

Abstract
Studies of the RNA-dependent RNA polymerase (RdRp) from poliovirus (PV), 3Dpol, have shown that Asn-297 permits this enzyme to distinguish ribose from 2′-deoxyribose. All animal RNA viruses have Asn at the structurally homologous position of their polymerases, suggesting a conserved function for this residue. However, all prokaryotic RNA viruses have Glu at this position. In the presence of Mg2+, the apparent affinity of Glu-297 3Dpol for 2′-deoxyribonucleotides was decreased by 6-fold relative to wild type without a substantial difference in the fidelity of 2′-dNMP incorporation. The fidelity of ribonucleotide misincorporation for Glu-297 3Dpol was reduced by 14-fold relative to wild type. A 4- to 11-fold reduction in the rate of ribonucleotide incorporation was observed. Glu-297 PV was unable to grow in HeLa cells due to a replication defect equivalent to that observed for a mutant PV encoding an inactive polymerase. Evaluation of the protein-(VPg)-primed initiation reaction showed that only half of the Glu-297 3Dpol initiation complexes were capable of producing VPg-pUpU product and that the overall yield of uridylylated VPg products was reduced by 20-fold relative to wild-type enzyme, a circumstance attributable to a reduced affinity for UTP. These studies identify the first RdRp derivative with a mutator phenotype and provide a mechanistic basis for the elevated mutation frequency of RNA phage relative to animal RNA viruses observed in culture. Although protein-primed initiation and RNA-primed elongation complexes employ the same polymerase active site, the functional differences reported here imply significant structural differences between these complexes.
RNA viruses cause a variety of acute and chronic diseases in humans: common cold, summer flu, hepatitis, severe acute respiratory syndrome, and liver cancer, to name a few (1–5). The genomes of these viruses are transcribed and replicated by a virus-encoded RNA-dependent RNA polymerase (RdRp)2 (4, 6, 7). Like other viral polymerases, the RdRp represents an important target for antiviral drug development (8–11). The RdRp from poliovirus (PV), 3Dpol, has emerged as an important model for understanding the structure, function, and mechanism of this class of nucleic acid polymerases (12–15).
PV 3Dpol will incorporate a ribonucleotide (rNMP) with an incorrect base at a frequency of ~10−7 to 10−4 (16, 17). However, this enzyme is much more tolerant of nucleotides with an incorrect sugar configuration. Both 2′- and 3′-deoxynucleotides (dNMPs) are incorporated at a frequency of ~10−2 (15). It is known that incorporation of more than one incorrect ribonucleotide per PV genome decreases the specific infectivity of the RNA (16, 18). Whether or not 2′-dNMP incorporation has an effect on viral RNA infectivity is not known.
Several factors likely limit 2′-dNMP incorporation into the genomes of RNA viruses of eukaryotes. First, dNTP pools in cells are thought to be low, in the 5–30 μM range (19). Low dNTP levels are maintained by regulating the activity and localization of ribonucleotide reductase (20, 21). In the G1-phase of the cell cycle, very little ribonucleotide reductase holoenzyme exists due to the localization of one subunit in the nucleus and the other in the cytoplasm. During the S-phase of the cell cycle, the ribonucleotide reductase holoenzyme is distributed in both cytoplasm and nucleus; allosteric control mechanisms would likely permit only the nuclear enzyme to function catalytically (20–22). If nuclear localization of active ribonucleotide reductase prevents eukaryotic RNA virus polymerases from having to select against utilization of 2′-dNTPs, then prokaryotic RNA virus polymerases may require a more stringent selection mechanism.
Asn-297 is the primary determinant of ribose specificity in PV 3Dpol (14). This residue is located in the ribose-binding pocket and hydrogen bonds to the ribose 2′-OH (Fig. 1A). The capacity for Ala-297 derivative to distinguish between ATP and 2′-dATP incorporation is reduced by 10-fold relative to wild-type 3Dpol (14). Asn is present at the structurally homologous position in all plant and animal RNA virus polymerases (Fig. 1B), suggesting that this residue functions in ribose selection for these enzymes as well. Curiously, RNA phage polymerases have a Glu at this position (Fig. 1B), consistent with a difference in ribose/2′-deoxyribose selection for these enzymes.
FIGURE 1. Differences in ribose/2′-deoxyribose specificity for RNA phage polymerases relative to animal and plant RNA virus polymerases predicted from structure/sequence comparison.
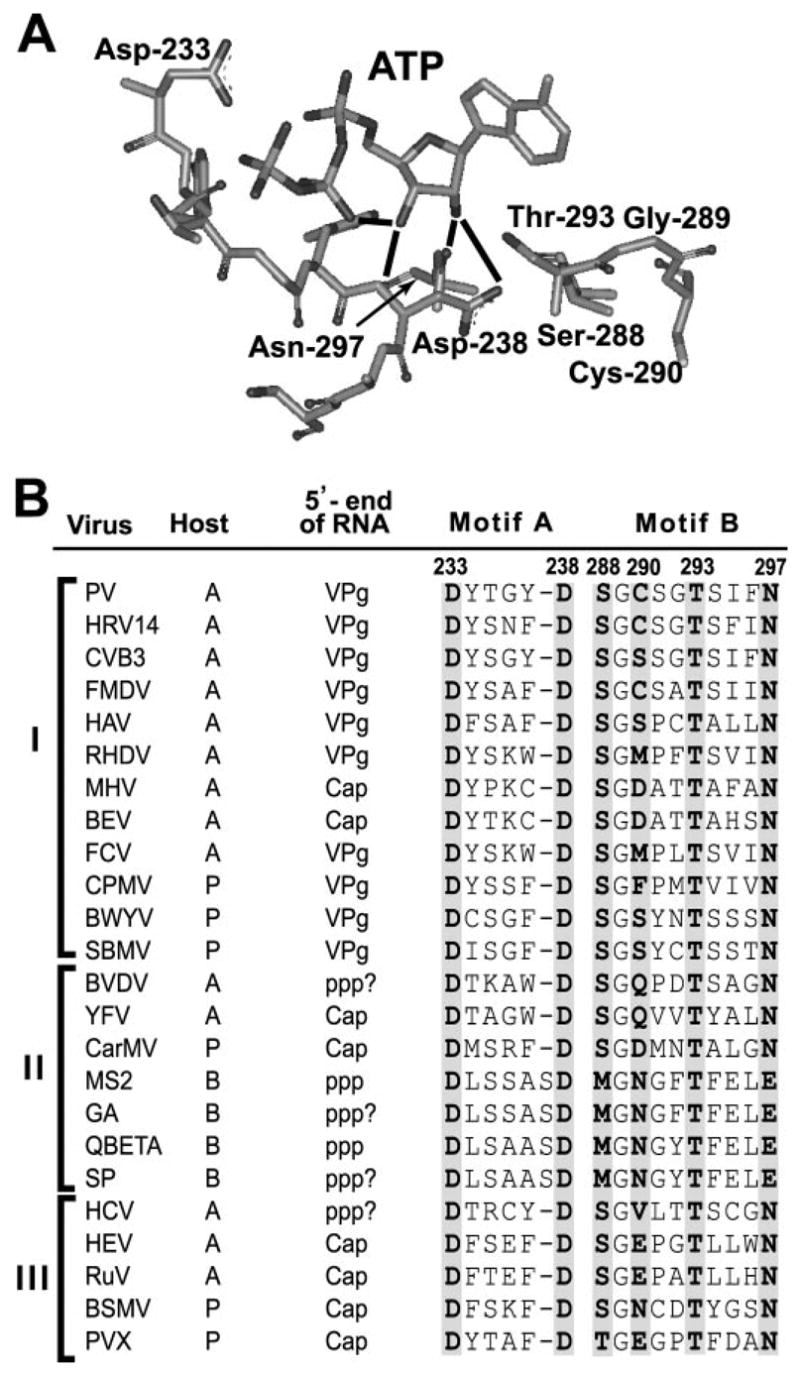
A, structural model for the ribose-binding pocket of the RdRp from PV, 3Dpol (14). The 3Dpol ribonucleotide-binding pocket is shown with bound ATP. This structural model is based on homology modeling, because a crystal structure for this complex is still not available for any RdRp (14). This pocket is composed of residues from conserved structural motifs A (Asp-238) and B (Ser-288 to Asn-297). The extensive hydrogen bonding network shown links the position of the triphosphate (catalytic efficiency) to the nature of the bound nucleotide, that is, whether or not a correct base and sugar configuration are present. Asn-297 permits the enzyme to distinguish between ribose and 2′-deoxyribose by hydrogen bonding to the 2′-OH group of ribose. A 3Dpol derivative containing Ala at this position instead of Asn fails to distinguish rNTPs from 2′-dNTP as well as wild-type 3Dpol (14). The image was created by using the program WebLab Viewer (Molecular Simulations Ins., San Diego, CA). B, conserved Asn in animal and plant RNA virus polymerases is a Glu in RNA phage polymerases. RNA virus polymerases can be organized into three supergroups: I, II, and III. Representative viruses, their host (A, animal; P, plant; and B, bacteria) and mechanism of initiation of RNA synthesis based on the structure of the 5′-end of the genome (VPg, protein primed; Cap and ppp, de novo) are indicated. In those instances in which the 5′-end has not been analyzed directly, the structure of the 5′-end appears with a question mark and represents the most logical structure based on available biochemical data or relationship to other viruses. Residues 233 and 238 (3Dpol numbering) of conserved structural motif A are conserved in all animal and plant RNA virus polymerases. Residues 288, 293, and 297 (3Dpol numbering) of conserved structural motif B are conserved in all animal and plant RNA virus polymerases. Variability exists for the RNA phage polymerases at positions 288 and 297. Abbreviations used are: PV, poliovirus; HRV14, human rhinovirus 14; CVB3, coxsackievirus B3; FMDV, foot-and-mouth disease virus; HAV, hepatitis A virus; RHDV, rabbit hemorrhagic disease virus; MHV, mouse hepatitis virus; BEV, Berne virus; FCV, feline calici virus; CPMV, cowpea mosaic virus; BWYV, beet western yellow virus; SBMV, southern bean mosaic virus; BVDV, bovine viral diarrhea virus; YFV, yellow fever virus; CarMV, carnation mottle virus; HCV, hepatitis C virus; HEV, hepatitis E virus; RuV, rubella virus; BSMV, barley stripe mosaic virus; and PVX, potato virus X. The bacteriophages were MS2, GA, SP, and QBETA. (Adapted from Refs. 51 and 52.)
