

Abstract
The recent discovery that histone demethylation can be catalyzed by the flavin-dependent amine oxidase LSD1 has ushered in a new chapter in the chromatin remodeling community. Herein we discuss the rapid progress of the histone demethylase field including: the recent identification of the nonheme iron-dependent histone demethylases (JmjC family), the basis for LSD1 substrate site-specificity, and the newly emerging potential for inhibition of these enzymes in structural and functional analysis.
Introduction
One of the surprises in determining the sequence of the human genome was how relatively few genes (ca. 25,000) are encoded by our DNA [1]. As a result, biologists have come to appreciate more than ever that the informational diversity that leads to our complexity stems from post-transcriptional mechanisms including RNA splicing and protein post-translational modifications. The latter allows for a rich variety of covalent protein structural changes that includes phosphorylation, acetylation, ubiquitylation, sulfation, glycosylation, lipidation, and methylation [2]. These changes in protein chemical structure often result in profound influences on the mechanism and function of proteins in cells.
Core histone proteins are highly conserved and play a critical role in modulating chromatin structure and DNA accessibility for replication, repair, and transcription [3]. There has been intense interest in mapping and characterizing the histone post-translational modifications that guide chromatin remodeling and account for epigenetics. In this regard, the protein lysine modifications have a special place in the history of gene regulation. Allfrey and colleagues demonstrated in the 1960's that histone lysine acetylation was often found in chromatin that was in an activated state [4]. Histone lysine methylation was proposed to be reciprocal with acetylation in structure and function. We now know that the effects of lysine acetylation and methylation are more variegated than these early concepts and depend on specific modification sites as well as additional factors at specific genes. The progress of research in this field has been catapulted by a series of discoveries of key enzymes that catalyze histone lysine acetylation and deacetylation [5, 6] on the one hand and methylation and demethylation [7, 8••] on the other.
The first specific histone lysine methyltransferases were reported in 2000 and as expected were S-adenosyl methionine dependent enzymes [7]. There are at least two classes of histone lysine methyltransferases, the SET domain containing protein family [9] and the non-SET domain protein analogues of DOT1 [10-12]. Several of these methyltransferases are quite specific for their targeted lysine and preferentially add one, two or three methyl groups. For many years, it was uncertain if methylation marks on histones were “permanent”. Unlike the reversibility of most post-translational modifications, which involve hydrolytic reactions, it seemed less likely that lysine methylation would be cleaved in this manner because of the kinetic stability of the N-Me bond. In 1973 Paik and colleagues demonstrated the enzymatic demethylation of N-methylated calf thymus histones, although they were unable to ultimately identify the corresponding enzyme [13].
Lysine Demethylases
In late 2004, it was reported that a flavin dependent amine oxidase that appeared to be a polyamine oxidase homolog, LSD1, was capable of selective demethylation Lys-4 of histone H3 [8••]. This study laid to rest the debate as to whether or not lysine methylation was a static epigenetic mark and opened up a new chapter in the chromatin field. Since the initial description of LSD1, there has been a frenzied pace of molecular biology discovery in reversible protein methylation. At least ten other lysine demethylases, belonging to the Jumonji C or JmjC family of iron(II)-alpha-ketoglutarate dependent demethylases have been described in the literature, with varying specificities for lysine site and the degree of methylation of the substrate [14-24] (Table 1). To some extent, the biochemical analysis of these JmjC proteins has lagged behind their genetic roles. Of some concern, there have been few kinetic studies reported for the JmjC catalyzed reactions, and estimated values deduced from published data suggest that they may have turnover numbers in the range of 0.01 min−1 with histone substrates, about 300-fold lower than LSD1 [25]. These unusually slow reaction rates may simply reflect the need for yet to be identified accessory proteins to stimulate JmjC activities but leave open the possibility that alternative substrates exist.
Table 1.
