

Abstract
Objective
Pathogenic antiphospholipid antibodies (aPL) bind the self antigen N-terminal domain (domain I) of β2-glycoprotein I (β2GPI), with residues G40–R43 being important. However, peptides homologous to other regions of domain I have also been shown to bind aPL. Furthermore, there are no published reports of the effects of altering R39, which has greater surface exposure than the G40–R43 residues.
Methods
We used a novel, efficient method of production and purification of human domain I by Escherichia coli to create multiple mutants of domain I. These domain I mutants were then screened for binding to a range of polyclonal IgG purified from patients with antiphospholipid syndrome, using both solid-phase and fluid-phase assays.
Results
E coli–expressed purified domain I selectively bound IgG derived from patients with antiphospholipid syndrome. In region R39–R43, the R39S mutation had the greatest effect in terms of reducing binding to a panel of aPL in the fluid phase (mean ± SD inhibition 14 ± 18.5% versus 44.1 ± 31.7% for G40E and 62.9 ± 25.7% for wild-type domain I). Conversely, altering both D8 and D9 to S8 and G9, respectively, had the effect of enhancing binding to aPL in the fluid phase. Adding the remainder of the domain I–II interlinker resulted in enhanced binding over wild-type in the solid phase but not the fluid phase.
Conclusion
The binding of aPL to β2GPI domain I is complex and likely to involve discontinuous epitopes that include R39 in addition to G40–R43, the domain I–II interlinker, and possibly D8 and D9. Domain I variants with enhanced binding to aPL compared with wild-type domain I may aid in the development of novel therapies.
The antiphospholipid syndrome (APS) is characterized by the clinical syndrome of arterial and venous thrombosis and/or recurrent pregnancy morbidity in association with the presence of antiphospholipid antibodies (aPL) (1). Antiphospholipid antibodies derived from patients with APS have been shown to promote a thrombophilic phenotype, as assessed using animal models of thrombosis and pregnancy morbidity and in vitro bioassays of human endothelial cell function and human platelet cell activation (2-7). In many cases, aPL derived from patents with APS bind phospholipids via the protein cofactor β2-glycoprotein I (β2GPI) (8-10). This protein consists of 5 domains. Domains I–IV each consist of ∼60 amino acids, while domain V is aberrant, consisting of 82 amino acids due to a 6-residue insertion and a 19-residue C-terminal extension crosslinked by an additional disulfide bond. Domain V is responsible for anchoring β2GPI to anionic phospholipids (11,12). Recent evidence also suggests that β2GPI binds the platelet receptor GPIbα-IX-V complex via domains II–V (7).
Given that β2GPI is the major antigenic target of pathogenic aPL, understanding the nature of the immunodominant binding epitopes located in this protein at the molecular level may aid in the development of therapies targeted at blocking this interaction. The bulk of the evidence favors aPL binding immunodominant epitopes located within the N-terminal (domain I) (13-15). The ability of anti-β2GPI antibodies purified from patients with autoimmune disease to bind domain I of β2GPI has been shown to be strongly correlated with the occurrence of thrombosis in those patients (16). Studies on expressed mutants of whole β2GPI with incorporated single-point substitution mutations within domain I identified the residues G40 and R43 as being important in conferring binding to aPL (14,17). De Laat et al (16) identified a population of anti-β2GPI antibodies derived from patients with thrombosis that bind domain I exclusively. This binding was considerably reduced by the introduction of mutations G40E and R43G. Clearly, epitope G40–R43 within domain I is important, but several questions regarding the nature of the domain I–anti-β2GPI antibody interaction remain unanswered.
Adjacent to the G40–R43 region is residue R39 (18). By studying the published crystal structure of β2GPI (19,20), complemented by computer modeling, it is apparent that the R39 residue has a considerably greater percentage (∼50%) of its surface area exposed for potential interaction with antibody than either G40 or R43. There are no published data regarding how altering R39 affects binding to aPL. Therefore, mutation analyses within this region need to be expanded to include R39. However, not all studies have identified the G40–R43 region as being the dominant epitope for binding to aPL. A peptide bearing homology to the N-terminal half of the domain I–II interlinker region (peptide A) has been shown to bind aPL derived from patients with APS and to inhibit aPL-induced thrombosis in mice (21,22). Peptide A bears no homology to region G40–R43. Vlachoyiannopoulos et al (23) demonstrated that a peptide with homology to a portion of the CD40 ligand and also to the P7–S13 region of the N-terminal portion of domain I was also capable of binding anti-β2GPI antibodies from patients with APS.
These observations, in combination with the knowledge that denatured β2GPI does not bind aPL (24), suggest that pathogenic aPL bind discontinuous epitopes within domain I, with region G40–R43 being crucial but not the sole determinant of binding to the majority of aPL. Giles and associates (25) from our unit demonstrated that specific R residues located within antigen-binding sites on monoclonal pathogenic aPL are important in determining binding to cardiolipin, whole β2GPI, and several other clinically relevant antigens (26). It is possible that these positively charged R residues on aPL may be interacting with anionic residues on domain I of β2GPI. Interestingly, a pair of negatively charged aspartic acid residues at positions 8 and 9 (D8 and D9) (18) are surface exposed (19,20) and are part of the P7–S13 region homologous to the peptide described by Vlachoyiannopoulos et al (23).