We show that Glu-297 3Dpol does not have a more stringent selection against 2′-dNMP incorporation than wild-type (WT) 3Dpol, although the affinity for 2′-dNTPs is reduced relative to WT 3Dpol. However, nucleotide incorporation by Glu-297 3Dpol was slower and less faithful than WT 3Dpol. Glu-297 3Dpol represents the first RdRp derivative with a mutator phenotype. Glu-297 PV was inviable due to an RNA synthesis defect in cells that was on par with that of a catalytically inactive 3Dpol derivative. This phenotype could be explained by a severe reduction in the yield of product produced by protein-primed initiation of RNA synthesis. We conclude that a requirement for decreased replication speed and/or incorporation fidelity imposes a constraint on the evolution of the polymerase active site that restricts the variety of initiation mechanisms that can be employed. Thus phage polymerases employ strictly a de novo mechanism for initiation, whereas animal RNA viruses can utilize de novo and protein-primed mechanisms for initiation (Fig. 1B). The capacity of a single 3Dpol derivative to exhibit significant differences between RNA- and protein-primed RNA synthesis suggests that the structural organization of the corresponding elongation complexes is different. We propose that inhibitors specific for the PV 3Dpol initiation complex may be more efficient than those targeting the elongation complex.
EXPERIMENTAL PROCEDURES
Materials
[γ-32P]ATP (>7000 Ci/mmol) was from MP Biomedicals (formerly ICN); [α-32P]UTP (>6000 Ci/mmol) was from Amersham Biosciences; DNA oligonucleotides were from Integrated DNA Technologies, Inc.; T4 polynucleotide kinase, Deep Vent DNA polymerase, and restriction enzymes were from New England Biolabs, Inc.; Sephadex G-25 was from Sigma; all nucleotides (ultrapure solutions) were from Amersham Biosciences; RNA oligonucleotides were from Dharmacon Research, Inc. (Boulder, CO); synthetic VPg peptide was from Alpha Diagnostic International (San Antonio, TX); all other reagents were of the highest grade available through Sigma, Fisher, or VWR.
Construction, Expression, and Purification of 3Dpol Derivatives
Mutations were introduced into a modified 3Dpol-coding sequence by using overlap-extension PCR and expressed in Escherichia coli by using a ubiquitin fusion system as described previously (23). In brief, the Glu-297 clone was produced by overlap-extension PCR with oligonucleotides 1 and 7, and 2 and 8 (Table 1), with pET26Ub-3D-BPKN-I92T as template. The final product of second PCR was purified and digested with PstI and NheI and ligated into pET26Ub-3D-BPKN-I92T, which had been digested with the same enzymes. Double and triple mutants (Met-288, Glu-297; Asn-290, Glu-297; and Met-288, Asn-290, Glu-297) were constructed in the same way using oligonucleotide primers 1, 2, and 9–14 (Table 1), with pET26Ub-3D-BPKNP-I92T plasmid used as template. This plasmid has a silent PmlI site in the 3Dpol coding sequence. Mutations were confirmed by DNA sequencing (Nucleic Acid Facility, Pennsylvania State University). 3Dpol derivatives were expressed and purified as described previously (23). The concentration of the Glu-297 3Dpol was determined by measuring absorbance at 280 nm in 6 M guanidine chloride solution, using a calculated WT 3Dpol extinction coefficient (71,840 M−1 cm−1). For double and triple mutants poly-r(U) activity assays were performed with partially purified enzymes recovered from ammonium sulfate pellets.
TABLE 1.
Oligonucleotides used in this study
| Oligonucleotide number | Oligonucleotide name | Sequencea,b |
|---|---|---|
| 1 | 3D-PV-AvrII-rev | 5′-CCT GAG TGT TCC TAG GAT CTT TAG T-3′ |
| 2 | 3D-PV-PstI-for | 5′-GGA GTG ATA ACA GGT TCT GCA GTG GGG TGC GAT-3′ |
| 3 | 3D-PV-AflII-for | 5′-AAC GAT CCC AGG CTT AAG ACA GAT TTT GAG-3′ |
| 4 | 3D-BamHI-rev | 5′-GCG GGA TCC TTA CTA AAA TGAGTC AAG CCA ACG GCG GTA-3′ |
| 5 | 3D-SacII-for | 5′-GCG CCG CGG TGG AGG TGA AAT CCA GTG GAT GAG A-3′ |
| 6 | 3Dseq500 | 5′-AGG TTG AGC AGG GGA AA-3′ |
| 7 | 3D-PV-N297E-for | 5′-GGT ACC TCA ATT TTT GAA TCA ATG ATT AAC-3′ |
| 8 | 3D-PV-N297E-rev | 5′-GTT AAT CAT TGA TTC AAA AAT TGA GGT ACC-3′ |
| 9 | 3D-PV-S288M-N297E-for | 5′-AAG GGC GGT ATG CCA ATG GGC TGT TCA GGT ACC TCA ATT TTT GAA TCA ATG ATT AAC AAC-3′ |
| 10 | 3D-PV-S288M-N297E-rev | 5′-GTT GTT AAT CAT TGA TTC AAA AAT TGA GGT ACC TGA ACA GCC CAT TGG CAT ACC GCC CTT-3′ |
| 11 | 3D-PV-C290N-N297E-for | 5′-GGT ATG CCA TCT GGC AAC TCA GGT ACC TCA ATT TTT GAA TCA ATG ATT AAC AAC-3′ |
| 12 | 3D-PV-C290N-N297E-rev | 5′-GTT GTT AAT CAT TGA TTC AAA AAT TGA GGT ACC TGA GTT GCC AGA TGG CAT ACC-3′ |
| 13 | 3D-PV-S288M-C290N-N297E-for | 5′-AAG GGC GGT ATG CCA ATG GGC AAC TCA GGT ACC TCA ATT TTT GAA TCA ATG ATT AAC AAC-3′ |
| 14 | 3D-PV-S288M-C290N-N297E-rev | 5′-GTT GTT AAT CAT TGA TTC AAA AAT TGA GGT ACC TGA GTT GCC CAT TGG CAT ACC GCC CTT-3′ |
Codons that introduce amino acid residue change in 3Dpol sequence are in bold.
Restriction enzymes sites are underlined.
Construction, Expression, and Purification of 3CD
Viral protein 3CD was expressed and purified as described previously (24).
Transcription of 61-nt 2C-cre
61-nt 2C-cre RNA was transcribed and gel-purified by denaturing PAGE as described previously (24).
Purification, 5′-32P Labeling and Annealing of sym/sub
RNA oligonucleotides were purified, labeled, and annealed as described previously (12).
Poly-r(U) Polymerase Activity Assay
Poly-r(U) activity assays were performed as described previously with minor modifications (14). Briefly, reactions contained 50 mM HEPES, pH 7.5, 10 mM 2-mercaptoethanol, 5 mM MgCl2, 60 μM ZnCl2, 500 μM UTP, 0.2 μCi/μl [α32-P]UTP, 2 μM dT15/0.2 μM poly-rA467 primer-template complex, and 500 ng of 3Dpol. Reactions were carried out in a total volume of 25 μl at 30 °C for 5 min and then quenched by mixing 5 μl with an equal volume of 0.1 M EDTA. 5 μl of the quenched reactions was spotted onto DE81 filter paper discs and dried completely. The discs were washed three times for 10 min in 250 ml of 5% dibasic sodium phosphate and rinsed in absolute ethanol. Bound radioactivity was quantitated by liquid scintillation counting in 5 ml of EcoScint scintillation fluid (National Diagnostics).
