at 10 μM hanatoxin was 0.4. This same plateau value was observed at lower concentrations (≈20 nM) for hanatoxin inhibition of the drk1 K+ channel. This result suggests that the binding affinity of hanatoxin for the Ca2+ channel is significantly lower than it is for the K+ channel (see Fig. 3 legend) because the plateau value reflects the fraction of unbound channels (13).
Figure 3.

Hanatoxin inhibition of the α1A voltage-gated Ca2+ channel. (A) Time course of inhibition of Ca2+ channel current by 10 μM hanatoxin. Amplitude of steady-state test current after depolarization to 0 mV plotted against time. Pulses were given every 5 sec. Holding voltage was −80 mV. (B) Traces showing currents elicited by various strength depolarizations in either the absence (○) or presence of 10 μM hanatoxin (•). Traces are shown unsubtracted. Within the voltage range examined, the leak currents observed after block of Ca2+ channels with Cd2+ were very small (<10 nA). (C) Steady-state current voltage relationship in either the presence or absence of 10 μM hanatoxin. Steady-state currents were measured 10 msec after depolarization. Holding voltage was −80 mV. Test pulses were 25 msec in duration. (D) Fraction of uninhibited current elicited by various strength depolarizations for 10 μM hanatoxin. Steady-state test current amplitude in the presence of grammotoxin (I) and steady-state test current in control (Io). Current amplitude was measured 10 msec after depolarization to the indicated test voltage. If a single site for hanatoxin exists on the α1A channel, then the plateau value for the fraction of uninhibited current (0.4) corresponds to a Kd for the toxin of ≈7 μM. Although unlikely (see Discussion), if there are four equivalent sites for hanatoxin, then the fraction of uninhibited current (0.4) would correspond to a Kd for the toxin of ≈40 μM.
The high degree of homology between hanatoxin and grammotoxin, taken together with the fact that both toxins interact with both voltage-gated Ca2+ and K+ channels, suggests that the two toxins share a very similar binding site on voltage-gated channels. However, the two toxins bind to the two types of channels with different affinities. For example, the affinity of hanatoxin is ≈200-fold higher than grammotoxin for the drk1 K+ channel (see Fig. 2E). This difference in affinity leaves open the possibility that the two toxins bind to different receptors. To address this directly, we asked whether mutations that have large effects on hanatoxin binding to the drk1 K+ channel (14) also alter the affinity of grammotoxin binding to the same channel. Mutation of three residues at the C-terminal edge of S3 (I273, F274, and E277) to Ala were previously found to lower hanatoxin binding affinity (14). Fig. 4 shows that the binding of grammotoxin to I273A, F274A, and E277A in the drk1 K+ channel occurred with significantly lower affinity than to the wild-type channel. This result provides evidence that hanatoxin and grammotoxin share a common binding site on the K+ channel.
Figure 4.

Mutation of residues in the S3–S4 linker alters the binding affinity of grammotoxin to the drk1 K+ channel. (A) Membrane folding model of a single K+ channel α subunit showing the location of three residues at the C-terminal end of S3 that influence hanatoxin-binding affinity (14). (B) Fraction of uninhibited tail current elicited by various strength depolarizations at 10 μM grammotoxin for F274A, E277A and the wild-type channel. Tail current amplitude in the presence of 10 μM grammotoxin (I) and tail current in control (Io). All tail currents were elicited by repolarization to −50 mV. Test depolarizations were 250 msec in duration from a holding voltage of −80 mV. (C) Equilibrium dissociation constants (Kd) for grammotoxin binding to mutant drk1 K+ channels shown as a fraction of the Kd for grammotoxin binding to the wild-type drk1 K+ channel. Kd values for grammotoxin (mean ± SEM; n = 3–5) were calculated from the fraction of uninhibited current at negative voltages as previously described for hanatoxin (13) and shown in Fig. 2 and legend. Data for hanatoxin from ref. 14 are shown for comparison.
