

Abstract
Our structure-based design strategies which specifically target the HIV-1 protease backbone, resulted in a number of exceedingly potent nonpeptidyl inhibitors. One of these inhibitors, darunavir (TMC114), contains a privileged, structure-based designed high-affinity P2 ligand, 3(R),3a(S),6a(R)-bis-tetrahydrofuranylurethane (bis-THF). Darunavir has recently been approved for the treatment of HIV/AIDS patients harboring multidrug-resistant HIV-1 variants that do not respond to previously existing HAART regimens.
Keywords: protease inhibitors, darunavir, design and synthesis
The AIDS (acquired immunodeficiency syndrome) epidemic has become one of the most pressing medical concerns of our time.1 The World Health Organization (WHO), as of 2006, estimated that over 40 million people are infected with HIV (human immunodeficiency virus), the causative agent of AIDS.2 During replication in the HIV life-cycle, gag and gag-pol gene products are produced as precursor polyproteins which are subsequently processed by a virally encoded protease to provide structural proteins (p17, p24, p9 and p7) and essential viral enzymes, including protease (PR), reverse transcriptase (RT) and integrase (IN).3 All three retroviral enzymes have been identified as potential drug targets. Specifically, the critical function of HIV protease has made it an important target for the treatment of HIV/AIDS. The approval of the first protease inhibitor (PI), saquinavir and its introduction into highly active antiretroviral therapy (HAART), with reverse transcriptase inhibitors, led to significantly enhanced HIV management and improved the quality of life of HIV/AIDS patients.4
Since the advent of the first PI, saquinavir, a number of PIs have been introduced in the regimens of highly active antiretroviral therapy or HAART. Thus improved HAART regimens have shown reduced viral load, increased CD4+ T-cell counts5 and drastically lowered AIDS-related deaths in the US and industrialized nations.6 While HAART proved to be a large step forward, there are still serious drawbacks with the first generation anti-protease therapeutics. These include: (1) severe side effects and drug toxicities, (2) higher therapeutic doses due to “peptide-like” character, (3) costly synthesis which leads to high treatment cost, and perhaps the most alarming, (4) the rapid emergence of drug resistance. Indeed, the emergence of multidrug-resistant HIV strains has greatly compromised current HAART regimens. It has been reported that treatment failure has ultimately occurred in at least 40-50% of patients, who initially achieved favorable viral suppression with HAART to undetectable levels.7 Furthermore, persistent viral replication (plasma HIV RNA >500 copies/mL) has been reported under HAART in 10-40% of anti-viral therapy-naive individuals as a result of transmission of drug-resistant HIV-1 variants.8 The management and effective treatment options for HIV/AIDS clearly depend upon the development of PIs and other novel anti-HIV therapeutics, which can effectively combat drug-resistant HIV strains, possess better pharmacokinetic properties, have no or less toxicities, and come at reduced costs of synthesis.
Design of nonpeptide ligands to eliminate peptidic character
Our initial investigation primarily focused on reducing peptidic features, molecular weight, and structural complexity of protease inhibitors. In this context, we have designed a number of nonpeptidic high-affinity ligands for the HIV protease substrate binding site based upon various available three-dimensional structures of the protein-ligand complex.9 Particularly, we planned to design conformationally constrained nonpeptidic molecules of a cyclic or heterocyclic nature to maximize the active site interactions. One of the important elements of our design strategy is the incorporation of a stereochemically defined and conformationally constrained cyclic ether template that could replace peptide bonds and mimic their biological mode of action by retaining critical interactions in the active site.9,10 The idea of designing cyclic ether-based ligands emerged from our observation that numerous bioactive natural products are comprised of these cyclic ether motifs. Of particular interest, ionophore antibiotic, monensin (1, Figure 1)11 and platelet activating factor antagonist, ginkgolide B (2),12 which feature these cyclic ether subunits, do not suffer from oral bioavailability problems inherent to peptide and peptidomimetic-based inhibitor drugs.
Figure 1.