3Dpol Kinetic Assays
Reactions contained 50 mM HEPES, pH 7.5, 10 mM 2-mercaptoethanol, 5 mM MgCl2, 60 μM ZnCl2, 500 μM NTPs (unless specified otherwise), 1 μM sym/sub (0.5 μM duplex), and 1 μM 3Dpol. Reactions were quenched by addition of EDTA to a final concentration of 50 mM 3Dpol was diluted immediately prior to use in 50 mM HEPES, pH 7.5, 10 mM 2-mercaptoethanol, 60 μM ZnCl2, and 20% glycerol. The volume of enzyme added to any reaction was always less than or equal to one-tenth of the total volume. All reactions were performed at 30 °C.
Rapid Chemical-Quench-Flow Experiments
Rapid mixing/quenching experiments were performed using a Model RQF-3 chemical-quench-flow apparatus (KinTek Corp., Austin, TX). All experiments were performed at 30 °C, with the temperature controlled by a circulating water bath. 3Dpol-sym/sub complex in 50 mM HEPES, pH 7.5, 10 mM 2-mercaptoethanol, 60 μM ZnCl2, and 5 mM MgCl2 was rapidly mixed with the nucleotide substrate in the same buffer, and the reactions were quenched either by addition of 0.5 M EDTA to a final concentration of 0.3 M or by addition of 2 N HCl to a final concentration of 1.2 N. Immediately after addition of HCl, the solution was neutralized by addition of 3 M KOH in 1 M Tris, pH 8.0.
Product Analysis
Products were analyzed by denaturing PAGE. An equal volume of loading buffer, 5 μl (90% formamide, 0.025% bromphenol blue, and 0.025% xylene cyanol, 1× TBE) (89 mM Tris, 89 mM boric acid, 2 mM EDTA) was mixed with 5 μl of the quenched reaction mixtures and heated at 65 °C for 2–5 min. prior to loading 5 μl on a denaturing 23% polyacrylamide gel containing 1× TBE and 7 M urea. Electrophoresis was performed at 90 watts. Gels were visualized by using a PhosphorImager and quantified by using ImageQuaNT software (Molecular Dynamics).
Data Analysis
Data were fit by non-linear regression using the program KaleidaGraph (Synergy Software, Reading, PA). Time courses at fixed nucleotide concentration were fit to Equation 1,
| (Eq. 1) |
where A is the maximal concentration of product formed, kobs is the observed first-order rate constant describing product formation, t is the time, and C is a constant. The apparent dissociation constant (Kd,app) and maximal rate for nucleotide incorporation (kpol) were determined using Equation 2.
| (Eq. 2) |
Kinetic Simulations
Kinetic simulations were performed by using KinTekSim version 2.03 (KinTek Corp., Austin TX). Agreement between the experimental data and kinetic simulations was determined by visual inspection.
Construction of Mutated Viral cDNA Clones and Mutated Replicons
To introduce a mutation into the 3Dpol coding sequence of viral cDNA, overlap extension PCR was performed with oligonucleotides 1 and 3 and the appropriate protein expression plasmid as template. PCR products were purified and digested with AvrII and AflII. The digested PCR product was ligated into pUC18-BglII-EcoRI-3CD vector, a subclone of the viral cDNA (14, 24). From this vector, the fragment between BglII and EcoRI was cloned into the viral cDNA plasmid (pMoRA) (25). To introduce mutations into the subgenomic replicon the fragment between the BglII and AvrII sites from the mutated pUC18-BglII-EcoRI-3CD subclone vector was ligated into pRLucRA (also known as pRLuc31-rib+polyAlong (25)). DNA sequencing was used to verify the integrity of all clones.
Infectious Center Assays
RNA transcripts were produced from the pMoRA-WT and pMoRA-Glu-297 plasmids after linearization with EcoRI. Transcription reactions, typically 20 μl, consisted of 350 mM HEPES, pH 7.5, 32 mM magnesium acetate, 40 mM dithiothreitol, 2 mM spermidine, 28 mM NTPs, 0.5 μg of template, and 0.5 μg of T7 RNA polymerase. Reactions were incubated at 37 °C for 3 h followed by removal of magnesium pyrophosphate. DNase treatment with RQ1 DNase (Promega, Madison, WI) was used to remove the template; lithium chloride precipitation was used to remove unincorporated nucleotides. RNA concentration was calculated by measuring absorbance at 260 nm, assuming an A260 of 1 was equivalent to 40 μg/ml. HeLa cells were propagated in Dulbecco’s modified Eagle’s medium/F-12 media (Invitrogen) supplemented with 10% (v/v) fetal bovine serum (Invitrogen), and 1% (v/v) penicillin and streptomycin (Invitrogen), always keeping the cultures between 20 and 80% confluency. Subconfluent monolayers of HeLa cells were detached from the culture flasks by trypsin treatment, washed with 1× phosphate-buffered saline, and cell number was adjusted to 1.2 × 106 cells/0.4 ml in phosphate-buffered saline. Cell suspensions (400 μl) were mixed with 5 μg of RNA in a microcentrifuge tube, transferred to an electroporation cuvette (0.2-cm gap width, Bio-Rad), and subjected to an electric pulse at 500 microfarads and 0.13 V using a GenePulser system (Bio-Rad). 400 μl of electroporated cells was plated onto appropriate wells of a 6-well plate containing a monolayer of HeLa cells plated with 5 × 105 cells the day before. For WT controls, 1/100, 1/1000, and 1/2000 dilutions were plated. Cells were allowed to adsorb to the plate for 1 h at 37 °C, and then the medium/phosphate-buffered saline was aspirated. Cells were then covered with 3 ml of a mixture of 1× Dulbecco’s modified Eagle’s medium/F-12 plus 10% fetal bovine serum, 1% penicillin and streptomycin, and 1% low melting point agarose (American Bioanalytical). Plates were then incubated at either 32 °C or 37 °C for 2–3 days. The agarose overlay was removed by using a spatula. Wells were stained with crystal violet, and viral plaques were counted.
Serial Passage and Reverse Transcription-PCR
For serial passage of Glu-297 PV 1 × 106 HeLa cells were electroporated with 5 μg of RNA (no RNA as negative control), essentially as described above for the infectious center assay. The electroporated cells were then plated onto a monolayer of HeLa cells (plated with 5 × 105 cells the previous day onto 6-well plates). Plates were incubated at 37 °C. Within 2 days, cells infected with WT RNA started showing signs of cytopathic effect, whereas for Glu-297 infected cells started rounding up only on day 4. Cells in mock control looked healthy at that time. For the first passage, 100 μl of media from the transfected cell were taken on day 2 and transferred onto a fresh monolayer of HeLa cells. Cells infected with the media from WT virus died within 1 day and for Glu-297 within 2 days. Mock control cells did not show any signs of cytopathic effect. 100 μl of media from day 2 (passage 1) was again transferred onto a fresh monolayer for passage 2. The infected cells from passage 2 showed the same kinetics for cytopathic effect. Passage 3 was done in the same manner as the first two. On the second day, when both WT and Glu-297 PV-infected cells were dead, 1 ml of media was collected and viral RNA was extracted with a NucleoSpin nucleic acid purification kit (Macherey-Nagel) according to the manufacturer’s manual. The 3Dpol cDNA was prepared from purified viral RNA by reverse transcription with Endo-Free RT (Ambion) with oligonucleotide 4 (Table 1). The resulting DNA product was then PCR-amplified using SuperTaq DNA polymerase (Ambion) and oligonucleotides 4 and 5 as primers. The PCR product for the N297E 3Dpol gene was sequenced with oligonucleotide 6 as primer. Codon GAA that encodes Glu-297 was mutated to GGA, encoding Gly.
Subgenomic Replicon Assays
RNA transcripts were produced from the pRLucRA plasmids after linearization with ApaI. RNA concentration was calculated as described above. HeLa cells (1.2 × 106) were electroporated with subgenomic replicon (5 μg) essentially as described above for infectious center assay. Immediately after electroporation 400 μl of cells was mixed with 6 ml of prewarmed media (Dulbecco’s modified Eagle’s medium/F-12, 10% fetal bovine serum, and 1% penicillin and streptomycin); 0.5 ml of this mixture was aliquoted into a 1.7-ml Eppendorf tube per single time point and transferred into either a 32 °C or 37 °C incubator. At fixed time points, cells were pelleted by centrifugation at 14,000 rpm for 2 min. Cells were lysed in 100 μl of 1× cell culture lysis reagent (Promega). Lysates were left on ice until luciferase and protein concentration assays were performed at which point the lysates were centrifuged for 2 min at 14,000 rpm to remove cellular debris and nuclei. Lysates were assayed for luciferase activity by mixing 10 μl of lysate supernatant with 10 μl of luciferase assay substrate (Promega) and quantifying light production for 10 s by using a Junior LB 9509 luminometer (Berthold). Relative light units were adjusted to total protein concentration in the samples. Protein concentration was measured at 595 nm using the Brad-ford reagent (Bio-Rad).