Identified histone lysine demethylases.
| Demethylase | Alternate Name | Site Specificity | Demethylates | Refs |
|---|---|---|---|---|
| Flavin dependent | ||||
| LSD1 | BHC110, AOF2 | H3K4 | mono, di | [8••] |
| Fe(II) α-ketoglutarate dependent | ||||
| JHDM1A | FBXL11 | H3K36 | mono, di | [14] |
| JMJD1A | JHDM2A, TSGA | H3K9 | mono, di | [15] |
| JMJD2A | JHDM3A | H3K9, H3K36 | tri, di | [16,17] |
| JMJD2B | KIAA0876 | H3K9 | tri, di | [18] |
| JMJD2C | GASC1 | H3K9, H3K36 | tri, di | [19,20] |
| JMJD2D | KIAA0780 | H3K9 | tri, di, mono | [20] |
| JARID1A | RBP2 | H3K4 | tri, di | [21] |
| JARID1B | PLU-1 | H3K4 | tri, di | [22] |
| JARID1C | SMCX | H3K4 | tri, di | [23] |
| JARID1D | SMCY | H3K4 | tri, di | [24] |
In contrast, the biochemical studies on LSD1 have moved at a somewhat brisker pace and leave little doubt at this juncture that LSD1 is a bona fide histone demethylase. LSD1's architecture contains a classical amine oxidase catalytic domain, an N-terminal SWIRM domain, which has been proposed to be involved in nucleosomal targeting [26], and an N-terminal extension [8••]. The catalytic turnover of LSD1 proceeds as expected for an FAD-dependent amine oxidase. Concurrent amine oxidation and flavin reduction are followed by the re-oxidation of flavin by one equivalent of molecular oxygen, producing stoichiometric hydrogen peroxide along with formaldehyde as the demethylation byproduct (Figure 1a). Since both hydrogen peroxide and formaldehyde have mutagenic potential and LSD1 is producing them in the nucleus, the stakes for detoxification pathways and tight control of enzymatic activity must be considerable. It has been proposed that the byproduct formaldehyde can be formally recycled and ultimately become the donated methyl of S-adenosyl methionine dependent histone methyltransferases [27].
Figure 1. Catalytic mechanisms of demethylase enzymes.

(a) The FAD dependent demethylation of Lys-4 of histone H3 proceeds through the hydrolysis of an iminium ion following a two electron oxidation of the amine by the flavin. R = ribosyl adenine dinucleotide (b) The iron(II) dependent demethylation of trimethyl-lysine substrates proceeds through an iron(II), α-ketoglutarate, and O2 derived hydroxyl radical oxidation of the methyl C-H bond.
LSD1 Reaction Mechanism
LSD1 is a member of the monoamine oxidase family based on sequence and recent X-ray crystal structures [8••, 28] and there has been a great deal of research into the detailed catalytic mechanisms of these enzymes. The FAD cofactor is oxidized by molecular oxygen, presumably by a single electron mechanism. Studies with LSD1 suggest that this flavin oxidation is rapid [29], based on changes in the flavin optical spectrum which show classic FAD absorbance maxima at approximately 460 and 380 nm in the resting state, assigned to fully oxidized flavin and a one electron reduced semiquinone form [30]. The precise mechanism of the oxidative conversion of the amine to an imine is controversial and may involve either hydride transfer or single electron mechanisms [31, 32]. Elegant recent studies on the flavin-dependent oxidase tryptophan 2-monooxygenase may shed light on the general reaction class. By measuring deuterium and 15N isotope effects on the amine to imine reaction, the authors found a large deuterium effect (6.0±0.5) and an inverse 15N isotope effect on CH bond cleavage (0.9917±0.0006) [31]. This was interpreted in favor of a hydride transfer mechanism rather than a radical mechanism for amine oxidation since such a large deuterium effect is consistent with irreversible CH bond cleavage. Whether this will hold for all other related amine oxidases will require further investigation. Following imine formation, hydration to the N,O-hemiacetal and subsequent collapse to formaldehyde and amine are presumed to be relatively rapid and spontaneous reactions. It is clear that the trimethylammonium functionality could not be a substrate for the flavin amine oxidases because of the absence of a nitrogen lone pair, which is integral to the reaction. In contrast, the iron(II) alpha-ketoglutarate oxygenases can hydroxylate alkyl groups directly through a hydroxyl radical, providing a chemically plausible path to demethylation of quaternary ammonium salts (Figure 1b).