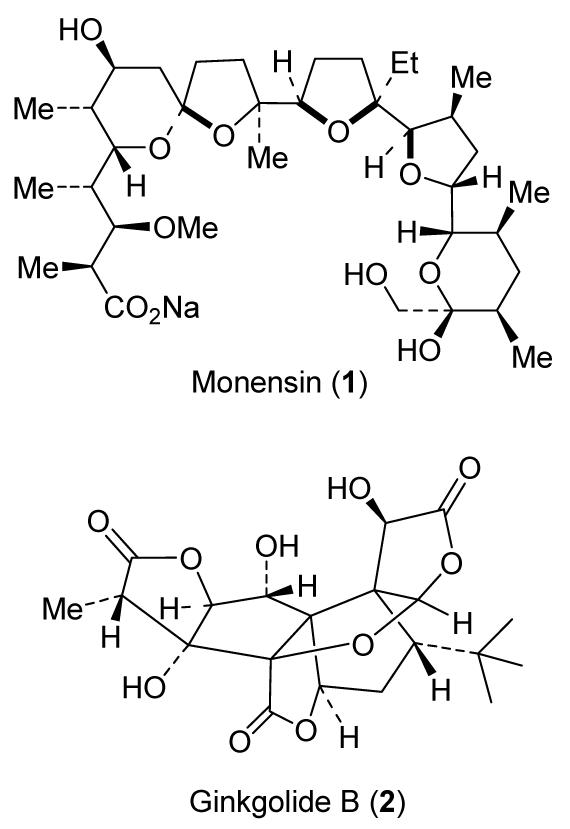
Structures of monensin and ginkgolide B
Indeed, our structure-based design strategy led to the development of a number of cyclic ether-derived nonpeptide P2-ligands for the HIV protease substrate binding site. We have documented an intriguing potency enhancing effect of 3(S)-tetrahydrofuranyl urethane in inhibitors containing a hydroxyethylene isostere or a hydroxyethylsulfonamide isostere.13 Incorporation of 3(S)-tetrahydrofuran into a (R)-(hydroxyethyl)sulfonamide isostere afforded a highly potent inhibitor which later became amprenavir.14 It is noteworthy to mention that the tetrahydrofuranyl subunit is inherent in both monensin and various ginkgolides (Figure 1).
Design of bis-THF: an inspiration from bioactive natural products
Analysis of a number of protein-ligand X-ray structures of 3(S)-tetrahydrofuranyl urethane-bearing inhibitors revealed weak hydrogen bonding between the tetrahydrofuranyl oxygen and the main chain aspartic acids (Asp-29 and Asp-30) as well as van der Waals interactions in the S2-site. Based upon this structure, our subsequent objective became to design a ligand that would maximize the hydrophobic and hydrogen bonding interactions with the residues in the S2-site. Our critical analysis of the saquinavir (3)-bound protease X-ray crystal structure led us to design and develop a stereochemically defined bicyclic tetrahydrofuran (bis-THF) ligand that appeared to effectively hydrogen bond with both Asp 29 and Asp 30 NHs. The bicyclic ring of bis-THF is also poised to offset the loss of P3 hydrophobic binding of the quinoline ring in saquinavir. Inhibitor 4 (enzyme IC50 of 1.8 nM and antiviral CIC95 of 46 nM, Figure 2) has shown improved aqueous solubility, reduced peptidic features and molecular weight compared to saquinavir.9 Subsequent detailed studies established that the stereochemistry, the position of the oxygen atoms, ring sizes and substituents are all essential for potency.9 The X-ray crystal structure of 4-bound protease revealed that the bis-THF ring oxygens are involved in effective hydrogen bonding interactions with both the backbone NH’s of Asp 29 and Asp 30 present in the S2 subsite. In essence, the bis-THF ligand, a subunit of ginkgolides (bicyclic acetal) remarkably mimics the binding of the P2 asparagine carboxamide and the P3 quinaldic amide carbonyls of saquinavir.
Figure 2.