VPg Uridylylation Assay
VPg uridylylation reaction mixtures contained 1 μM 3Dpol, 1 μM 61-nt 2C-cre, and 5 μM VPg in reaction buffer (50 mM HEPES, pH 7.5, 5 mM magnesium acetate, 10 μM UTP, and 0.04 μM [α-32P]UTP (6000 Ci/mmol)). All reactions were adjusted to a final NaCl concentration of 20 mM 3Dpol and 3CD were diluted immediately prior to use in buffer containing 50 mM HEPES, 10 mM 2-mercaptoethanol, and 20% glycerol. First, VPg was mixed with 2C-cre RNA in 10 μl of the reaction buffer described above. This mixture was incubated at 30 °C for 5 min prior to addition of 3CD to a final concentration of 1 μM. After addition of 3CD the reaction mixture was incubated at 30 °C for 5 min prior to adding 3Dpol to a final concentration of 1 μM. The uridylylation reaction was allowed to proceed for 30 min, after which 5 μl of reaction were quenched by addition of an equal volume of 100 mM EDTA in a loading dye (90% formamide, 1× TBE, 0.025% bromphenol blue). Products were separated by Tris-Tricine SDS-PAGE. Gels contained 15% acrylamide and 0.4% bisacrylamide. The cathode buffer (upper chamber) contained 0.1 M Tris, 0.1 M Tricine, and 0.1% (w/v) SDS; the anode buffer contained 0.2 M Tris, pH 8.9. Gels were run at 80 watts. Products were visualized by using a PhosphorImager and quantified by using Image-Quant software.
RESULTS
Rationale
Previous studies identified Asn-297 as a key player in ribonucleotide selection by the RdRp from PV, 3Dpol. Asn-297 hydrogen bonds to the 2′-hydroxyl of the incoming ribonucleotide (rNTP) (Fig. 1A). Changing Asn-297 to Ala prevented 3Dpol from distinguishing an rNTP with correct base pairing capacity from a 2′-dNTP with correct base pairing capacity, a lethal change for PV (Table 2) (14). Asn-297 is conserved in all plant and animal RNA virus polymerases; however, this residue is a Glu in RNA phage polymerases (Fig. 1B). This study was initiated to determine the consequence(s) of this difference in the ribose binding pocket of eukaryotic and prokaryotic RNA viruses.
TABLE 2.
Glu-297 3Dpol incorporates 2′-deoxyribonucleotides as efficiently as WT 3Dpol
| Substrates
|
Kinetic parameters
|
||||
|---|---|---|---|---|---|
| Enzyme | Nucleic acid | Nucleotide | Kd,app | kpol | kpol/Kd,app |
| μM | s−1 | s−1/μM | |||
| sym/sub-U GCAUGGGCCC CCCGGGUACG | |||||
| WTa | ATP | 130 ± 20 | 90 ± 10 | 0.7 | |
| Glu-297 | 530 ± 120 | 8 ± 1 | 0.02 | ||
| Ala-297b | 180 ± 30 | 5 ± 1 | 0.03 | ||
| WTa | dATP | 280 ± 60 | 0.8 ± 0.1 | 0.003 | |
| Glu-297 | 1700 ± 400 | 0.4 ± 0.02 | 0.0002 | ||
| Ala-297b | 260 ± 30 | 0.2 ± 0.05 | 0.0008 | ||
| WTa | GTP | 210 ± 70 | 0.01 ± 0.001 | 0.00005 | |
| Glu-297 | 290 ± 90 | 0.005 ± 0.001 | 0.00002 | ||
| sym/sub-A GCUAGGGCCC CCCGGGAUCG | |||||
| WTa | UTP | 100 ± 10 | 270 ± 20 | 2.7 | |
| Glu-297 | 380 ± 80 | 70 ± 6 | 0.2 | ||
| WT | dUTP | 190 ± 40 | 6 ± 1 | 0.03 | |
| Glu-297 | 1100 ± 200 | 2 ± 0.2 | 0.002 | ||
Glu-297 3Dpol Incorporates 2′-Deoxyribonucleotides as Efficiently as WT 3Dpol
3Dpol-coding sequence was changed to encode a glutamic acid at position 297 (Glu-297 3Dpol). This derivative was purified to >90% purity exactly as described for WT 3Dpol, suggesting no significant change in the structure of the enzyme (23). In addition, this derivative retained at least half of the poly(rU) polymerase activity of WT 3Dpol (Table 3).
TABLE 3. Activity of Glu-297 3Dpol is not enhanced by introduction of residues conserved in phage RdRps.
Poly-r(U) polymerase activity was measured in purified or partially purified preparations of the indicated 3Dpol derivatives as described under “Experimental Procedures.” The specific activity is reported to one significant figure; the standard error of the measurement was <20%.
| Enzyme | Specific activity |
|---|---|
| pmol/min/3g | |
| WT | 800 |
| WTa | 400 |
| Glu-297 | 300 |
| Met-288 and Glu-297a | <5 |
| Asn-290 and Glu-297a | 10 |
| Met-288, Asn-290, and Glu-297a | 5 |
This enzyme was only purified through the ammonium sulfate precipitation step.
To evaluate the kinetics and mechanism of 3Dpol, we use a 10-nt symmetrical RNA substrate referred to as sym/sub (S/S). S/S-U, for example, has uracil as the first templating base (Fig. 2A) (12). 3Dpol binds tightly to the duplex portion of this substrate and can utilize either 3′-end as a primer for nucleotide addition. The rate constant for dissociation of Glu-297 3Dpol was 0.0001 s−1, corresponding to a half-life for the complex of ~2 h as observed for WT 3Dpol (12).
FIGURE 2. Kinetic analysis of Glu-297 3Dpol.

A, RNA primer-template substrate employed. A 10-nt self-complementary RNA referred to as sym/sub (S/S). Shown is S/S-U. The “U” indicates that uracil is the templating base. B, product analysis. In this experiment, 2 μM Glu-297 3Dpol was incubated with 2 μM sym/sub (1 μM duplex) and rapidly mixed with 200 μM ATP. Reactions were quenched at various times up to 1 s by addition of EDTA. Reaction products were resolved by electrophoresis on a denaturing 23% polyacrylamide gel and visualized by using a PhosphorImager (Amersham Biosciences). C, concentration dependence of the kinetics of nucleotide incorporation. Experiments were performed as described above in the presence of: 50 (●), 100 (○), 250 (■), 500 (□), 1000 (▲), or 2000 μM (△) ATP. The solid line represents the fit of the data to a single exponential (Equation 1), yielding kobs values of 0.6 ± 0.03, 1.3 ± 0.1, 2.3 ± 0.15, 3.53 ± 0.3, 5.6 ± 0.5, and 5.7 ± 0.4 s−1, for 50 –2000 μM ATP, respectively. D, kinetic constants for AMP incorporation by Glu-297 3Dpol. Values for kobs were plotted as a function of ATP concentration. The solid line represents the fit of data to a hyperbola (Equation 2), yielding values for Kd,app for ATP of 530 ± 120 μM and kpol of 8 ± 1 s−1.
We evaluated the dependence of the rate constant for AMP incorporation on ATP concentration. For this experiment, 2 μM 3Dpol was first incubated with 2 μM 5′-32P-labeled S/S-U (1 μM duplex) in reaction buffer to establish the enzyme-RNA complex. The assembled complex was then rapidly mixed with ATP of the appropriate concentration in reaction buffer, as described under “Experimental Procedures.” Mixing resulted in a 2-fold dilution. The reaction was stopped at various times by addition of EDTA to 0.3 M final concentration. The product, 11-mer RNA, was separated from the substrate, 10-mer RNA, by electrophoresis through a denaturing, 23% polyacrylamide gel. Substrate and product were visualized by using a Phosphor-Imager and quantitated by using the ImageQuant software (Molecular Dynamics) (Fig. 2B).