We believe that the interaction of aPL with domain I is likely to be complex and to involve multiple epitopes. A convenient, rapid, and efficient system of recombinant human domain I production would aid in extending the previous mutation studies of domain I to investigate these epitopes. We developed a novel platform of expression and purification of recombinant human domain I, using Escherichia coli as the expression host (27). This system of production was used to create multiple variants of recombinant human domain I with single-point or double-point mutations. The key areas targeted for investigation were G40–R43, as identified by other investigators, and we extended this area to include R39 as well as the negatively charged residues D8, D9, and the domain I–II interlinker region. We affinity purified IgG from serum of patients with APS alone, patients with APS and systemic lupus erythematosus (SLE), patients with SLE but no APS, and from healthy controls. We tested binding of these IgG samples to wild-type recombinant human domain I and to recombinant human domain I variants, in both solid-phase and fluid-phase assays.
Here, we demonstrate for the first time that residue R39, in addition to G40–R43, is important for binding to aPL, with R39 being the most important residue. In addition, we present data suggesting that D8 and D9, as well as the interlinker region, are also important and use computer modeling studies to explain how these results support the theory that aPL may bind discontinuous epitopes incorporating these areas.
PATIENTS AND METHODS
Materials
Automated sequencing was carried out by personnel at MWG Biotech (Ebersburg, Germany). Ninety-six–well irradiated or polysorb plates were purchased from VWR International (Leicester, UK), and nickel chelate plates were purchased from VH Bio (Gateshead, UK).
Expression and purification of wild-type and mutant recombinant human domain I by E coli
The system used for wild-type recombinant human domain I expression by E coli and purification using nickel chromatography was described previously (27). The purity of eluted recombinant human domain I was confirmed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
For the production of mutant recombinant human domain I proteins, synthetic genes encoding for the mutant recombinant human domain I proteins were individually synthesized using recursive polymerase chain reaction (PCR), as described previously for wild-type domain I (27). Each synthetic mutant domain I gene was then cloned into the expression plasmid pET-26b(+), the sequence was checked, and target protein was expressed and purified as for wild-type recombinant human domain I. The correct folding of each expressed protein was confirmed by the ability to bind murine anti–domain I antibodies that recognize conformational epitopes of domain I, as described previously (27).
Human polyclonal IgG
Polyclonal IgG was purified from 22 patients who satisfied the American College of Rheumatology (ACR) revised classification criteria for APS (1,28). IgG was also purified from 20 patients with SLE satisfying the ACR classification criteria for SLE (29,30) (disease controls) and from 10 healthy control subjects. The patients with SLE did not have features of APS and did not have persistent positivity for aPL, as defined by the revised classification criteria for APS proposed by Miyakis et al (1). Ethical approval for the study was granted by the University College London Hospital Research Ethics Committee. Protein G beads (Amersham, Bucks, UK) were used to purify IgG from all 3 groups, as described previously (27). The amount of IgG was quantified using a direct IgG enzyme-linked immunosorbent assay (ELISA), as described previously (31,32). Results of all subsequent direct ELISAs (described below) are expressed as the percentage binding of an in-house IgG APS–positive control sample known to strongly bind recombinant human domain I, whole β2-GPI, and cardiolipin.
Direct binding ELISA of aPL binding to purified wild-type and mutant recombinant human domain I
A nickel plate was coated (50 μl per well) with 10 μg/ml of wild-type or mutant recombinant human domain I on the test half and with phosphate buffered saline (PBS) alone on the control half. Human polyclonal IgG samples were diluted to a concentration of 20 μg/ml in SEC (sample, enzyme, and conjugate) buffer (100 mM Tris HCl [pH 7], 100 mM NaCl, 0.02% Tween 20, and 0.2% bovine serum albumin [BSA]). Fifty-microliter aliquots of these samples were tested for binding to recombinant human domain I by direct ELISA, as previously described (27).
In addition, the density of recombinant human domain I and selected mutants on nickel chelate plates was measured as follows: nickel chelate plates were coated in triplicate with native recombinant human domain I and mutants, which were chosen based on their pattern of binding to polyclonal aPL at 10 μg/ml, and blocked as described above. After washing, an anti-His–tagged horseradish peroxidase–conjugated antibody (HisDetector Nickel-HRP; KPL, Gaithersburg, MD) was added to each well for 30 minutes at room temperature, developed with tetramethylbenzidine substrate (KPL), and read at 450 nm.
Anticardiolipin ELISA
Human polyclonal IgG samples were diluted to a concentration of 20 μg/ml in PBS containing 10% fetal calf serum. Fifty-microliter aliquots of these samples were tested (in duplicate) for binding to cardiolipin by direct ELISA, as previously described (31).
Binding to β2GPI coated on an irradiated plate
Human polyclonal IgG samples were diluted in 0.1% BSA/PBS to 40 μg/ml. Fifty-microliter aliquots of these samples were tested (in duplicate) for binding to purified native β2GPI by direct ELISA, as previously described (31).
Binding to β2GPI in the presence of cardiolipin
A polysorb ELISA plate was coated with 50 μl of cardiolipin (diluted to 2.5 μg/ml in ethanol). The plate was incubated overnight at 4°C. After washing with PBS, 100 μl of 0.25% gelatin/PBS was added to each well. The plate was incubated at room temperature for 1 hour and washed with PBS. Fifty microliters of 10 μg/ml human β2GPI in PBS was added to each well on one side of the plate (the test side). PBS alone (50 μl per well) was added to each well on the other side of the plate (the control side). The plate was incubated at room temperature for 1 hour and washed with PBS. Human polyclonal IgG was diluted to 40 μg/ml in 0.1% BSA/PBS, and 50-μl aliquots were added to wells in the test side and to the corresponding wells in the control side. All patient samples were tested in duplicate.