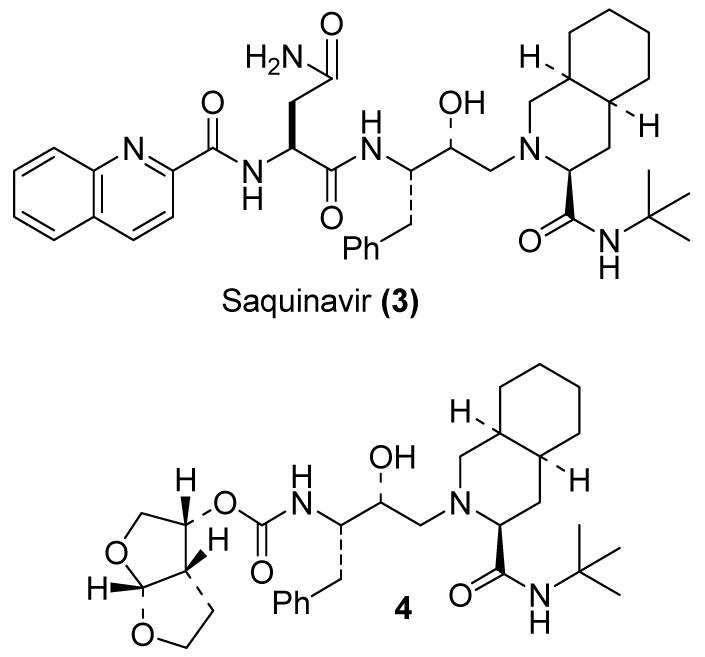
Structures of saquinavir and a bis-THF inhibitor 4
Design and development of ‘darunavir’ to combat drug resistance
Our analysis of protein-ligand complexes of wild-type and mutant proteases and an overlay of the corresponding protein backbones showed only minimal distortion of the backbone conformation, particularly in the active site of the protease.15 This is also apparent in numerous other reported high resolution X-ray structures of related inhibitor/ligand complexes.16 This observation led us to speculate that an inhibitor making extensive hydrogen bonding interactions with the protein backbone of the wild-type enzyme will also maintain potency against mutant strains. Our inhibitor design strategy to combat drug-resistance then focused on optimization of the ligand-binding site interactions so as to make maximum interactions in the active site, including hydrophobic, electrostatic and most critically hydrogen bonding with the backbone atoms located in the S2 to S2′-subsites of protease. As mentioned previously, an inhibitor 4-bound X-ray structure of HIV protease revealed that while the P2 bis-THF ligand makes extensive interactions including backbone hydrogen bondings in the S2-subsite, similar hydrogen bondings in the S2′-site are mostly absent.
With the objective of designing an inhibitor that can make robust hydrogen bonding throughout the S2-S2′-subsites, we investigated the effect of a P2 bis-THF ligand with a number of different isosteres.9 Incorporation of the P2 bis-THF in (R (hydroxyethyl) sulfonamide isosteres led to a number of potent PIs, with impressive drug resistance profiles. For instance, inhibitor 5 (UIC-PI or UIC94003 and later TMC-126), incorporating a P2 bis-THF and a p-methoxysulfonamide as the P2′ ligand exhibited remarkably potent enzyme inhibitory potency (Ki value 14 pM) and antiviral activity (ID50 value of 1.2 nM) in CEM cell lines.17 It was also profiled against numerous mutant HIV proteases. The Ki was less than 100 pM in every case and the Kimut/Kiwt was no greater than five. This indicates a low level of resistance even for enzymes with multiple mutations which have been shown to be resistant to clinically active inhibitors. TMC-126 has shown a remarkable drug resistance profile and has maintained high potency in the presence of human serum albumin.17
Inhibitor 6 (UIC 94017, later known as TMC-114) with a bis-THF as the P2 ligand and p-aminosulfonamide as the P2′ ligand has also exhibited very impressive inhibitory properties. It displayed an enzyme inhibitory potency (Ki) of 16 pM and an antiviral ID50 of 4.7 nM in CEM cell lines. It showed an antiviral ID90 of 10 nM, and a TD50>100μM in a cell culture assay.18 Inhibitor-bound X-ray structure analysis revealed that both P2 and P2′-ligands of inhibitor 6 are involved in extensive hydrogen bonding with the protein backbone. This may be responsible for its potency and wide-spectrum activity against multi-PI-resistant HIV-1 variants. It was tested against a panel of 20 HIV variants resistant to current PI’s, but there was no greater than a 5-fold increase in ID50 values, indicating it remained active against the resistant strains. In addition, the P2′-amine group provided more favorable pharmacokinetic properties compared to the P2′-methoxy group in inhibitor 5. Subsequently, it was selected for clinical development and renamed darunavir.19