The concentration of 11-nt product formed was plotted as a function of time and fit to a single exponential equation (Equation 1), yielding the observed rate constant for AMP incorporation templated by U (Fig. 2C). The observed rate constants were then plotted as a function of ATP concentration, and the data were fit to a hyperbola (Equation 2), yielding an apparent dissociation constant for ATP, Kd,app, of 530 ± 120 μM, and a maximal rate constant for AMP incorporation, kpol, of 8 ± 1 s−1 (Fig. 2D and Table 2). The ability to fit the data to a hyperbola is in good agreement with the minimal kinetic mechanism proposed for WT 3Dpol (Scheme 1) (26). The Kd,app value for ATP was increased by 4-fold, and the maximal rate constant for AMP incorporation by Glu-297 3Dpol was reduced by 11-fold relative to WT 3Dpol (Table 2).
SCHEME 1. Minimal kinetic mechanism for 3Dpol-catalyzed nucleotide incorporation.

ERn, 3Dpol-sym/sub substrate complex; NTP, nucleotide; ERnNTP, catalytically competent ternary complex; and ERn+1, 3Dpol-sym/sub product complex.
Next, we evaluated the kinetics of dAMP incorporation for Glu-297 3Dpol. The Kd,app and kpol values for dAMP incorporation into S/S-U were 1700 ± 400 μM and 0.4 ± 0.02 s−1, respectively (Table 2). Relative to WT 3Dpol, these values reflect a 6-fold increase in the Kd,app value for dATP and a 2-fold decrease in the kpol value for dAMP incorporation. These data are consistent with Glu-297 3Dpol selecting against dAMP incorporation better than WT 3Dpol (Table 2). To show that this observation was not unique to dAMP incorporation, we evaluated dUMP incorporation into S/S-A. For Glu-297 3Dpol, the Kd,app value for dUTP increased by 6-fold, and the kpol value decreased by 3-fold relative to WT 3Dpol (Table 2), consistent again with this derivative selecting against dNMP incorporation better than WT 3Dpol. If we define the overall efficiency of nucleotide incorporation as kpol/Kd,app, then the efficiency of dNMP incorporation relative to the efficiency of AMP incorporation for Glu-297 3Dpol was essentially the same as WT 3Dpol (Table 2).
Glu-297 3Dpol exhibited a 14- to 35-fold reduction in the efficiency of rNMP incorporation relative to WT 3Dpol (Table 2). Unexpectedly, the fidelity of base selection (GMP incorporation into S/S-U) by Glu-297 3Dpol was decreased by 10-fold relative to WT 3Dpol. This is the first RdRp derivative with a mutator phenotype.
Glu-297 PV Is Quasi-infectious
The rate constant for rNMP incorporation by Glu-297 3Dpol was decreased by 4- to 11-fold relative to WT 3Dpol. Therefore, this mutant virus should reveal the impact, if any, of replication speed and fidelity on virus multiplication and fitness. Glu-297-encoding changes were engineered into the infectious cDNA for PV. RNA was produced by in vitro transcription, and the infectivity of the RNA was evaluated by infectious center assay at both 37 °C and 32 °C (14). Glu-297 PV RNA did not produce virus at either temperature (Fig. 3A). Under the same conditions, WT PV RNA was able to establish a productive infection (Fig. 3A).
FIGURE 3. Glu-297 PV is quasi-infectious.

A, PV genomic RNA encoding Glu-297 3Dpol is not infectious. An infectious center assay (see “Experimental Procedures”) was performed to evaluate the infectivity of WT or Glu-297 PV RNA after electroporation into HeLa cells. Virus was not recovered from cells containing Glu-297 PV at 37 °C or 32 °C. B, isolation of Gly-297 PV as a pseudo-revertant of Glu-297. After transfection of Glu-297 PV RNA into HeLa cells, a fraction of the media was transferred to a fresh monolayer every 2 days for 6 days at which point cytopathic effect was observed. Therefore, Glu-297 PV is quasi-infectious. Shown is the electropherogram and sequence for cDNA prepared from viral RNA by using reverse transcription-PCR. The underlined codon is for position 297 and encodes a Gly. The original sequence was GAA.
If the change of Asn-297 to Glu is indeed lethal, then viable virus will never be isolated. However, if this change permits some low level of replication (quasi-infectious), then viable virus may be recovered after serial passage. In this experiment, HeLa cells were electroporated with Glu-297 PV RNA and plated onto a fresh monolayer of HeLa cells. Plates were incubated at 37 °C. Every 2 days, a fraction of the media was used to infect a fresh monolayer. This process was repeated reiteratively until the monolayer was killed in 2 days. This took three serial passages. The 3Dpol gene from this virus was amplified by using reverse transcription-PCR and sequenced. A single nucleotide substitution was observed that converted the GAA codon encoding Glu-297 to a GGA codon encoding Gly, a virus isolated by us previously (Fig. 3B) (27). Together, these data permit us to conclude that Glu-297 PV is quasi-infectious.
Glu-297 PV Is Severely Impaired for RNA Synthesis in Cells
The polymerase domain (3D) has functions in the context of precursors that are unrelated to RNA elongation. For example, the 3CD precursor is a protease that processes the polyprotein into “mature” forms required for virus multiplication (7, 24, 28, 29). 3CD is an RNA-binding protein that binds to the cis-acting element required for initiation of genome replication, possibly recruiting 3Dpol to this location (30–32). Finally, 3CD appears to function after RNA synthesis as well in a step related to virion maturation that does not require 3CD protease activity (33). To determine to what extent RNA synthesis was attenuated for Glu-297 PV, mutations encoding this change were engineered into the PV subgenomic replicon. This replicon consists of a PV genome in which capsid-coding sequence has been replaced by a luciferase reporter gene (Fig. 4A), permitting RNA synthesis to be monitored indirectly by measuring luciferase activity (14).
FIGURE 4. Replication of Glu-297 PV in cells is as debilitated as a PV encoding a catalytically inactive polymerase.

A, PV subgenomic replicon (14). This replicon has the capsid-coding sequence replaced by a luciferase reporter gene. After transfection of the RNA into HeLa cells, translation of the open reading frame produces a luciferase-P2-P3 polyprotein from which luciferase is released by the 2A protease activity encoded by the P2 region of the genome. In the absence of replication, translation occurs, yielding a 1–2 log increase in luciferase activity. Mutations that affect translation or RNA stability would be scored in this manner. Replication is monitored indirectly by luciferase activity that accumulates to levels higher than observed for translation. B, Glu-297 PV replicon translates but does not replicate. In this experiment the Glu-297 (●) PV replicon was compared with WT (■) and GAA (○) PV replicons at 37 °C. The GAA replicon encodes an inactive polymerase and serves as a control for translation and RNA stability in the absence of RNA synthesis. This experiment was performed three times; a representative experiment is shown.
WT and Glu-297 PV replicons were evaluated at 37 and 32 °C (Fig. 4B and data not shown). Exponential replication of the WT PV replicon was observed for 5 h after transfection, leading to a >105-fold increase in luciferase activity. In contrast, only a 2-log increase in luciferase activity was observed for the Glu-297 PV replicon. This level of luciferase activity was also observed for the GAA PV replicon that encodes a polymerase completely lacking polymerase activity due to a change in the active site GDD motif to GAA (34, 35). Luciferase activity observed for the GAA PV replicon derives from translation of the transfected RNA. Such a substantial replication defect was not expected for Glu-297 PV given the significant polymerase activity associated with this Glu-297 3Dpol (Tables 2 and 3).
The Product Yield and Processivity of VPg-primed RNA Synthesis by Glu-297 3Dpol Is Severely Impaired
Another aspect of PV genome replication in which 3Dpol plays a crucial role is VPg uridylylation. VPg is a 22-amino acid-long peptide that is found covalently attached to the 5′-end of PV RNA. VPg or some precursor thereof is used to prime RNA synthesis (35–38). Uridylylation of VPg leads to production of VPg-pUpU, which serves as a primer for viral RNA synthesis (31, 36, 39). This reaction is catalyzed by 3Dpol, templated by an adenylate residue in the 2C-cre (cis-replicating element) by using a slide-back mechanism, and is stimulated by protein 3CD (31, 40, 41). This reaction can be mimicked in vitro as shown in Scheme 2.
SCHEME 2. VPg uridylylation reaction.

Incorporation of two UMP residues into VPg by 3Dpol is templated by a single AMP residue in the 2C-cre RNA using a slide-back mechanism (40). The reaction is capable of processive synthesis of VPg-pUpU but under sub-optimal conditions VPg-pU also accumulates (31, 41). The reaction is stimulated by 3CD, presumably as a recruitment/retention factor for 3Dpol (31, 35, 41).