LSD1 Substrate Specificity
LSD1 is most closely related to the polyamine oxidase superfamily and LSD1 was originally tested with various small molecule polyamines as potential substrates, but these compounds showed minimal or undetectable turnover under the reaction conditions [8••]. Various methyl-lysine peptides derived from histones were then investigated as possible substrates, including mono- di- and tri-methylated Lys-4 H3 tail peptides, full-length Lys-4-methylated histone H3, and dimethyl-Lys-9, 36, and 79 of H3 and Lys-20 of H4 containing peptides [8••]. A high level of substrate specificity for Lys-4 of H3 was suggested since no other methyl-lysine sites were processed during this in vitro analysis [8••]. Forneris et al. demonstrated that histone H3 tail peptides greater than 16 amino acids in length are necessary to achieve high demethylase efficiency, furthermore LSD1 does not have a strong kinetic preference for the mono- or di-methyl forms of Lys-4 based on histone H3 synthetic tail peptides [33•]. Epigenetic post-translational modifications of Lys-9 (methylation, acetylation), Ser-10 (phosphorylation), Lys-14 (acetylation), Lys-18 (acetylation), Arg-2 (methylation), Arg-8 (methylation), and Arg-17 (methylation) of histone H3 and their influence on LSD1 Lys-4 demethylation of an H3 tail peptide have been explored [29,33•]. Interestingly, nearly all of these modifications led to a drop in enzymatic demethylation efficiency by LSD1, revealing the high specificity for the native histone tail sequence and the potential importance for electrostatic interactions between histone and LSD1. Consistent with the important role for electrostatic interactions, demethylation rate is very sensitive to ionic strength [33•]. This selectivity correlates nicely with the known function of LSD1 as part of a general transcriptional repressor complex along with CoREST and HDAC1/2 [34-35]. Since methylation of Lys-4 is a mark of gene activation, methyl removal at this site is gene silencing. Interestingly, there are reports that the LSD1 substrate specificity can be modulated in vivo by protein-protein interactions [36,37], but this has yet to be confirmed in vitro.
Structural Studies on LSD1 in Complex with a Histone Analog Suicide Inhibitor
Insertion of various functional groups including cyclopropyls and propargyls into monoamine oxidase substrates convert these compounds into mechanism-based inactivators [38,39]. Substitutions at the epsilon position of Lys-4 of histone H3 peptides including propargylamine, aziridine, and cyclopropylamine have been synthesized and the propargylamine containing peptide behaves as a classical mechanism-based inactivator [40•]. Based on a combination of mass spectrometry and optical and NMR spectroscopy, an inactivation mechanism in which the propargylamine is initially oxidized to the conjugated imine which undergoes Michael addition with the flavin cofactor has been proposed [24, 40•] (Figure 2a). The resultant N5 flavin adduct generated leads to a stably bound enzyme-inhibitor complex. Taking advantage of this behavior, a recent 2.7 Å crystal structure was obtained of LSD1 after treatment with a N-methylpropargylamine analog induced inactivation [41•]. In order to see electron density for the peptide moiety, it was necessary to treat the inactivated complex with sodium borohydride prior to crystallization, which presumably led to reduction of the linker double bonds between the flavin and peptide inactivator (Figure 2b). The importance of borohydride treatment was rationalized as increasing the resistance of the adduct to hydrolysis as well as eliminating the potential heterogeneity of the double bond stereoisomers associated with the parent adduct.
Figure 2. Mechanism-based inactivation of LSD1.