Clinical development of darunavir was conducted by Tibotec-Virco, Belgium.18b POWER 1 and POWER 2 clinical trials of ritonavir-boosted darunavir (DRV/r) were carried out with treatment-experienced patients who were no longer benefiting from available PIs. Over a period of six months, both studies showed that combination therapy using DRV/r led to a reduction in viral load below the 50 copies/ mL in 45% of participants compared with only 12% of participants given another available PI. CD4+ cell counts in the DRV/r group rose by an average of 92 cells/mm3 over the six months period compared with an average increase of 17 CD4+ cells/mm3 for participants receiving another PI during this time.20 POWER 3, a non-randomized, open-label trial was conducted to assess the long-term efficacy and safety of DRV/r 600/100mg BID in treatment-experienced patients. The primary efficacy endpoint was the proportion of patients with ≥ 1 log10 reduction in HIV RNA by week 24. Reduction of HIV RNA with an efficacy endpoint of ≥ 1 log10 was observed in 65% of patients. Reductions in HIV RNA levels to <400 copies/mL and <50 copies/mL were observed in 57% and 40% of patients, respectively. DRV received accelerated approval by the FDA on June 23, 2006.20 Recent studies have shown that darunavir, when used in combination with the fusion inhibitor FUZEON, can substantially increase the chances of reaching undetectable viral load.21
Recent PIs based on the bis-THF ligand
The success of Darunavir and the evidence indicating the importance of the bis-THF P2 ligand has led to an expansion of the “backbone binding concept”, and produced several novel and active PIs. Ritonavir has recently been modified with the addition of the P2-bis-THF ligand, and initial SAR results revealed a new potent inhibitor.22 Introduction of a fused benzodioxolane and other related functionalities as P2′ sulfonamides have shown significant potency enhancement and drug-resistance properties.23 GlaxoSmithKline researchers have investigated various structural modifications at the P1 and P1′ positions of inhibitors containing a bis-THF group as the P2-ligand and a benzodixoane sulfonamide as the P2’-ligand.24 One of the inhibitors has shown IC50 values in the single digit nanamolar range as well as Ki’s in the femtomolar range.25 This inhibitor was later renamed brecanavir and had undergone extensive clinical studies. However brecanavir development was terminated later reportedly due to its difficulty to formulate. Tibotec researchers have discovered a new series of fused benzoxazole and benzothiazole ligands to fit the S2′ domain. These new inhibitors have shown broad-spectrum antiviral activity against PI resistant mutants, as well as excellent pharmacokinetic properties.26
Beyond the bis-THF ligand and darunavir: Recent developments in protease inhibitors
Recently, Procyon pharmaceuticals has reported a new class of protease inhibitors based on L-lysine. Uniquely, two novel sulfonamide based PIs (PL-100, 7 and PPL-100, 8, Figure 4) have shown good protease inhibitory activity.27 They have shown enzyme inhibitory activity less than 20 nM, but their preliminary cross-resistance results are very impressive. Against 14 viral stains from highly PI-experienced patients, PL-100 showed a 4.5 average fold-increase in IC50 values.28 The phosphate ester prodrug, PPL-100 (8), showed a superior pharmacokinetic profile.29 AG-001859, 9 is another recently identified compound which exhibited potency against resistant strains of HIV.30 This new compound is an allophenylnorstatin-based PI, and has shown Ki’s for wild-type and mutant proteases as low as 0.1 nM. When AG-001859 was tested against 44 PI-resistant HIV-1 isolates, it displayed excellent potency with a median EC50 of 34 nM (range 5.3-420 nM).30 AG-001859 was selected for further testing and has started a phase 1 clinical trial.