For WT 3Dpol, both VPg-pU and VPg-pUpU products are observed at UTP concentrations <5 μM. Above 5 μM UTP, however, VPg-pUpU is the primary product (Fig. 5A). For Glu-297 3Dpol, VPg-pU is the primary product at low UTP concentrations, and both VPg-pU and VPg-pUpU are still equimolar at concentrations of UTP as high as 300 μM (Fig. 5B). Quantitative evaluation of the UTP concentration dependence of UMP incorporation into VPg showed that the maximal value of UMP incorporated by Glu-297 3Dpol was reduced by approximately half relative to WT 3Dpol (Fig. 5, C and D). Moreover, the concentration of UTP required for half-maximal UMP incorporation (K0.5) by Glu-297 3Dpol was increased by 14-fold relative to WT 3Dpol (Fig. 5, C and D). These data suggest that the capacity for the Glu-297 3Dpol uridylylation complex to bind UTP is compromised significantly relative to WT 3Dpol, whereas the incorporation rate of Glu-297 3Dpol uridylylation complex was much less affected. These observations are different from those made for the Glu-297 3Dpol RNA elongation complex where the incorporation rate constant (kpol) was affected more than UTP binding (Kd,app) (Table 2).
FIGURE 5. VPg-primed RNA synthesis by Glu-297 3Dpol is impaired.

A and B, VPg uridylylation assay. The reaction described in Scheme 2 was performed as described under “Experimental Procedures.” Tyr-3 of the VPg peptide is used as the nucleophile for nucleotidyl transfer of UMP templated by the 2C-cre to yield VPg-pU, which undergoes another round of nucleotidyl transfer to yield VPg-pUpU. Uridylylated products are separated from labeled UTP by electrophoresis through a 15% Tris-Tricine gel and visualized by phosphorimaging. WT (A) and Glu-297 (B) 3Dpol uridylylation activity was monitored as a function of UTP concentration. Both VPg-pU and VPg-pUpU were produced by both enzymes. C and D, decreased affinity of Glu-297 3Dpol for UTP relative to WT 3Dpol. Total product formed was plotted as a function of UTP concentration and fit to a hyperbola (Equation 2, solid line) for WT (C) and Glu-297 (D) 3Dpol. The K0.5 values for UTP were 3.9 ± 1.2 μM for WT 3Dpol and 56 ± 11 μM for Glu-297 3Dpol. The maximum values for UMP incorporated were 5.0 ± 0.5 μM UMP incorporated for WT 3Dpol and 3 ± 0.2 μM UMP incorporated for Glu-297 3Dpol. E and F, decreased processivity for Glu-297 3Dpol. Processivity is defined as the fraction of product that appears as VPg-pUpU relative to all products. Processivity is plotted as a function of UTP concentration and fit to a hyperbola (Equation 2, solid line) for WT (E) and Glu-297 (F) 3Dpol. The K0.5 values for UTP were 0.8 ± 0.2 μM for WT 3Dpol and 5.7 ± 1.6 μM for Glu-297 3Dpol. The maximum values for processivity were 1.0 ± 0.02 for WT 3Dpol and 0.6 ± 0.04 μM UMP for Glu-297 3Dpol.
The 5′-ends of PV plus and minus strands contain VPg-pUpU, likely the result VPg-pUpU produced at the 2C-cre being used as the primer for extension from the 3′-end of PV plus and minus strand RNAs (36, 37, 39, 42). Although WT 3Dpol produced as much VPg-pUpU product at saturating concentrations of UTP (Fig. 5E), Glu-297 3Dpol produced half as much VPg-pUpU product, even at apparently saturating concentrations of UTP (Fig. 5F). Moreover, the K0.5 value for UTP for production of VPg-pUpU by Glu-297 3Dpol was still 7-fold higher than that observed for WT 3Dpol. We conclude that both the overall product yield and the processivity of the Glu-297 3Dpol uridylylation complex is reduced relative to WT 3Dpol and that, when coupled with the reduced elongation rate of Glu-297 3Dpol, provides a biochemical explanation for the severe RNA synthesis defect observed biologically.
The Processivity of RNA-primed RNA Synthesis by Glu-297 3Dpol Appears Normal
The finding that processive VPg-primed RNA synthesis was reduced for Glu-297 3Dpol suggested the possibility that processive RNA-primed RNA synthesis might also be reduced, contributing to the lack of RNA synthesis observed in cells. Processive RNA synthesis was evaluated by using S/S-U, the same RNA substrate utilized for single-nucleotide-addition experiments, in the presence of all four ribonucleotides (125 μM each). Upon rapid mixing of Glu-297 3Dpol-S/S-U complex with all four nucleotides, formation of 11-, 12-, 13-, and 14-mer RNA products were observed (Fig. 6). The net rate constants for sequential incorporation of all four nucleotides were comparable, and no strong stops (Fig. 6A) or long lags (inset to Fig. 6B) were observed. We conclude that any change in processivity of RNA-primed RNA synthesis by Glu-297 3Dpol is below the limit of detection of this assay. Because the rate constant for dissociation of 3Dpol from S/S-U is <0.0001 s−1 (12), an incorporation rate constant in the 0.0001–0.001 s−1 regime would be required to observe a difference in processivity by using this assay.
FIGURE 6. Processive RNA-primed RNA synthesis by Glu-297 3Dpol is normal.

Nucleotide-addition experiments were performed as described in the legend to Fig. 2 in the presence of all four nucleotides. The phosphorimaging of the gel (A) and quantification thereof (B) are shown. The kinetics of formation and disappearance of 11-mer (○), 12-mer (□), 13-mer (●), and 14-mer (■) were simulated (solid lines) to net rate constants of 1 s−1, 4 s−1, 7 s−1, and 2 s−1 for formation of each of the individual products. The inset is an enlargement of the data between 0 and 2.5 s.
Efficient and Processive Uridylylation of VPg by Gly-297 3Dpol
Serial passage of Glu-297 PV uncovered a pseudo-revertant virus, Gly-297 PV, that we isolated previously (27). This virus exhibits delayed growth kinetics but reaches titers within one log of wild type (27). Interestingly, growth of this virus could be stimulated at least 5-fold by growth in the presence of Mn2+ (27). RNA-primed RNA synthesis by Gly-297 3Dpol was similar to WT 3Dpol; VPg-primed RNA synthesis was not evaluated (27). If the reduced yield of uridylylated VPg products and/or processivity of VPg uridylylation by Glu-297 3Dpol was indeed the reason that Glu-297 PV did not replicate, then Gly-297 3Dpol should exhibit enhanced production of uridylylated VPg products, processivity, or both. Indeed, VPg uridylylation by Gly-297 3Dpol was essentially indistinguishable from WT 3Dpol (Fig. 7).
FIGURE 7. VPg-primed RNA synthesis by Gly-297 3Dpol is normal.
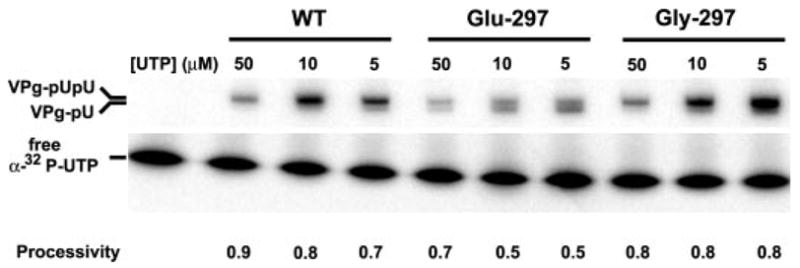
The yield of uridylylated VPg products and processivity of VPg uridylylation by Gly-297 3Dpol was compared with that of the WT and Glu-297 enzymes as described in the legend to Fig. 5. Gly-297 3Dpol uridylylation activity is qualitatively and quantitatively indistinguishable from WT 3Dpol. The standard error on the values for processivity was no greater than 10%.
Attempts to Rescue Glu-297 3Dpol RNA-primed Elongation Rate by Rational Design
The initial observation that the rate constant for AMP incorporation into S/S-U by Glu-297 3Dpol was 10-fold slower than WT 3Dpol was first interpreted to mean that the context into which Glu-297 was placed was suboptimal. Structure/sequence comparisons identified two other changes in the ribose-binding pocket between RdRps from RNA viruses of eukaryotes and prokaryotes. These changes were at positions 288 and 290 using the PV 3Dpol numbering (Fig. 1). We changed Ser-288 to methionine and Cys-290 to asparagine individually and in combination in the context of Glu-297 3Dpol. None of these derivatives rescued the activity of Glu-297 3Dpol, because all changes yielded derivatives that retained <3% of the poly(rU) polymerase activity observed for WT 3Dpol (Table 3).
DISCUSSION
Previous studies of the RdRp from PV, 3Dpol, established a role for Asn-297 in ribonucleotide selection (14). By changing Asn-297 to Ala, a 3Dpol derivative was created that was incapable of distinguishing an rNTP with correct base pairing capacity from a 2′-dNTP with the same capacity (14). A structural model for the 3Dpol elongation complex has been developed based on homology modeling, because a crystal structure for this complex is still not available (14). Many of the features of this model are consistent with biochemical data, in particular a hydrogen bonding interaction between Asn-297 and the 2′-hydroxyl of the rNTP (Fig. 1A) (14). Interestingly, PV containing substitutions at position 297 of 3Dpol, including Ala, Gln, and Asp, were all inviable, despite the fact that all of these 3Dpol derivatives exhibited demonstrable levels of RNA-primed elongation activity (14). Consistent with the essential nature of this residue was the finding that all animal and plant RNA virus polymerases have an Asn at this position (Fig. 1B). Therefore, it was quite surprising to find that RNA phage polymerases have a Glu at this position (Fig. 1B).