After 90 minutes of incubation at room temperature and washing with PBS, bound antibody was detected by adding goat anti-human IgG alkaline phosphatase conjugate followed by appropriate chromogenic substrate. The net optical density (OD) was calculated using the following equation: OD in test well – OD in corresponding control well. Given that the whole plate was coated with cardiolipin and subsequently only one-half was coated with whole β2GPI, the net OD represents the difference between binding to β2GPI in the presence of cardiolipin and binding to cardiolipin alone, which is relevant clinically.
Competitive inhibition ELISA
Binding of polyclonal IgG to purified whole human β2GPI in the presence of cardiolipin was measured as described above. For each patient sample, the concentration of purified IgG required to achieve ∼50% maximum binding in that assay was determined, and this concentration was mixed with test inhibitors in the inhibition assay. Samples of wild-type recombinant human domain I and mutant recombinant human domain I were diluted to 3.25 μM in PBS and used as test inhibitors. Each test inhibitor was incubated with IgG purified from APS serum for 2 hours at room temperature. Duplicate samples were tested in each case. Binding to β2GPI on a cardiolipin-coated surface was then assessed as described above. The percent inhibition for each inhibitor with each patient sample was determined using the following formula: % inhibition = (A0 − A/A0) × 100, where A is the OD from the well containing the inhibitor (corrected for background) and A0 is the OD from a well containing no inhibitor (corrected for background).
Statistical analysis
For each of the direct binding ELISAs, the mean strength of binding of purified IgG samples from the different study groups was calculated. The means for the different study groups were compared using the Mann-Whitney 2-tailed U test, to determine whether there were significant differences between patients with APS, patients with SLE but no APS, and healthy controls in each of the assays. The Mann-Whitney U test was also used to compare the mean strength of binding of each recombinant human domain I mutant to aPL with the mean strength of binding of wild-type recombinant human domain I to the same panel of aPL.
Computer modeling
The Swiss-Model automated protein structure homology-modeling server (GlaxoSmithKline R&D and the Swiss Institute of Bioinformatics; online at http://swissmodel.expasy.org/spdbv/) was used to create a 3-dimensional template of the structure of domain I from its amino acid sequence (33,34). This program was used to predict the effects of altering single amino acids on hydrogen-bond formation, surface residue exposure, and overall protein conformation.
RESULTS
Clinical characteristics of patients from whom purified polyclonal IgG was obtained
Polyclonal IgG was purified from serum samples from 22 patients with APS, 20 patients with SLE (without APS or consistent aPL positivity, as defined by the most recent classification criteria [1]), and 10 healthy volunteers. Twenty-one of the 22 patients with APS, 19 of the 20 patients with SLE, and 7 of the 10 healthy volunteers were women. The mean ± SD ages of the subjects in the 3 groups were comparable (for patients with APS, 45.1 ± 9.9 years; for patients with SLE, 36.5 ± 10.6 years; for healthy controls, 32 ± 6.4 years). All individuals in both the APS group and the SLE disease control group were white. Among the healthy control group, 70% of individuals were white, 20% were of southeast Asian origin, and 10% were African Caribbean.
The clinical features of patients with APS were as follows: 16 patients (72.7%) had APS alone, and 6 patients (27.3%) also had SLE. Among the 18 women with a prior pregnancy, 15 (83.3%) had experienced at least 1 miscarriage. Seventeen patients (77.3%) had a history of 1 or more thrombotic events. The most common venous thrombotic event was deep vein thrombosis (7 of 22 patients [31.8%]) followed by pulmonary embolism (4 patients [18.2%]). The most common arterial thrombotic event was stroke (7 patients [31.8%]) followed by myocardial infarction (2 patients [9.1%]). Four patients (18.2%) had a history of both arterial and venous thrombosis. The clinical features of patients in the APS group were representative of the features of patients in larger epidemiologic studies of APS (35).
Binding to recombinant human domain I in the solid phase was stronger for IgG purified from patients with APS than that for IgG purified from disease controls or healthy controls. Figure 1 demonstrates that IgG purified from patients with APS bound recombinant human domain I in the solid phase significantly better than IgG derived from patients in the SLE disease control group (P = 0.0004) and subjects in the healthy control group (P < 0.0001). However, no significant difference between the 2 control groups was observed (P = 0.39).
Figure 1.
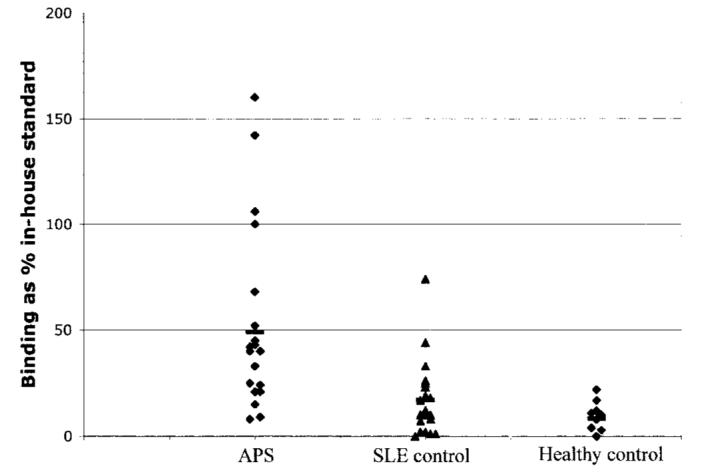
Binding of polyclonal IgG from patients with antiphospholipid syndrome (APS) to Escherichia coli–expressed recombinant human domain I in the solid phase. APS, n = 22; systemic lupus erythematosus (SLE) controls, n = 20; healthy controls, n = 10. For APS patients versus SLE/autoimmune controls, P = 0.0004; for APS patients versus healthy controls, P < 0.0001; for SLE/autoimmune controls versus healthy controls, P = 0.39. Bars show the mean.