(a) The proposed mechanism of inactivation of LSD1 by an N-methylpropargylamine containing H3 peptide proceeds through conjugate addition of the flavin N5 to the gamma carbon of the electrophile following a two electron oxidation to the iminium ion. (b) Reduction of the trimethine linkage of the FADinactivator conjugate with NaBH4 was necessary for crystallographic studies. (c) The proposed catalytic mechanism of inactivation of LSD1 by tranylcypromine proceeds through radical recombination and subsequent dehydration following a one electron oxidation and ring opening on the amine. R = ribosyl adenine dinucleotide in a-c
The structure of the LSD1-inactivator complex, obtained in the presence of CoREST, was noteworthy in several respects. First, only residues 1-7 of the histone tail peptide gave sufficiently strong electron density to be modeled (Figure 3a). Weak density for H3 residues 8-21 may have been detected extending toward the SWIRM domain but it was insufficient to accurately model. The importance of residues beyond 7 for demethylase action is established [33•] so it is likely that the LSD1-inactivator structure is incomplete in detailing biologically important recognition. Nevertheless, the H3 residues visualized formed a very unusual series of three gamma turns in the secondary structure [41•] (Figure 3b). Gamma turn structure involves backbone hydrogen bond interactions between the i and i+2 residues in a protein chain providing a pseudo 7-membered ring [42] (Figure 3c). Based on related structural studies, most enzymes involved in post-translational modifications recognize extended conformations of peptide substrates proximal to the targeted residue. To our knowledge, this interaction between LSD1 and the tail of H3 is unique among characterized enzyme-peptide substrate complexes.
Figure 3. LSD1 substrate specificity revealed by an LSD1-inactivator crystal structure.
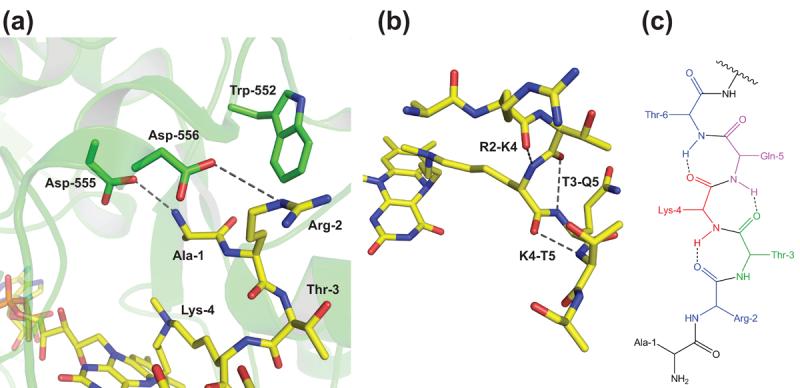
(a) Active site view of the key interactions between Asp-556 and Ala-1, the N-terminus of the H3 peptide, and both Trp-552 and Asp-556 with Arg-2 of the H3 peptide. (b) Active site view of the configuration of the three consecutive gamma turns that the H3 peptide backbone adopts between Arg-2 and Lys-4, Thr-3 and Gln-5, and Lys-4 and Thr-6. (c) Rendering of a H3 peptide containing three consecutive gamma turns to illustrate the pseudo 7-membered rings inferred by the i, i+2 backbone hydrogen bonding.
Given this unusual conformation observed in the LSD1 complex, it is appropriate to question its biological relevance, especially since it was obtained with a suicide inactivator. However, the crystal structure reveals a series of hydrogen bond and van der Waals interactions between the side chains of H3 residues and those of LSD1, effectively predicting the behavior of enzyme mutants and alternative substrates [41•]. For example, the LSD1 structure shows that the free N-terminal amino group of H3 makes a key electrostatic interaction with Asp-555 of LSD1. Replacement of the amino group by a methyl group, charge masking by an acetyl group, or mutagenesis of Asp-555 all result in a major loss in substrate processing. Analogous experiments confirm the key interactions between Arg-2 of histone H3 and LSD1 residues Asp-556 and Trp-552. Overall, the LSD1 inactivated structure reveals an elegant basis for the highly specific demethylation for Lys-4 of histone H3. The ability of amino terminal acetylation to reduce processing efficiency by LSD1 may have biological significance for interpretation of the histone code since histone N-terminal acetylation is likely to occur reversibly in vivo.