We hypothesized that the cytosolic availability of 2′-dNTPs in prokaryotes may account for this difference. Perhaps a Glu at this position would confer upon the polymerase the capacity to be more stringent in its selection against 2′-dNTP utilization and perhaps even permit the virus to grow. A Glu-297 3Dpol was indeed active, and a decrease in 2′-dNTP affinity was noted. However, a decrease in efficiency of 2′-dNMP incorporation relative to WT 3Dpol was not observed (Table 2). Glu-297 3Dpol exhibited two differences relative to WT 3Dpol in the RNA-primed synthesis of RNA. The observed rate constant for correct rNMP incorporation was decreased by 4- to 11-fold to the 10–100 s−1 regime (Table 2). The efficiency of GMP mis-incorporation was increased by 14-fold, corresponding to a mutation frequency of 1 × 10−3.
Recent studies of the kinetics of nucleotide incorporation by the phage Qβ RdRp have shown that the average rate constant for nucleotide incorporation by this enzyme is ~10 s−1 (43), with projected maximal rates being on the order of 30 s−1.3 In addition, Drake (44) has shown that the mutation rate of Qβ in cells is 1.5 × 10−3, 10- to 100-fold higher than measured for PV in the same study. Our data would suggest that the presence of Glu instead of Asn in the ribose-binding pocket contributes to both the reduction in replication speed and fidelity.
Do RNA bacteriophage require a slower replication rate, greater population diversity, both, or neither? Given the substantially faster doubling time of prokaryotic cells relative to mammalian cells, it appears unlikely that a slower replication rate for RNA bacteriophage would increase the yield of progeny virus per cell at a given time. Unlike mammalian host cells, prokaryotic host cells exhibit substantial phenotypic diversity, for example the presence of phage-resistant cells (45). It is possible that the increased mutation frequency of the RNA phage permits phage evolution to keep pace with host cell evolution, thus precluding virus extinction caused by the selection of phage-resistant cells. Because viral population dynamics theory predicts a log-linear relationship between virus titer and mutation frequency, RNA phage will be able to absorb the increased mutational load and avoid extinction catastrophe by growing to high titer (46).
To our knowledge, Glu-297 3Dpol represents the first RdRp derivative with a mutator phenotype. As discussed below, studying the biological consequences of decreased fidelity is not possible in the PV system. However, construction of this derivative in the context of other animal RNA viruses may be possible. Similarly, construction of a Qβ mutant encoding an RdRp with an Asn in the ribose-binding pocket instead of Glu should alter phage evolutionary capacity relative to the host, providing important insight into the role of parasite/pathogen-evolving ability in host-parasite co-evolution.
Our previous biochemical and biological studies of position 297 of PV 3Dpol led us to conclude that subtle (2.5- to 5-fold) decreases in the elongation rate measured in vitro cause substantial RNA replication defects in vivo due to tight coupling between translation and replication (14). Since these initial studies were published, there have been two advances in the field. First, an in vitro reaction that mimics VPg-primed initiation of PV genome replication has been developed (Scheme 2) (31, 47). Second, it has become clear that not all 3Dpol alleles that confer a reduced elongation rate in vitro confer a substantial defect to RNA replication in vivo (48).
Evaluation of Glu-297 3Dpol in the VPg uridylylation reaction revealed that this derivative had a substantially reduced affinity for UTP (14-fold) relative to WT 3Dpol (Fig. 5, compare C to D), without a significant difference in the observed rate of uridylylation, perhaps a reflection of the fact that nucleotidyl transfer is not rate-limiting in this reaction. In addition, there was a decrease in uridylylation processivity in that only approximately half of the Glu-297 3Dpol complexes yielded VPg-pUpU even under apparently saturating concentrations of UTP (Fig. 5F). In the Glu-297 3Dpol elongation complex, affinity for UTP was only affected minimally (Table 2), elongation rate was decreased substantially (10-fold) (Table 2), and processivity was not affected at all (Fig. 6).
The opposite effects of the Glu-297 substitution on the two different reactions, VPg uridylylation and RNA elongation, suggest differences in the structural role and biochemical function of Asn-297 in these reactions. The structure of the foot-and-mouth disease virus 3Dpol-VPg-pU complex has been solved (49). Based upon this structure, Asn-297 of PV 3Dpol would interact with the 3′-OH in the uridylylation complex (Fig. 8) but with the 2′-OH in the elongation complex (Fig. 1A). The finding, that relatively modest reductions in yield of uridylylated VPg (10- to 15-fold) and processivity (2-fold) have such dramatic effects on genome replication and virus production, suggests that drugs targeting initiation may be more efficacious than those targeting RNA elongation.
FIGURE 8. Asn-297 interacts with the 3′-OH of the VPg-pU product.

The model for 3Dpol-VPg-pU complex shown is that published for the FMDV enzyme (PDB entry 2F8E). PV 3Dpol numbering has been employed to preclude confusion. In this complex, Asn-297 hydrogen bonds to the 3′-OH instead of the 2′-OH observed for the RNA-primed elongation complex shown in Fig. 1B (the structural model shown in Fig. 1 is based on homology modeling, because a crystal structure for this complex is still not available for any RdRp (14)). Biochemical/biological validation of this complex has not been performed.
The inability of Glu-297 3Dpol to support both protein-primed RNA synthesis and RNA-primed RNA synthesis may suggest that the pressure to be more selective against dNTP utilization also limits the nature of the initiation mechanisms employed by RNA phages. For example, both plant and animal RNA viruses employ both protein-primed (VPg in Fig. 1B) and de novo (Cap and ppp in Fig. 1B) mechanisms of initiation; however, RNA phages employ only a de novo mechanism (Fig. 1B).
Glu-297 PV was quasi-infectious as a pseudo-revertant PV (Gly-297 PV) was isolated (Fig. 3B). This virus was also isolated in the past and shown to be an Mn2+-stimulated PV mutant (27). Gly-297 3Dpol restored the defects to uridylylation observed for Glu-297 3Dpol, suggesting that the uridylylation defect is the cause of the severe RNA synthesis defect in cells observed here and with other 297 alleles (14). Why do Gly-297 3Dpol and PV function (27) and Ala-297 3Dpol and PV not function (14)? We propose that a water molecule can be accommodated in the pocket formed by Gly-297 but not in the pocket formed by Ala-297, thus permitting hydrogen bonding as observed for Asn-297. If our interpretation is correct, then the temperature sensitivity of RNA synthesis observed for Asp-297 PV (14) and the Mn2+ dependence of RNA synthesis observed for Gly-239 and Ala-297 PV (27) are a result of an initiation (2C-cre-dependent VPg uridylylation) defect. To our knowledge, these PV mutants represent the first polymerase mutants with a specific defect to VPg uridylylation for any picornavirus or any RNA virus that employs a protein-primed mechanism of initiation. These mutants will be very useful for studying initiation and clarifying some of the controversies surrounding the role of the 2C-cre-templated uridylylation in picornavirus minus-strand and plus-strand syntheses (37, 50).
Acknowledgments
We thank Dr. Harsh B. Pathak for purification of the 3CD and 2C-cre RNA.
Footnotes
This work was supported by NIAID, National Institutes of Health Grant AI053531 (to C. E. C.).
The abbreviations used are: RdRp, RNA-dependent RNA polymerase; PV, poliovirus; WT, wild type; nt, nucleotide(s); cre, cis-replicating element; Tricine, N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine; S/S, sym/sub; rNMP, ribonucleotide monophosphate; dNMP, deoxyribonucleotide monophosphate.