Solid-phase binding to cardiolipin, β2GPI coated on an irradiated plate, and β2GPI coated on cardiolipin
A summary of the solid-phase binding data is shown in Table 1. In all 3 assays, IgG samples from patients with APS showed the strongest binding. For the assays testing binding to cardiolipin and β2GPI coated on an irradiated plate, binding was significantly greater in samples from the SLE control group as compared with samples from the healthy control group. This was not true of the assay of binding to β2GPI coated on cardiolipin, in which samples from both control groups showed very low binding. The presence of elevated levels of IgG anticardiolipin antibodies in patients with SLE in the absence of clinical features of APS is a recognized finding (36). The finding of nonpathogenic anti-β2GPI antibodies in non-APS patients with autoimmune disease has also been described (16). Our results suggest that both the anti–recombinant human domain I assay and the assay for binding to β2GPI coated on cardiolipin do not detect these nonpathogenic antibodies but do detect antibodies derived from patients with APS that are hence more likely to be pathogenic.
Table 1.
Summary of solid-phase binding data*
| Study group |
P, by Mann-Whitney 2-tailed U test |
|||||
|---|---|---|---|---|---|---|
| Assay | APS (n = 22) |
SLE (n = 20) |
Healthy (n = 10) |
APS vs. SLE |
APS vs. healthy |
SLE vs. healthy |
| Cardiolipin | 53.3 ± 63.9 | 11.4 ± 13.4 | 1.7 ± 2.4 | 0.005 | <0.0001 | <0.0001 |
| β2GPI on an irradiated plate surface | 51.0 ± 49.0 | 24.1 ± 13.1 | 5.4 ± 3.2 | 0.02 | <0.0001 | <0.0001 |
| β2GPI on cardiolipin-coated surface | 69.3 ± 85.8 | 0.62 ± 1.25 | 0.4 ± 1.0 | <0.0001 | <0.0001 | 0.81 |
Values are the mean ± SD percent binding activity compared with in-house standard. APS = antiphospholipid syndrome; SLE = systemic lupus erythematosus; β2GPI = β2-glycoprotein I.
Binding of domain I mutants in the solid phase to purified IgG from patients with APS
The 8 different mutants were designed to investigate 3 potential epitopes: the negatively charged residues D8 and D9, the region G40–R43 extended to include R39, and the domain I–II interlinker region. D8 was changed to serine (D8S), and D9 was changed to glycine (D9G). Serine and glycine were chosen because they are small and neutral, and because computer-modeling studies predicted that these changes would have the least effect on the tertiary structure of the protein. Thus, 3 recombinant human domain I mutants were made, 2 of which had a single amino acid substitution (D8S and D9G) and 1 in which both D residues were mutated (termed Mut 8+9).
In region R39–R43, we studied the mutations G40E and R43G, which were previously identified as being important for binding aPL derived from patients with APS (14,16,17). However, we studied the effects of these mutations in isolated domain I of β2GPI, whereas previous studies incorporated these mutation within whole β2GPI. We also studied 2 mutations at R39, 1 with an altered charge (R39S) and 1 with a conserved positive charge (R39K). Last, we expressed recombinant human domain I with the remainder of the domain I–II interlinker added at the C-terminal (residues TPRVPF) and termed this extended domain I. This was done to test whether addition of the interlinker enhanced binding to aPL. As a control, we attempted to express recombinant human domain I with the domain II–III interlinker added instead, but this protein was unstable and could not be expressed in sufficient quantities in the bacterial system to permit further analysis.
Figure 2 demonstrates the ability of these recombinant human domain I variants to bind a panel of IgG samples purified from patients with APS, all of which bound cardiolipin, whole β2GPI, and wild-type recombinant human domain I. Figure 2 shows that small changes in the recombinant human domain I amino acid sequence had significant effects on binding to these purified IgG samples. The effects were either to reduce or enhance binding as compared with wild-type recombinant human domain I. The D8S and D9G mutations in isolation resulted in reduced binding, but the combination of these mutations caused enhanced binding to all samples tested. Altering R39 also reduced binding, and there was no difference between R39K and R39S. The G40E and R43G mutations in the solid phase enhanced binding as compared with wild-type recombinant human domain I. The extended domain I variant in the solid phase also enhanced binding as compared with wild-type recombinant human domain I.
Figure 2.
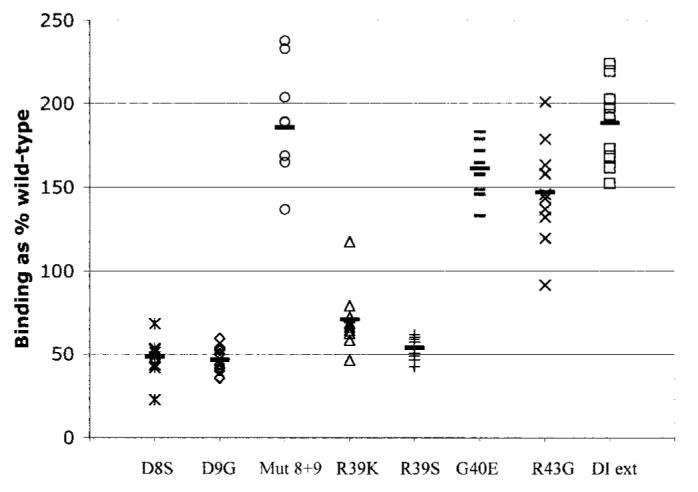
Binding of polyclonal IgG from patients with antiphospholipid syndrome (APS) to mutant recombinant human domain I in the solid phase. A panel of purified IgG samples derived from 10 patients with APS was selected. Enhanced binding as compared with wild-type recombinant human domain I was greatest with the domain I mutant in which both D residues were mutated (Mut 8+9) and the extended domain I (DI ext) mutant. The greatest reduction in binding compared with wild-type domain I was observed with the single-point D8 and D9 mutants and the R39 mutant. Bars show the mean.