Figure 4.
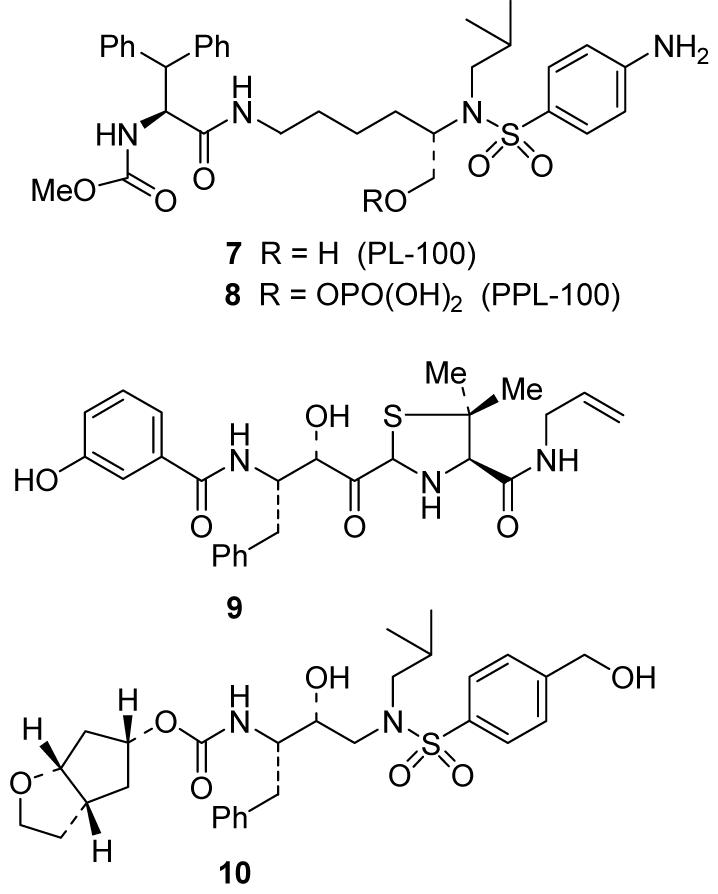
Structures of recent protease inhibitors
We recently developed a new P2 ligand, based upon the “backbone binding” design concepts. Inhibitor 10 contains a stereochemically defined bicyclic hexahydrocyclopentanofuran as a P2 ligand and a hydroxymethylphenylsulfonamide group as the P2’ ligand (Figure 4) It has shown potent antiviral activity (Ki = 4.5 pM, IC50 = 1.8 nM) and effective drug-resistance properties against a panel of multi-PI-resistant HIV-1 isolates with IC50 values ranging from 4-52 nM.31
Future directions of anti-protease treatment
The future management of HIV/AIDS should rely upon the development of therapies that are less toxic and more effective in combating drug-resistance. Since protease inhibitors are very important components of current HAART regiments, design and development of new PIs with improved pharmacological properties and better drug-resistance profiles are of great importance. In this context, our design strategies target the active site protein-backbone as there is minimal change in the backbone conformations of the wild-type and mutant proteases. Of particular note, we have developed a new generation of PIs bearing a structure-based designed bis-THF ligand that effectively fills in the hydrophobic pocket and maximizes hydrogen bonding interactions with the backbone atoms of the S2-site. A number of bis-THF-derived inhibitors are exceedingly potent and have maintained very impressive potency against multidrug- resistant HIV-1 variants. One of these inhibitors, darunavir, has been recently approved by the FDA as the first treatment of drug-resistant HIV. Our detailed structural analysis of darunavir-bound wild-type and mutant proteases have documented extensive hydrogen bonding interactions with the active site backbone atoms. This design concept targeting the backbone may serve as an important guide to combat drug resistance. Further development of novel PIs with designed functionalities is currently the focus of our ongoing investigation.