There are likely to be some very interesting differences between the interaction of hanatoxin and grammotoxin with voltage-gated K+ and Ca2+ channels. K+ channels are formed by the assembly of four identical subunits. The hanatoxin-binding site is eccentrically located at least 10–15 Å from the central pore axis, and four hanatoxins can bind to the channel at once (13, 14). The homology between hanatoxin and grammotoxin, and the influence of I273, F274, and E277 on both toxins, strongly suggests that grammotoxin interacts with four eccentrically located receptors on K+ channels. The concentration dependence for grammotoxin occupancy of K+ channels is consistent with this stoichiometry (Fig. 2). What about the toxin receptors on voltage-gated Ca2+ channels? The four repeats of a voltage-gated Ca2+ channel α subunit are homologous but not identical. This repeat (pseudosubunit) asymmetry suggests that there may not be four identical hanatoxin and grammotoxin receptors on each Ca2+ channel. Fig. 5 shows an alignment of a number of voltage-gated ion channels in the region spanning from S3 through S4. The dark shading highlights the identity of residues at positions equivalent to 273, 274, and 277 in the drk1 K+ channel. The general conservation in this region very likely underlies the conservation of toxin-binding sites between the drk1 K+ channel and the α1A Ca2+ channel. However, the differences seen between repeats of the Ca2+ channel in this region implies that there is a heterogeneous population of toxin receptors on each Ca2+ channel.
Figure 5.

(A) Alignment of various voltage-gated ion channels in a region spanning from S3 through S4. Light shading indicates proposed transmembrane segments. Dark shading highlights the position of residues that when mutated in the drk1 K+ channel alters hanatoxin- and grammotoxin-binding affinity (14, Fig. 4). Amino acid sequence for α1A is from ref. 23, and the sequence for αIIA is from ref. 42. (B) Diagram illustrating voltage-sensing domains (with toxin receptors contained within) and some differences that likely exist between voltage-gated K+, Ca2+, and Na+ channels. Voltage-sensing domains are drawn as modules that surround the pore domain. K+ channels studied experimentally are tetramers of four identical subunits. Each subunit contains a voltage-sensing domain to which a toxin binds. The toxin receptor is located at least 10–15 Å from the central pore axis. Ca2+ and Na+ channels are heterotetramers that contain four homologous but different pseudosubunits, each containing a voltage-sensing domain that likely binds gating modifier toxins with different energetics. Toxin receptors are arbitrarily drawn as contained within each subunit; receptors could be at the interface between subunits.
There seems to be a fundamental connection between the gating modifiers of voltage-gated K+ and Ca2+ channels examined in the present study and a number of gating modifiers of voltage-gated Na+ channels. α-Scorpion toxin and sea anemone toxin are gating modifiers of voltage-gated Na+ channels that bind to the channel in a voltage dependent manner and produce a dramatic slowing of inactivation (28–32). Of the residues examined so far, mutation of E1613 in repeat four of the Na+ channel has the largest effect on the binding affinity of α-scorpion toxin and sea anemone toxin (33). An alignment of this region (Fig. 5) shows that E1613 corresponds to E277 in the drk1 K+ channel, a residue that has a large effect on the binding affinity of both hanatoxin (14) and grammotoxin (Fig. 4). (Repeat IV of the Na+ channel also has a Phe at the equivalent position to F274 in drk1, another residue that has a large influence on hanatoxin and grammotoxin binding to K+ channels, but not yet examined in the Na+ channel). Thus, a glutamate residue present at the homologous position at the outside edge of S3 has a profound effect on hanatoxin and grammotoxin binding to K+ channels and on α-scorpion toxin and sea anemone toxin binding to Na+ channels. This comparison suggests that all these gating modifiers interact with the same region of voltage-gated ion channels.