It is conceivable that the mutations in recombinant human domain I alter interactions with the nickel chelate plates and thus the coating density of each mutant recombinant human domain I; however, the fact that they bind the plate through a His tag that is not part of domain I itself makes this less likely. To exclude this possibility, we measured the density of recombinant human domain I and that of 2 selected mutants that displayed enhanced (Mut 8+9) or reduced (R39K) binding to purified IgG. To determine the amount of recombinant human domain I bound on the microwells, we took advantage of the fact that each peptide contains a His tag and thus, using an anti-His–tagged antibody, measured the relative amounts of each bound peptide on the ELISA plates. We observed <10% variation in coating density, as shown by the mean ± SD OD from triplicate wells, between recombinant human domain I (0.51 ± 0.03) and Mut 8+9 (0.56 ± 0.02) and R39K (0.5 ± 0.02). The background binding of the anti-His–tagged antibody to empty nickel chelate wells was minimal (0.08 ± 0.002). Therefore, we found no evidence that mutations in recombinant human domain I altered the coating density on the nickel chelate plates.
Inhibition studies of wild-type and recombinant human domain I mutants in the fluid phase
We investigated the ability of recombinant human domain I to inhibit binding to whole β2GPI (16). Our results in the solid-phase assays (Table 1) suggested that binding to β2GPI coated on cardiolipin was a more specific assay for pathogenic antibodies from patients with APS than either the anticardiolipin ELISA or the standard anti-β2GPI ELISA. We therefore tested the ability of 5 recombinant human domain I proteins to inhibit binding to a panel of 8 purified IgG samples from patients with APS in this assay, all of which strongly bind β2GPI coated on cardiolipin.
The recombinant human domain I proteins chosen to test binding in the fluid phase were wild-type recombinant human domain I, 2 proteins that demonstrated good binding in the solid-phase ELISA (Mut 8+9 and extended domain I), a poor binder (R39S), and 1 mutant that had been shown in the context of whole β2GPI to cause marked reduction in binding to clinically relevant aPL (16,17) (G40E), although our solid-phase ELISA had shown an opposite effect of this mutation. Figure 3 shows that results for the fluid phase differed from those for the solid phase. Wild-type recombinant human domain I inhibited binding of the 8 IgG samples tested, to variable extents (mean ± SD inhibition 62.9 ± 25.7%). However, the binding of 2 samples was inhibited by >90%, whereas inhibition for another 2 samples was <35%. The results were much clearer for Mut 8+9, which inhibited binding of all IgG samples (the lowest level of inhibition was 72.6% [mean ± SD 83.1 ± 8.9%]). Therefore, the combination of D8S and D9G mutations consistently enhanced binding to all IgG samples from patients with APS, in both solid-phase and fluid-phase assays.
Figure 3.
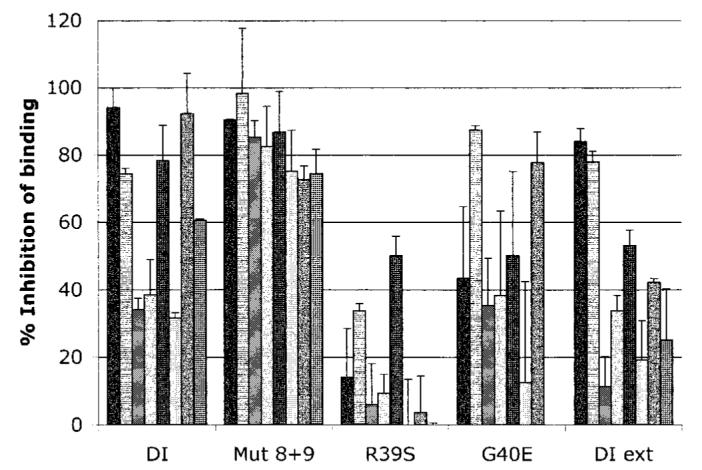
Binding of polyclonal IgG from patients with antiphospholipid syndrome to mutant recombinant human domain I (DI) in the fluid phase. The graph shows the degree to which wild-type and mutant recombinant human domain I (at 3.25 μM) can inhibit polyclonal IgG antiphospholipid antibodies (aPL) from binding to whole β2-glycoprotein I coated on cardiolipin. Variable inhibition was observed with wild-type recombinant human domain I and extended domain I (DI ext). The domain I mutant in which both D residues were mutated (Mut 8+9) had the effect of enhancing binding to the majority of aPL tested. In contrast, the R39S mutation resulted in a significant reduction in binding as compared with wild-type (P = 0.001). G40E had a variable effect. Values are the mean and SD of 8 samples.