In Vivo and Nucleosomal Targeting
Relatively little is known about the structural basis of demethylation of chromatin by LSD1 in vivo. However, it appears that CoREST plays an important role in targeting histone H3 demethylation in the context of nucleosomes [34,35]. Knockdown of CoREST greatly reduces the efficiency of LSD1-mediated histone demethylation in vivo and biochemical studies with nucleosome substrates have confirmed this behavior. Based on NMR and mutagenesis experiments it has been proposed that CoREST may target LSD1 to DNA [28].
In separate experiments, it has been proposed that LSD1 can demethylate Lys-9 of histone H3 in different physiologic contexts [36, 37]. In this model, androgen receptor interactions with LSD1 are thought to dictate this substrate specificity change. If correct, these data suggest that LSD1 can have a gene activation function as well as silencing effect, depending on promoter context. These data have not yet been supported by in vitro biochemical studies, nor does the LSD1-H3 crystal structure provide a clue as to how this could be accomplished [41•]. However, recent reports that the S. pombe homologues of LSD1, splsd1 and spsld2, may target Lys-9 for demethylation adds plausibility to the altered substrate specificity in mammalian systems [43].
LSD1 Small Molecule Inhibitors
LSD1 as a therapeutic target for small molecule inhibitors is plausible in analogy to the now clinically validated epigenetic HDAC enzyme class. Along these lines, the cyclopropylamine containing monoamine oxidase inhibitor tranylcypromine was proposed to inhibit LSD1 [44•] and further characterized as a mechanism-based inactivator of LSD1 [45, 46]. It is noteworthy that a cyclopropylamine substrate analog of histone H3 failed to induce inactivation of LSD1, suggesting tranylcypromine adopts a distinctive set of enzyme interactions [25]. The recent kinetic and structural studies have investigated the mechanism of inactivation by tranylcypromine; the authors of both studies propose similar mechanisms to that of Figure 2c. While less potent against LSD1 than against monoamine oxidase B, tranylcypromine thus appears to represent a promising lead agent for further development. Recently, a series of bisguanidine and biguanide polyamine analogs were tested for inhibitory activity against LSD1. The in vitro analysis characterized these compounds as being noncompetitive in nature [47]. Interestingly, these compounds proved successful in reactivating the expression of silenced genes associated with tumor suppression, hinting at the therapeutic potential of LSD1 inhibition in the treatment of cancer [47].
Conclusion and Future Challenges
It is difficult to overstate the briskness of research in the histone demethylation field set-off by the initial report of LSD1 less than three years ago [8••]. The flavin- and iron-dependent demethylases appear to play key roles in gene regulation and cellular growth and differentiation. Still, there are many remaining challenges. How precisely do these demethylases catalyze reactions and recognize their substrates and how does the in vivo context modify and regulate these interactions? Recently, several crystal structures of the JmjC containing protein JMJD2A bound to substrate analogs have been reported and these should help clarify the basis for specificity and catalytic action [48-50]. Also, a second structure of the LSD1-CoREST complex with a substrate mimic has been shown, introducing the possibility of multiple substrate binding conformations [51]. An increased effort on structural and enzymatic studies which take into account partner proteins and nucleosomal assemblies will be needed. Will there be therapeutic potential in the recognition of small molecule gamma turn mimics or will other small molecule scaffolds offer greater promise? Here the synthetic chemist will have an important role to play. Are there other flavin-dependent protein demethylases lurking in the mammalian genome and are there bona fide protein substrates outside of histones? Chemical proteomic approaches involving mechanism-based inactivators may play a useful role in this regard [25]. How do the redox chemistries of the flavin- and iron-dependent demethylases affect the cell nucleus and what protective measures are most critical in preventing DNA damage? There has been a growing interest in the role that environmental-toxin induced oxidative damage can play in disease progression, and it would seem that attention to endogenous sources of chemically reactive species should also be considered. Are there bona fide CpG island DNA demethylases? The chemical challenge of this class of reaction is even more difficult than for protein demethylation, but it remains possible that such DNA-targeted enzymes have yet to be uncovered. We look forward to the years ahead and the likelihood of many more exciting discoveries and developments in the oxidative demethylation field which will rely heavily on the involvement and ingenuity of chemical biologists.