What is the function of this toxin-binding region and why has its structure been conserved in the voltage-gated ion channel superfamily? Perhaps the simplest answer is that the toxins recognize a voltage-sensing domain or module that has a highly conserved three-dimensional structure in voltage-gated K+, Ca2+, and Na+ channels. What part of the channel protein comprises the voltage-sensing module? The S4 segment, which contains a high density of basic residues, clearly is an important component of the voltage-sensing domain (34–39). However, a growing body of evidence suggests that the voltage-sensing domain also includes residues in the region spanning from S2 through S4 (34–41). This idea is reinforced by the fact that the binding sites for the gating modifer toxins are located in this region (including residues in the C-terminal end of S3 and possibly the N-terminal end of S4) (Fig. 4; refs. 14 and 33). Distant sequence conservation among voltage-gated K+, Ca2+, and Na+ channels clearly identifies these channels as members of the same gene superfamily. The promiscuity of the gating modifier toxins underscores conservation of the three-dimensional structure of voltage-gated channels. It is as if voltage-gated ion channels follow a multidomain architecture whereby the ion selective pore domain has diverged to confer selectivity for K+, Ca2+, or Na+, and a separate voltage-sensing domain, which is conserved in its structure, is shared by all of them.
Acknowledgments
We thank Roderick MacKinnon and Zhe Lu for helpful discussions. This work was supported by a grant from the National Institutes of Health (GM43949) to Roderick MacKinnon and by the National Institute of Neurological Disorders and Stroke intramural research program.
References
- 1.MacKinnon R, Miller C. J Gen Physiol. 1988;91:335–349. doi: 10.1085/jgp.91.3.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller C. Neuron. 1988;1:1003–1006. doi: 10.1016/0896-6273(88)90157-2. [DOI] [PubMed] [Google Scholar]
- 3.MacKinnon R, Miller C. Science. 1989;245:1382–1385. doi: 10.1126/science.2476850. [DOI] [PubMed] [Google Scholar]
- 4.Park C-S, Miller C. Neuron. 1992;9:307–313. doi: 10.1016/0896-6273(92)90169-e. [DOI] [PubMed] [Google Scholar]
- 5.Stocker M, Pongs O, Hoth M, Heinemann S H, Stuhmer W, Schroter K H, Ruppersberg J P. Proc R Soc London. 1991;245:101–107. doi: 10.1098/rspb.1991.0094. [DOI] [PubMed] [Google Scholar]
- 6.Hurst R S, Busch A E, Kavanaugh M P, Osborne P B, North R A, Adelman J P. Mol Pharmacol. 1991;40:572–576. [PubMed] [Google Scholar]
- 7.Cahalan M D. J Physiol. 1975;244:511–534. doi: 10.1113/jphysiol.1975.sp010810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanck D A, Sheets M F. J Gen Physiol. 1995;106:601–616. doi: 10.1085/jgp.106.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Catterall W A. J Gen Physiol. 1979;74:375–391. doi: 10.1085/jgp.74.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang G K, Strichartz G. J Gen Physiol. 1985;86:739–762. doi: 10.1085/jgp.86.5.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonoi T, Hille B. J Gen Physiol. 1987;89:253–274. doi: 10.1085/jgp.89.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swartz K J, MacKinnon R. Neuron. 1995;15:941–949. doi: 10.1016/0896-6273(95)90184-1. [DOI] [PubMed] [Google Scholar]
- 13.Swartz K J, MacKinnon R. Neuron. 1997;18:665–673. doi: 10.1016/s0896-6273(00)80306-2. [DOI] [PubMed] [Google Scholar]
- 14.Swartz K J, MacKinnon R. Neuron. 1997;18:675–682. doi: 10.1016/s0896-6273(00)80307-4. [DOI] [PubMed] [Google Scholar]
- 15.Lampe R A, Defeo P A, Davison M D, Young J, Herman J L, Spreen M B, Horn M B, Mangano T J, Keith R A. Mol Pharmacol. 1993;44:451–460. [PubMed] [Google Scholar]