In contrast, altering the R39 residue (R39S) had the effect of significantly reducing binding to the majority of aPL in the fluid phase (mean ± SD inhibition 14 ± 18.5%) as compared with wild-type recombinant human domain I (P = 0.001). The G40E mutation also showed evidence of overall reduced binding to aPL as compared with wild-type recombinant human domain I (mean ± SD inhibition 44.1 ± 31.7%), but the effect was more variable than that observed with R39S. Although adding the remainder of the interlinker region onto the C-terminal of recombinant human domain I had the effect of enhancing binding in the solid phase, this effect was not observed in the fluid phase (mean ± SD inhibition 43.3 ± 26.7%).
Computer modeling studies of domain I
It was apparent from the binding studies that minor changes within domain I could cause marked alterations in binding to different IgG samples from patients with APS. Of the residues R39, G40, and R43, residue R39 seemed to be crucial in conferring binding. In contrast, altering both D8 and D9 had the opposite effect of enhancing binding to the majority of aPL tested. Extending recombinant human domain I to include all of the domain I–II interlinker had the effect of enhancing binding as compared with wild-type recombinant human domain I in the solid phase while having a variable effect in the fluid phase.
Figure 4a demonstrates that the domain I–II interlinker region is closely approximated to the R39–R43 region on one side and the D8–D9 region on the other. R39 is only 4Å from the domain I–II interlinker and has a considerable proportion (60%) of its surface area exposed to the surface, both in isolated domain I, as predicted by Swiss-Model, and as part of the whole β2GPI protein (19). The marked reduction in binding to aPL in the fluid phase seen with R39S suggests that aPL bind this R residue. Figures 4c and d show that altering R39 to S39 results in the loss of a protruding side chain and a change in the electrostatic charge within that area. R39S binds 2 murine monoclonal anti–domain I antibodies that recognize conformationally restricted epitopes (17) as strongly as wild-type domain I (data not shown), suggesting that this mutation did not significantly alter the overall protein tertiary structure. This finding is in accordance with the computer model demonstrating that altering R39 to S39 alters the exposure of this residue while retaining the same overall protein conformation (Figures 4a-d).
Figure 4.
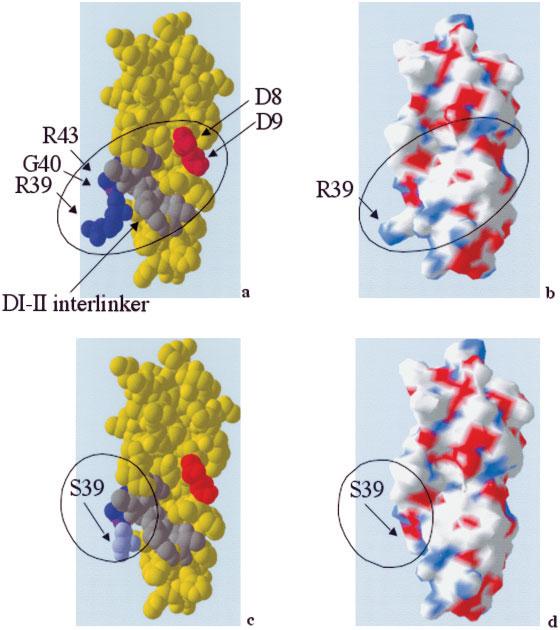
Computer models generated by the Swiss-Model automated protein structure homology-modeling server. a, Molecular structure of the wild-type recombinant domain I molecule. b, Electrostatic charge on the surface of the wild-type recombinant domain I molecule. c, Molecular structure of R39S. d, Electrostatic charge on the R39S molecular surface. Residues R39 and R43 are shown in dark blue, G40E in purple, the domain I–II interlinker region in gray, and residues D8 and D9 in red. Mutating R39 to S39 (light blue) results in marked alteration of surface epitope conformation, with no significant effect on the overall protein tertiary structure.
DISCUSSION
We have developed a novel platform for the expression and purification of recombinant human domain I of human β2GPI, using E coli (27). Our results show that recombinant human domain I produced by this method can be used as the antigen in a polyclonal IgG ELISA assay that distinguishes patients with APS (with or without an additional autoimmune disease) from patients with SLE alone and healthy controls. Using a limited number of patient samples, the anti–recombinant human domain I assay performed better than either the anticardiolipin or anti-β2GPI (coated on an irradiated plate) assay, because the latter 2 assays also picked up IgG from some patients who had SLE but not APS. When β2GPI was coated on cardiolipin, only IgG from patients with APS showed strong binding, suggesting that β2GPI, upon binding anionic phospholipids, undergoes a conformational change, exposing epitopes that specifically bind pathogenic aPL.
Our results support those of a recent study by de Laat et al (37), which suggest that this conformational change leads to an alteration in the position of glycosylated chains attached to domain III and domain IV, preventing them from covering epitopes on domain I. If glycosylated chains extend up and cover the G40–R43 region, as suggested by de Laat et al (37), it is likely that other adjacent residues such as R39 and neighboring epitopes within the tertiary protein structure of domain I (such as the domain I–II interlinker) may also be covered. Our studies, however, shed further light on the nature of the critical epitopes in domain I.
We used our E coli expression system to create mutants designed specifically to test 3 hypotheses regarding these epitopes. These hypotheses were generated through the study of the known domain I structure (19), previous binding studies performed by other investigators using variants of whole β2GPI containing mutations in domain I (16,17), and mutation studies performed on monoclonal aPL in our laboratory (25,26). In comparing our results with those of previous groups of investigators, it is important to stress that we studied isolated recombinant human domain I, whereas the investigators in most previous studies examined whole β2GPI containing mutations in domain I. Despite this difference in methodology, we confirmed the previous finding that region G40–R43 is likely to be important in binding to aPL, but our results lead to the new idea that the R39 residue plays a particularly important role.