Acknowledgements
We are grateful to our colleagues and collaborators for helpful advice and discussions including Yang Shi, Hongtao Yu, Maojun Yang, Xin Liu, Ronen Marmorstein, Larry Szewczuk, Rong Huang, and Marc Holbert. We thank the NIH for financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Paabo S. The human genome and our view of ourselves. Science. 2001;291:1219–1220. doi: 10.1126/science.1056972. [DOI] [PubMed] [Google Scholar]
- 2.Walsh CT. Posttranslational modification of proteins: expanding nature's inventory. B. Roberts; Colorado: 2005. [Google Scholar]
- 3.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 4.Allfrey VG, Faulkner R, Mirsky AG. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA. 1964;51:786–94. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, Allis CD. Tetrehymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- 6.Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast regulator Rpd3p. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 7.Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 8. ••.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. (This study identifies for the first time an enzyme responsible for enzymatic protein demethylation.) [DOI] [PubMed] [Google Scholar]
- 9.Jenuwein T. Re-SET-ting heterochromatin by histone methyltransferases. Trends Cell Biol. 2001;11:266–273. doi: 10.1016/s0962-8924(01)02001-3. [DOI] [PubMed] [Google Scholar]
- 10.van Leeuwen F, Gafken PR, Gottschling DE. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002;109:745–756. doi: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- 11.Ng HH, Feng Q, Wang H, Erdjument-Bromage H, Tempst P, Zhang Y, Struhl K. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 2002;16:1518–1527. doi: 10.1101/gad.1001502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lacoste N, Utley RT, Hunter J, Poirier GG, Cote J. Disruptor of telomeric silencing 1 is a chromatin-specific histone H3 methyltransferase. J Biol Chem. 2002;277:30421–30424. doi: 10.1074/jbc.C200366200. [DOI] [PubMed] [Google Scholar]
- 13.Paik WK, Kim S. Enzymatic demethylation of calf thymus histones. Biochem Biophys Res Commun. 1973;51:781–788. doi: 10.1016/0006-291x(73)91383-1. [DOI] [PubMed] [Google Scholar]
- 14.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 15.Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell. 2006;125:483–495. doi: 10.1016/j.cell.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 16.Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006;442:312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- 17.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 18.Fodor BD, Kubicek S, Yonezawa M, O'Sullivan RJ, Sengupta R, Perez-Burgos L, Opravil S, Mechtler K, Schotta G, Jenuwein T. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006;20:1557–1562. doi: 10.1101/gad.388206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cloos PAC, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 20.Shin S, Janknecht R. Diversity within the JMJD2 histone demethylase family. Biochem Biophys Res Comm. 2007;353:973–977. doi: 10.1016/j.bbrc.2006.12.147. [DOI] [PubMed] [Google Scholar]
- 21.Klose RJ, Yan Q, Tothoya Z, Yamane K, Erdjument-Bromage H, Tempst P, Gilliland DG, Zhang Y, Kaelin WG., Jr The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell. 2007;128:889–900. doi: 10.1016/j.cell.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 22.Seward DJ, Cubberley G, Kim S, Schonewald M, Zhang L, Tripet B, Bentley DL. Demethylation of trimethylated histone Lys4 in vivo by JARID1 JmjC proteins. Nat Struct Mol Biol. 2007;14:240–242. doi: 10.1038/nsmb1200. [DOI] [PubMed] [Google Scholar]