Figure 3.
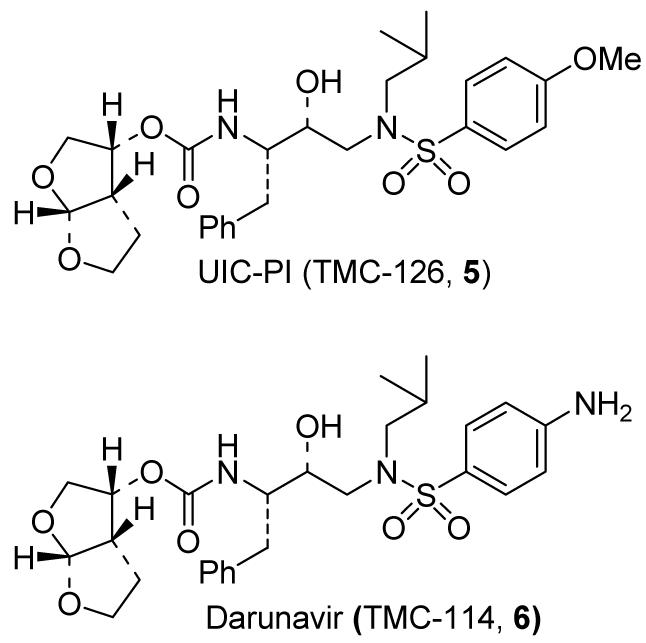
Structure of darunavir and inhibitor 5
Acknowledgment
Financial support by the National Institutes of Health (GM 53386; AKG), the Japan Health Sciences Foundation (International Research Grant SA14801; HM and AKG), and the Intramural Research Program of Center for Cancer Research, National Cancer Institute, NIH (HM) is gratefully acknowledged. We also thank Dr. Bruno Chapsal for helpful discussion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.UNAIDS/WHO Report on Annual AIDS Epidemic Update, December 2005. http://www.unaids.org/epi/2005/
- 2(a).Barre-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, Dauguet C, Axler-Blin C, Vezinet-Brun F, Rouzioux C, Rozenbaum W, Montagnier L. Science. 1983;220:868. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]; (b) Gallo RC, Salahuddin SZ, Popovic M, Shearer GM, Kaplan M, Haynes BF, Palker TJ, Redfield R, Oleske J, Safai B, White G, Foster P, Markham PD. Science. 1984;224:500. doi: 10.1126/science.6200936. [DOI] [PubMed] [Google Scholar]
- 3.Graves MC, Lim JJ, Heimer EP, Kramer RA. Proc. Natl. Acad. Sci. USA. 1988;85:2449. doi: 10.1073/pnas.85.8.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Farmerie WG, Leob DD, Casavant NC, Hutchison CA, Edgell MH, Swanstorm R. Science. 1987;236:305. doi: 10.1126/science.2436298. [DOI] [PubMed] [Google Scholar]
- 4.Palella FJ, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD. N. Engl. J. Med. 1998;338:853. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 5.Bartlett JA, DeMasi R, Quinn J, Moxham C, Rousseau F. AIDS. 2001;15:1369. doi: 10.1097/00002030-200107270-00006. [DOI] [PubMed] [Google Scholar]
- 6.Sepkowitz KA. N. Engl. J. Med. 2001;344:1764. doi: 10.1056/NEJM200106073442306. [DOI] [PubMed] [Google Scholar]
- 7.Grabar S, Pradier C, Le Corfec E, Lancar R, Allavena C, Bentata M, Berlureau P, Dupont C, Fabbro-Peray P, Poizot-Martin I, Costagliola D. Factors. AIDS. 2000;14:141. doi: 10.1097/00002030-200001280-00009. [DOI] [PubMed] [Google Scholar]
- 8.Staszewski S, Morales-Ramirez J, Tashima KT, Rachlis A, Skiest D, Stanford J, Stryker R, Johnson P, Labriola DF, Farina D, Manion DJ, Ruiz NM. N. Engl. J. Med. 1999;341:1865. doi: 10.1056/NEJM199912163412501. [DOI] [PubMed] [Google Scholar]
- 9.Ghosh AK, Sridhar P, Kumaragurubaran N, Koh Y, Weber IT, Mitsuya H. ChemMedChem. 2006;1:937. doi: 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