K. Hosada, personal communication.
References
- 1.Hayden FG. Curr Opin Infect Dis. 2006;19:169–178. doi: 10.1097/01.qco.0000216628.51563.b1. [DOI] [PubMed] [Google Scholar]
- 2.Weiss SR, Navas-Martin S. Microbiol Mol Biol Rev. 2005;69:635–664. doi: 10.1128/MMBR.69.4.635-664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim WR. Hepatology. 2002;36(Suppl 1):S30–S34. doi: 10.1053/jhep.2002.36791. [DOI] [PubMed] [Google Scholar]
- 4.Behrens SE, Tomei L, De Francesco R. EMBO J. 1996;15:12–22. [PMC free article] [PubMed] [Google Scholar]
- 5.Hayden FG. Adv Exp Med Biol. 1999;458:55–67. doi: 10.1007/978-1-4615-4743-3_6. [DOI] [PubMed] [Google Scholar]
- 6.Kitamura N, Semler BL, Rothberg PG, Larsen GR, Adler CJ, Dorner AJ, Emini EA, Hanecak R, Lee JJ, van der Werf S, Anderson CW, Wimmer E. Nature. 1981;291:547–553. doi: 10.1038/291547a0. [DOI] [PubMed] [Google Scholar]
- 7.Semler BL, Wimmer E, editors. Molecular Biology of Picornaviruses. ASM Press; Washington, DC: 2002. [Google Scholar]
- 8.Vignuzzi M, Stone JK, Andino R. Virus Res. 2005;107:173–181. doi: 10.1016/j.virusres.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 9.Harki DA, Graci JD, Korneeva VS, Ghosh SK, Hong Z, Cameron CE, Peterson BR. Biochemistry. 2002;41:9026–9033. doi: 10.1021/bi026120w. [DOI] [PubMed] [Google Scholar]
- 10.Harki DA, Graci JD, Galarraga JE, Chain WJ, Cameron CE, Peterson BR. J Med Chem. 2006;49:6166–6169. doi: 10.1021/JM060872x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jochmans D, Deval J, Kesteleyn B, Van Marck H, Bettens E, De Baere I, Dehertogh P, Ivens T, Van Ginderen M, Van Schoubroeck B, Ehteshami M, Wigerinck P, Gotte M, Hertogs K. J Virol. 2006;80:12283–12292. doi: 10.1128/JVI.00889-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arnold JJ, Cameron CE. J Biol Chem. 2000;275:5329–5336. doi: 10.1074/jbc.275.8.5329. [DOI] [PubMed] [Google Scholar]
- 13.Thompson AA, Peersen OB. EMBO J. 2004;23:3462–3471. doi: 10.1038/sj.emboj.7600357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gohara DW, Crotty S, Arnold JJ, Yoder JD, Andino R, Cameron CE. J Biol Chem. 2000;275:25523–25532. doi: 10.1074/jbc.M002671200. [DOI] [PubMed] [Google Scholar]
- 15.Gohara DW, Arnold JJ, Cameron CE. Biochemistry. 2004;43:5149–5158. doi: 10.1021/bi035429s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crotty S, Cameron CE, Andino R. Proc Natl Acad Sci U S A. 2001;98:6895–6900. doi: 10.1073/pnas.111085598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castro C, Arnold JJ, Cameron CE. Virus Res. 2005;107:141–149. doi: 10.1016/j.virusres.2004.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crotty S, Maag D, Arnold JJ, Zhong W, Lau JY, Hong Z, Andino R, Cameron CE. Nat Med. 2000;6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- 19.Traut TW. Mol Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 20.Lee YD, Elledge SJ. Genes Dev. 2006;20:334–344. doi: 10.1101/gad.1380506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao R, Zhang Z, An X, Bucci B, Perlstein DL, Stubbe J, Huang M. Proc Natl Acad Sci U S A. 2003;100:6628–6633. doi: 10.1073/pnas.1131932100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.An X, Zhang Z, Yang K, Huang M. Genetics. 2006;173:63–73. doi: 10.1534/genetics.105.055236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gohara DW, Ha CS, Kumar S, Ghosh B, Arnold JJ, Wisniewski TJ, Cameron CE. Protein Expr Purif. 1999;17:128–138. doi: 10.1006/prep.1999.1100. [DOI] [PubMed] [Google Scholar]
- 24.Pathak HB, Ghosh SK, Roberts AW, Sharma SD, Yoder JD, Arnold JJ, Gohara DW, Barton DJ, Paul AV, Cameron CE. J Biol Chem. 2002;277:31551–31562. doi: 10.1074/jbc.M204408200. [DOI] [PubMed] [Google Scholar]
- 25.Herold J, Andino R. J Virol. 2000;74:6394–6400. doi: 10.1128/jvi.74.14.6394-6400.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arnold JJ, Cameron CE. Biochemistry. 2004;43:5126–5137. doi: 10.1021/bi035212y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crotty S, Gohara D, Gilligan DK, Karelsky S, Cameron CE, Andino R. J Virol. 2003;77:5378–5388. doi: 10.1128/JVI.77.9.5378-5388.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parsley TB, Cornell CT, Semler BL. J Biol Chem. 1999;274:12867–12876. doi: 10.1074/jbc.274.18.12867. [DOI] [PubMed] [Google Scholar]
- 29.Cornell CT, Semler BL. Virology. 2002;298:200–213. doi: 10.1006/viro.2002.1481. [DOI] [PubMed] [Google Scholar]
- 30.Xiang W, Harris KS, Alexander L, Wimmer E. J Virol. 1995;69:3658–3667. doi: 10.1128/jvi.69.6.3658-3667.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paul AV, Rieder E, Kim DW, van Boom JH, Wimmer E. J Virol. 2000;74:10359–10370. doi: 10.1128/jvi.74.22.10359-10370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Y, Rijnbrand R, Watowich S, Lemon SM. J Biol Chem. 2004;279:12659–12667. doi: 10.1074/jbc.M312992200. [DOI] [PubMed] [Google Scholar]
- 33.Franco D, Pathak HB, Cameron CE, Rombaut B, Wimmer E, Paul AV. Virol J. 2005;2:86. doi: 10.1186/1743-422X-2-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lyle JM, Clewell A, Richmond K, Richards OC, Hope DA, Schultz SC, Kirkegaard K. J Biol Chem. 2002;277:16324–16331. doi: 10.1074/jbc.M112429200. [DOI] [PubMed] [Google Scholar]
- 35.Richards OC, Spagnolo JF, Lyle JM, Vleck SE, Kuchta RD, Kirkegaard K. J Virol. 2006;80:7405–7415. doi: 10.1128/JVI.02533-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ambros V, Baltimore D. J Biol Chem. 1978;253:5263–5266. [PubMed] [Google Scholar]
- 37.Murray KE, Barton DJ. J Virol. 2003;77:4739–4750. doi: 10.1128/JVI.77.8.4739-4750.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tellez AB, Crowder S, Spagnolo JF, Thompson AA, Peersen OB, Brutlag DL, Kirkegaard K. J Mol Biol. 2006;357:665–675. doi: 10.1016/j.jmb.2005.12.044. [DOI] [PubMed] [Google Scholar]
- 39.Crawford NM, Baltimore D. Proc Natl Acad Sci U S A. 1983;80:7452–7455. doi: 10.1073/pnas.80.24.7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paul AV, Yin J, Mugavero J, Rieder E, Liu Y, Wimmer E. J Biol Chem. 2003;278:43951–43960. doi: 10.1074/jbc.M307441200. [DOI] [PubMed] [Google Scholar]
- 41.Rieder E, Paul AV, Kim DW, van Boom JH, Wimmer E. J Virol. 2000;74:10371–10380. doi: 10.1128/jvi.74.22.10371-10380.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lyons T, Murray KE, Roberts AW, Barton DJ. J Virol. 2001;75:10696–10708. doi: 10.1128/JVI.75.22.10696-10708.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakaishi T, Iio K, Yamamoto K, Urabe I, Yomo T. J Biosci Bioeng. 2002;93:322–327. doi: 10.1263/jbb.93.322. [DOI] [PubMed] [Google Scholar]
- 44.Drake JW. Proc Natl Acad Sci U S A. 1993;90:4171–4175. doi: 10.1073/pnas.90.9.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jain R, Knorr AL, Bernacki J, Srivastava R. Biotechnol Prog. 2006;22:1650–1658. doi: 10.1021/bp060161s. [DOI] [PubMed] [Google Scholar]
- 46.Bull JJ, Sanjuan R, Wilke CO. J Virol. 2007;81:2930–2939. doi: 10.1128/JVI.01624-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paul AV, van Boom JH, Filippov D, Wimmer E. Nature. 1998;393:280–284. doi: 10.1038/30529. [DOI] [PubMed] [Google Scholar]
- 48.Arnold JJ, Vignuzzi M, Stone JK, Andino R, Cameron CE. J Biol Chem. 2005;280:36417–36428. doi: 10.1074/jbc.M503444200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferrer-Orta C, Arias A, Agudo R, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N. EMBO J. 2006;25:880–888. doi: 10.1038/sj.emboj.7600971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharma N, O’Donnell BJ, Flanegan JB. J Virol. 2005;79:3565–3577. doi: 10.1128/JVI.79.6.3565-3577.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koonin EV. J Gen Virol. 1991;72:2197–2206. doi: 10.1099/0022-1317-72-9-2197. [DOI] [PubMed] [Google Scholar]
- 52.Buck KW. Adv Virus Res. 1996;47:159–251. doi: 10.1016/S0065-3527(08)60736-8. [DOI] [PMC free article] [PubMed] [Google Scholar]