- 23.Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Oi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077–1088. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 24.Lee MG, Norman J, Shilatifard A, Shiekhattar R. Physical and functional association of a trimethyl H3K4 demethylase and Ring6a/MBLR, a polycomb-like protein. Cell. 2007;128:877–887. doi: 10.1016/j.cell.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 25.Szewczuk LM, Culhane JC, Yang M, Majumdar A, Yu H, Cole PA. Mechanistic analysis of a suicide inactivator of histone demethylase LSD1. Biochemistry. 2007;46:6892–6902. doi: 10.1021/bi700414b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Da G, Lenkart J, Zhao K, Shiekhattar R, Cairns BR, Marmorstein R. Structure and function of the SWIRM domain, a conserved protein module found in chromatin regulatory complexes. Proc Natl Acad Sci USA. 2006;103:2057–2062. doi: 10.1073/pnas.0510949103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tyihak E, Trezl L, Szende B. Formaldehyde cycle and the phases of stress syndrome. Ann N Y Acad Sci. 1998;851:259–270. [Google Scholar]
- 28.Yang M, Gocke C, Luo X, Borek D, Tomchick D, Machius M, Otwinowski Z, Yu H. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol Cell. 2006;23:377–387. doi: 10.1016/j.molcel.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 29.Forneris F, Binda C, Dall'Aglio A, Fraaije MW, Battaglioli E, Mattevi A. A highly specific mechanism of histone H3-K4 recognition by histone demethylase LSD1. J Biol Chem. 2006;281:35289–35295. doi: 10.1074/jbc.M607411200. [DOI] [PubMed] [Google Scholar]
- 30.Woo JCG, Silverman RB. Observation of two different chromophores in the resting state of monoamine oxidase B by fluorescence spectroscopy. Biochem Biophys Res Comm. 1994;202:1574–1578. doi: 10.1006/bbrc.1994.2111. [DOI] [PubMed] [Google Scholar]
- 31.Ralph EC, Anderson MA, Cleland WW, Fitzpatrick PF. Mechanistic studies of the flavoenzyme tryptophan 2-monooxygenase: deuterium and 15N kinetic isotope effects on alanine oxidation by an L-amino acid oxidase. Biochemistry. 2006;45:15844–15852. doi: 10.1021/bi061894o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silverman RB, Hoffman SJ, Catus WB., III A mechanism for mitochondrial monoamine oxidase catalyzed amine oxidation. J Am Chem Soc. 1980;102:7126–7128. [Google Scholar]
- 33. •.Forneris F, Binda C, Vanoni MA, Battaglioli E, Mattevi A. Human histone demethylase LSD1 reads the histone code. J Biol Chem. 2005;280:41360–41365. doi: 10.1074/jbc.M509549200. (This study describes the first detailed enzymatic study of a protein demethylase.) [DOI] [PubMed] [Google Scholar]
- 34.Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437:432–435. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- 35.Shi Y, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell. 2005;19:857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 36.Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 37.Wissmann M, Yin N, Muller JM, Greschik H, Fodor BD, Jenuwein T, Vogler C, Schneider R, Gunther T, Buettner R, Metzger E, Schule R. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol. 2007;9:347–353. doi: 10.1038/ncb1546. [DOI] [PubMed] [Google Scholar]
- 38.Walsh CT. Suicide substrates, mechanism-based enzyme inactivators: recent developments. Annu Rev Biochem. 1984;53:493–535. doi: 10.1146/annurev.bi.53.070184.002425. [DOI] [PubMed] [Google Scholar]