- 10.Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. J. Med. Chem. 2006;49:5252. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- 11.Clark MR, Mohandas N, Shohet SB. J. Clin. Invest. 1982;70:1074. doi: 10.1172/JCI110695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakanishi K. Bioorg. Med. Chem. 2005;13:4987. doi: 10.1016/j.bmc.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh AK, Thompson WJ, Holloway MK, McKee SP, Duong TT, Lee HY, Munson PM, Smith AM, Wai JM, Darke PL, Zugay JA, Emini EA, Schleif WA, Huff JR, Anderson PS. J. Med. Chem. 1993;36:2300. doi: 10.1021/jm00068a006. [DOI] [PubMed] [Google Scholar]
- 14.Kim EE, Baker CT, Dwyer MD, Murcko MA, Rao BG, Tung RD, Navia MA. J. Am. Chem. Soc. 1995;117:1181. [Google Scholar]
- 15(a).Kovalevsky AY, Liu F, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. J. Mol. Biol. 2006;363:161. doi: 10.1016/j.jmb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tie Y, Kovalevsky AY, Boross P, Wang YF, Ghosh AK, Tozser J, Harrison RW, Weber IT. Proteins. 2007;67:232. doi: 10.1002/prot.21304. [DOI] [PubMed] [Google Scholar]
- 16(a).Hong L, Zhang X, Hartsuck JA, Tang J. Protein Sci. 2000;9:1898. doi: 10.1110/ps.9.10.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Laco GS, Schalk-Hihi C, Lubkowski J, Morris G, Zdanov A, Olson A, Elder JH, Wlodawer A, Gustchina A. Biochemistry. 1997;36:10696. doi: 10.1021/bi9707436. [DOI] [PubMed] [Google Scholar]
- 17(a).Yoshimura K, Kato R, Kavilck MF, Nguyen A, Maroun V, Maeda K, Hussain KA, Ghosh AK, Gulnik SV, Erickson JW, Mistuya H. J. Virol. 2002;76:1349. doi: 10.1128/JVI.76.3.1349-1358.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh AK, Pretzer E, Cho H, Hussain KA, Duzgunes N. Antiviral Res. 2002;54:29. doi: 10.1016/s0166-3542(01)00209-1. [DOI] [PubMed] [Google Scholar]
- 18(a).Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang Y-F, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Antimicrob. Agents Chemother. 2003;47:3123. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) De Meyer S, Azijn H, Surleraux D, Jochmans D, Tahri A, Pauwels R, Wigerinck P, de Bethune M-P. Antimicrob. Agents Chemother. 2005;49:2314. doi: 10.1128/AAC.49.6.2314-2321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoetelmans R, van der Sandt I, De Pauw M, Struble K, Peeters M, van der Geest R. 10th Conference on Retroviruses and Opportunistic Infections (CROI); Boston, MA (USA). February 2003. [Google Scholar]
- 20.Molina J-M, Cohen C, Katlama C, Grinsztejn B, Timerman A, Pedro R, et al. Abstract P4, 12th Annual BHIVA; Brighton, UK. 29 March-1 April 2006. [Google Scholar]
- 21. http://www.findarticles.com/p/articles/mi_m0EIN/is_2006_June_23/ai_n16498426.