Altering residue R39 to serine had a greater effect on reducing binding to aPL in the fluid phase than either the G40E or R43G changes that have previously been studied (16,17). The R39S change reduced binding to aPL in both the solid-phase and fluid-phase ELISAs, whereas G40E reduced binding only in the fluid phase. Iverson and colleagues (17) studied 10 mutants of whole β2GPI with changes in domain I, but none of these had changes at R39. Their selection of mutations was not hypothesis driven but was based on an initial screen of a phage peptide display library of domain I fragments in which mutations had been incorporated by a random process through error-prone PCR.
We observed some evidence for involvement of an epitope in the domain I–II interlinker region, because the extended domain I molecule bound better than wild-type domain I to aPL in the solid-phase ELISA. However, these results were not confirmed in the fluid-phase assay. We have also demonstrated, for the first time, the production of a mutagenized variant of domain I (Mut8+9) in which the mutations (D8S and D9G) are associated with markedly enhanced binding to the majority of aPL tested, in both solid-phase and fluid-phase assays. In the study by Iverson et al, the majority of mutants showed reduced binding to most sera tested by direct ELISA. Two mutants, M42K and M42V, did show enhanced binding to affinity-purified aPL in the solid phase, but the increases were not as large or as repeatable by fluid-phase ELISA as those seen with Mut 8+9 in this study.
The observation of enhanced binding upon altering both D8 and D9 to S8 and G9, respectively, was unexpected and did not concur with our original hypothesis that interactions between R residues in aPL and D residues in domain I contribute to the antigen–antibody interaction. We speculate that the N-terminal region of domain I is likely to play a complex role in ensuring a stable aPL–domain I interaction, and altering this area seems to have a variable effect on binding, depending on the alteration. The complexity is underlined by the fact that the single mutations D8S and D9G both reduced binding to aPL even though the combination of D8S and D9G increased binding to the same panel of aPL. Although the cause of the binding properties of Mut 8+9 remains unclear, the fact that this product can potentially be expressed in large quantities in bacterial cultures and can inhibit binding of polyclonal aPL to whole β2GPI could have implications regarding therapy.
Although the G40E and R43G mutations showed poor binding in the fluid phase as compared with wild-type domain I, enhanced binding was observed in the solid phase. The observation that recombinant human domain I in isolation may behave differently in solid-phase and fluid-phase ELISAs is similar to previous observations by other investigators using whole native β2GPI (37-39). One possible explanation for this would be that some mutations in recombinant human domain I alter its ability to bind to the nickel plate, thus affecting the density of antigen available for binding. Such alterations in density would affect binding to aPL in the solid-phase but not the fluid-phase ELISA. We believe this is an unlikely explanation for our results, because binding to the plate occurred via a His tag that was the same in all mutants, and because our direct measurements of binding of wild-type recombinant human domain I, Mut 8+9, and R39K using anti-His–tagged antibody showed that these mutants coated the plate at similar densities despite giving widely divergent results in assays of binding to aPL.
An alternative explanation is that some of the mutations alter the way in which the recombinant human domain I molecules pack together on the plate, thus affecting the way in which the epitopes are exposed to aPL. This underlines the importance of carrying out both solid-phase and fluid-phase ELISAs, because the latter are not affected by these plate-based considerations. The fact that the R39S mutant exhibited reduced binding to aPL in both the solid-phase and fluid-phase assays supports our hypothesis that this residue is particularly crucial for conferring binding to aPL due to its considerable degree of surface exposure, as shown in the computer models.
How can we explain the fact that discontinuous epitopes at D8–D9 and R39, as well as the adjacent G40 and R43 residues and the domain I–II interlinker, all seem to contribute to binding to aPL from patients with APS, with alterations in R39 having the greatest effect? Computer modeling (Figure 4a) shows that these 3 epitopes are in close proximity within the tertiary structure of domain I, with considerable surface exposure of the R39 residue. R43 forms hydrogen bonds with R39 and T57 located in the interlinker, which are lost upon altering R43 to G43. Changes at any of these residues (R39, R43, and T57) might be expected to alter this hydrogen-bonded structure and thus to affect binding of aPL. Similarly, if R39 is important, then altering the neighboring residue G40 to a negatively charged E residue is likely to affect binding. Figures 4c and d demonstrate the surface epitope change that occurs when R39 is altered to S39, although the overall protein conformation is not greatly altered. This observation is consistent with our finding that changes at R39 have a particularly marked effect on binding of aPL to domain I.
The domain I–II interlinker is located just 4Å from R39 (Figure 4a); hence, it is likely that this region also participates in binding to aPL, as suggested by the observation that a peptide homologous to this region inhibits the thrombogenic effects of aPL in mice (22). Single-point mutation studies in this area would help further define the importance of this region. It is interesting to note that in the study by Iverson et al (17), the N56T mutation within the N-terminal portion of the interlinker resulted in a significant reduction in binding to some aPL in the fluid phase, although not to as many samples as seen with the G40E and R43G mutants. It is also worth noting that a study of the amino acid sequence of β2GPI showed that the domain I–II interlinker bears marked homology to the domain III–IV region (18). Because some groups of investigators have suggested that domain IV harbors aPL-binding epitopes (40,41), it is conceivable that some aPL may bind homologous epitopes located in both the domain I–II and domain III–IV interlinker region, if this region is adequately exposed (37). However, the crystal structure of β2GPI suggests that epitopes within domain IV would be hidden and inaccessible to aPL, thus favoring domain I as harboring the aPL-binding epitopes (19).