- 39.Silverman RB. Mechanism-based enzyme inactivators. Methods Enzymol. 1995;249:240–283. doi: 10.1016/0076-6879(95)49038-8. [DOI] [PubMed] [Google Scholar]
- 40. •.Culhane JC, Szewczuk LM, Liu X, Da G, Marmorstein R, Cole PA. A mechanism-based inactivator for histone demethylase LSD1. J Am Chem Soc. 2006;128:4536–4537. doi: 10.1021/ja0602748. (This paper is the first report of high potency inhibitors of LSD1.) [DOI] [PubMed] [Google Scholar]
- 41. •.Yang M, Culhane JC, Szewczuk LM, Gocke CB, Brautigam CA, Tomchick DR, Machius M, Cole PA, Yu H. Structural basis for histone demethylation by LSD1 revealed by suicide inactivation. Nat Struct Mol Biol. 2007;14:535–539. doi: 10.1038/nsmb1255. (This paper reveals the first structural analysis of a histone demethylase in complex with its substrate with the surprising observation of gamma turns.) [DOI] [PubMed] [Google Scholar]
- 42.Rose GD, Gierasch LM, Smith JA. Turns in peptides and proteins. Adv Protein Chem. 1985;37:1–109. doi: 10.1016/s0065-3233(08)60063-7. [DOI] [PubMed] [Google Scholar]
- 43.Lan F, Zaratiegui M, Villen J, Vaughn MW, Verdel A, Huarte M, Shi Y, Gygi SP, Moazed D, Martienssen RA, Shi Y. S. pombe LSD1 homologs regulate heterochromatin propagation and euchromatic gene transcription. Mol Cell. 2007;26:89–101. doi: 10.1016/j.molcel.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 44. •.Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhatter R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol. 2006;13:563–567. doi: 10.1016/j.chembiol.2006.05.004. (This paper is the first report that a clinically used monoamine oxidase inhibitor, despite its dissimilarity to a histone, can inhibit a histone demethylase.) [DOI] [PubMed] [Google Scholar]
- 45.Schmidt DM, McCafferty DG. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry. 2007;46:4408–4416. doi: 10.1021/bi0618621. [DOI] [PubMed] [Google Scholar]
- 46.Yang M, Culhane JC, Szewczuk LM, Jalili P, Ball HL, Machius M, Cole PA, Yu H. Structural basis for inhibition of the LSD1 histone demethylase by the antidepressant trans-2-Phenylcyclopropylamine. Biochemistry. 2007;46:8058–8065. doi: 10.1021/bi700664y. [DOI] [PubMed] [Google Scholar]
- 47.Huang Y, Greene E, Stewart TM, Goodwin AC, Baylin PM, Casero RA., Jr Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci USA. 2007;104:8023–8028. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ng SS, Kavanagh KL, McDonough MA, Butler D, Pilka ES, Lienard BMR, Bray JE, Savitsky P, Gileadi O, von Delft F, Rose NR, Offer J, Scheinost JC, Borowski T, Sundstrom M, Schofield CJ, Oppermann U. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature. 2007;448:87–91. doi: 10.1038/nature05971. [DOI] [PubMed] [Google Scholar]
- 49.Couture JF, Collazo E, Ortiz-Tello PA, Brunzelle JS, Trievel RC. Specificity and mechanism of JMJD2A, a trimethyllysine-specific histone demethylase. Nat Struct Mol Biol. 2007 doi: 10.1038/nsmb1273. In Press. [DOI] [PubMed] [Google Scholar]
- 50.Chen Z, Zang J, Kappler J, Hong X, Crawford F, Wang Q, Lan F, Jiang C, Whetstine J, Dai S, Hansen K, Shi Y, Zhang G. Structural basis of the recognition of a methylated histone tail by JMJD2A. Proc Natl Acad Sci USA. 2007;104:10818–10823. doi: 10.1073/pnas.0704525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Forneris F, Binda C, Adamo A, Battaglioli E, Mattevi A. Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J Biol Chem. 2007;282:20070–20074. doi: 10.1074/jbc.C700100200. [DOI] [PubMed] [Google Scholar]