- 22a).Chen X, Kempf DJ, Li L, Sham HL, Vasavanonda S, Wideburg NE, Saldivar A, Marsh KC, McDonald E, Norbeck DW. Bioorg. Med. Chem. Lett. 2003;13:3657. doi: 10.1016/j.bmcl.2003.08.043. [DOI] [PubMed] [Google Scholar]; (b) Chen X, Li L, Kempf DJ, Sham H, Widebury NE, Saldivar A, Vasavanonda S, Marsh KC, McDonald E, Norbeck DW. Bioorg. Med. Chem. Lett. 1996;6:2847. doi: 10.1016/s0960-894x(98)00653-2. [DOI] [PubMed] [Google Scholar]
- 23.Amano M, Koh K, Das D, Li J, Leschenko S, Wang Y-F, Boross PI, Weber IT, Ghosh AK, Mitsuya H. Antimicrob. Agents Chemother. 2007;51:2143. doi: 10.1128/AAC.01413-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24(a).Miller JF, Furfine ES, Hanlon MH, Hazen RJ, Ray JA, Robinson L, Samano V, Spaltenstein A. Bioorg. Med. Chem. Lett. 2004;14:959. doi: 10.1016/j.bmcl.2003.12.008. [DOI] [PubMed] [Google Scholar]; (b) Sherrill RG, Furfine ES, Hazen RJ, Miller JF, Reynolds DJ, Sammond DM, Spaltenstein A, Wheelan P, Wright LL. Bioorg. Med. Chem. Lett. 2005;15:3560. doi: 10.1016/j.bmcl.2005.05.101. [DOI] [PubMed] [Google Scholar]
- 25.Miller JF, Andrews CW, Brieger M, Furfine ES, Hale MR, Hanlon MH, Hazen RJ, Kaldor I, McLean EW, Reynolds D, Sammond DM, Spaltenstein A, Tung R, Turner EM, Xu RX, Sherrill RG. Bioorg. Med. Chem. Lett. 2006;16:1788. doi: 10.1016/j.bmcl.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 26(a).Surleraux DLNG, Tahri A, Verschueren WG, Pille GME, de Kock HA, Jonckers THM, Peeters A, De Meyer S, Azijn H, Pauwels R, de Bethune M-P, King NM, Jeyabalan MP, Schiffer CA, Wigerinck PBTP. J. Med. Chem. 2005;48:1813. doi: 10.1021/jm049560p. [DOI] [PubMed] [Google Scholar]; (b) Surleraux DLNG, de Kock BA, Verschueren WG, Pille GME, Maes LJR, Peeters A, Vendeville S, De Meyer S, Azijn H, Pauwels R, de Bethune M-P, King NM, Jeyabalan MP, Schiffer CA, Wigernick PBTP. J. Med. Chem. 2005;48:1965. doi: 10.1021/jm049454n. [DOI] [PubMed] [Google Scholar]
- 27.Stranix BR, Sauve G, Bouzide A, Cote A, Sevigny G, Yelle J, Perron V. Bioorg. Med. Chem. Lett. 2004;14:3971. doi: 10.1016/j.bmcl.2004.05.049. [DOI] [PubMed] [Google Scholar]
- 28.Sévigny G, Stranix BR, Parkin N, Lie Y, Yelle J. XIII International HIV Drug Resistance Workshop; Tenerife Sur - Costa Adeje, Canary Islands, Spain. June 8-12 2004. [Google Scholar]
- 29.Wu JJ, Sevigny G, Stranix BR, Dandache S, Petrella M, Ge M, Milot G, Yelle J, Panchal C, Parkin N, Shapiro JM, Wainberg MA. XIV International HIV Drug Resistance Workshop [Abstract #77]; Quebec City, Canada. June 2005. [Google Scholar]
- 30.Hammond J, Jackson L, Graham J, Knowles S, Digits J, Tatlock JH, Jewell T, Canan-Koch S, Patick AK. 13th International HIV Drug Resistance Workshop; Tenerife Sur-Costa Adeja, Canary Islands, Spain. June 8-12 2004. [Google Scholar]
- 31.Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. J. Med. Chem. 2006;49:5252. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]