- 16.McDonough S I, Lampe R A, Keith R A, Bean B P. Mol Pharmacol. 1997;52:1095–1104. doi: 10.1124/mol.52.6.1095. [DOI] [PubMed] [Google Scholar]
- 17.McDonough S I, Mintz I M, Bean B P. Biophys J. 1997;72:2117–2128. doi: 10.1016/S0006-3495(97)78854-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller C. Neuron. 1995;15:5–10. doi: 10.1016/0896-6273(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 19.Lucchesi K, Ravindran A, Young H, Moczydlowski E. J Membr Biol. 1989;109:269–281. doi: 10.1007/BF01870284. [DOI] [PubMed] [Google Scholar]
- 20.Escobar L, Root M J, MacKinnon R. Biochemistry. 1993;32:6982–6987. doi: 10.1021/bi00078a024. [DOI] [PubMed] [Google Scholar]
- 21.Lu Z, MacKinnon R. Biochemistry. 1997;36:6936–3940. doi: 10.1021/bi9702849. [DOI] [PubMed] [Google Scholar]
- 22.Aggarwal S K. Dissertation. Cambridge, MA: Harvard University; 1996. [Google Scholar]
- 23.Mori Y, Friedrich T, Kim M S, Mikami A, Nakai J, Ruth P, Bosse E, Hofmann F, Flockerzi V, Furuichi T, et al. Nature (London) 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- 24.Pragnell M, Sakamoto J, Jay S D, Campbell K P. FEBS Lett. 1991;291:253–258. doi: 10.1016/0014-5793(91)81296-k. [DOI] [PubMed] [Google Scholar]
- 25.Ellis S B, Williams M E, Ways N R, Brenner R, Sharp A H, Leung A T, Campbell K P, McKenna E, Koch W J, Hui A, et al. Science. 1988;241:1661–1664. doi: 10.1126/science.2458626. [DOI] [PubMed] [Google Scholar]
- 26.Wei X Y, Perez-Reyes E, Lacerda A E, Schuster G, Brown A M, Birnbaumer L. J Biol Chem. 1991;266:21943–21947. [PubMed] [Google Scholar]
- 27.Frech G C, VanDongen A M, Schuster G, Brown A M, Joho R H. Nature (London) 1989;340:642–645. doi: 10.1038/340642a0. [DOI] [PubMed] [Google Scholar]
- 28.Catterall W A. J Biol Chem. 1977;252:8660–8668. [PubMed] [Google Scholar]
- 29.Catterall W A, Beress L. J Biol Chem. 1978;253:7393–7396. [PubMed] [Google Scholar]
- 30.Catterall W A. J Gen Physiol. 1979;74:357–391. doi: 10.1085/jgp.74.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang G K, Strichartz G. J Gen Physiol. 1985;86:739–762. doi: 10.1085/jgp.86.5.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gonoi T, Hille B. J Gen Physiol. 1987;89:253–274. doi: 10.1085/jgp.89.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rogers J C, Qu Y, Tanada T N, Scheuer T, Catterall W A. J Biol Chem. 1996;271:15950–15960. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- 34.Yang N, George A L, Horn R. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]
- 35.Larsson H P, Baker O S, Dhillon D S, Isacoff E Y. Neuron. 1996;16:387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- 36.Yusaf S P, Wray D, Sivaprasadarao A. Pflügers Arch. 1996;433:91–97. doi: 10.1007/s004240050253. [DOI] [PubMed] [Google Scholar]
- 37.Mannuzzu L M, Moronne M M, Isacoff E Y. Science. 1996;271:213–216. doi: 10.1126/science.271.5246.213. [DOI] [PubMed] [Google Scholar]
- 38.Aggarwal S K, MacKinnon R. Neuron. 1996;16:1169–1177. doi: 10.1016/s0896-6273(00)80143-9. [DOI] [PubMed] [Google Scholar]
- 39.Seoh S-A, Sigg D, Papazian D M, Bezanilla F. Neuron. 1996;16:1159–1167. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- 40.Cha A, Bezanilla F. Neuron. 1997;19:1127–1140. doi: 10.1016/s0896-6273(00)80403-1. [DOI] [PubMed] [Google Scholar]
- 41.Papazian D M, Shao X M, Seoh S-A, Mock A F, Yu H, Wainstock D H. Neuron. 1995;14:1293–1301. doi: 10.1016/0896-6273(95)90276-7. [DOI] [PubMed] [Google Scholar]
- 42.Auld V J, Goldin A L, Frafte D S, Marshall J, Dunn J M, Catterall W A, Lester H A, Davidson N, Dunn R J. Neuron. 1988;1:449–461. doi: 10.1016/0896-6273(88)90176-6. [DOI] [PubMed] [Google Scholar]