In conclusion, our findings suggest that pathogenic aPL bind discontinuous epitopes neighboring R39–R43 located within the tertiary protein structure of domain I, most likely the domain I–II interlinker region and the D8–D9 region. Thus, peptides representing only single epitopes may have limited success as therapeutic agents in APS. Molecules that resemble domain I itself, containing all 3 epitopes in close proximity, may be more appropriate for this purpose. The variant Mut 8+9 described in the current study may be an example of such a molecule.
It has been suggested that wild-type domain I may be a useful B cell toleragen, given its lack of T cell epitopes (42). That theory is similar to that for the development of abetimus, which is currently being used in therapeutic trials in SLE (43,44). Abetimus carries nucleotide epitopes allowing strong binding to anti–double-stranded DNA (anti-dsDNA) IgG antibodies on the surface of B cells but has no T cell epitopes, so that T cell help is not recruited and the B cell dies. A drug similar to abetimus, but carrying wild-type domain I rather than nucleotides, has been developed (42). It is conceivable that replacement of domain I in this drug with Mut 8+9 would increase its ability to bind and tolerize B cells producing anti-β2GPI antibody. Furthermore, the Mut 8+9 mutation of recombinant human domain I is the first reported domain I variant to show reproducible enhanced binding to polyclonal aPL from patients with APS and can inhibit binding of these aPL to whole native β2GPI.
ACKNOWLEDGMENTS
We thank Drs. M. Iverson and M. Linnik for donating the murine monoclonal anti–domain I antibodies.
REFERENCES
- 1.Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome. J Thromb Haemost. 2006;4:295–306. doi: 10.1111/j.1538-7836.2006.01753.x. [DOI] [PubMed] [Google Scholar]
- 2.Pierangeli SS, Harris EN. In vivo models of thrombosis for the antiphospholipid syndrome. Lupus. 1996;5:451–5. doi: 10.1177/096120339600500524. [DOI] [PubMed] [Google Scholar]
- 3.Pierangeli SS, Liu X, Espinola R, Olee T, Zhu M, Harris NE, et al. Functional analyses of patient-derived IgG monoclonal anticardiolipin antibodies using in vivo thrombosis and in vivo microcirculation models. Thromb Haemost. 2000;84:388–95. [PubMed] [Google Scholar]
- 4.Raschi E, Testoni C, Bosisio D, Borghi MO, Koike T, Mantovan A, et al. Role of the MyD88 transduction signaling pathway in endothelial activation by antiphospholipid antibodies. Blood. 2003;101:3495–500. doi: 10.1182/blood-2002-08-2349. [DOI] [PubMed] [Google Scholar]
- 5.Pierangeli SS, Colden-Stanfield M, Liu X, Barker JH, Anderson GL, Harris EN. Antiphospholipid antibodies from antiphospholipid syndrome patients activate endothelial cells in vitro and in vivo. Circulation. 1999;99:1997–2002. doi: 10.1161/01.cir.99.15.1997. [DOI] [PubMed] [Google Scholar]
- 6.Opara R, Robbins DL, Ziboh VA. Cyclic-AMP agonists inhibit antiphospholipid/β2-glycoprotein I induced synthesis of human platelet thromboxane A2 in vitro. J Rheumatol. 2003;30:55–9. [PubMed] [Google Scholar]
- 7.Shi T, Giannakopoulos B, Yan X, Yu P, Berndt MC, Andrews RK, et al. Anti–β2-glycoprotein I antibodies in complex with β2-glycoprotein I can activate platelets in a dysregulated manner via glycoprotein Ib-IX-V. Arthritis Rheum. 2006;54:2558–67. doi: 10.1002/art.21968. [DOI] [PubMed] [Google Scholar]
- 8.McNeil HP, Simpson RJ, Chesterman CN, Krilis SA. Anti-phospholipid antibodies are directed against a complex antigen that includes a lipid-binding inhibitor of coagulation: β2-glycoprotein I (apolipoprotein H) Proc Natl Acad Sci U S A. 1990;87:4120–4. doi: 10.1073/pnas.87.11.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galli M, Comfurius P, Maassen C, Hemker HC, de Baets MH, van Breda-Vriesman PJ, et al. Anticardiolipin antibodies directed not to cardiolipin but to a plasma protein cofactor. Lancet. 1990;335:1544–7. doi: 10.1016/0140-6736(90)91374-j. [DOI] [PubMed] [Google Scholar]
- 10.Matsuura E, Igarashi Y, Fujimoto M, Ichikawa K, Koike T. Anticardiolipin cofactor(s) and differential diagnosis of auto-immune disease. Lancet. 1990;336:177–8. doi: 10.1016/0140-6736(90)91697-9. [DOI] [PubMed] [Google Scholar]
- 11.Hunt J, Krilis S. The fifth domain of β2-glycoprotein I contains a phospholipid binding site (Cys281-Cys288) and a region recognized by anticardiolipin antibodies. J Immunol. 1994;152:653–9. [PubMed] [Google Scholar]
- 12.Sheng Y, Sali A, Herzog H, Lahnstein J, Krilis SA. Site-directed mutagenesis of recombinant human β2-glycoprotein I identifies a cluster of lysine residues that are critical for phospholipid binding and anti-cardiolipin antibody activity. J Immunol. 1996;157:3744–51. [PubMed] [Google Scholar]
- 13.Iverson GM, Victoria EJ, Marquis DM. Anti-β2 glycoprotein I autoantibodies recognize an epitope on the first domain of β2GPI. Proc Natl Acad Sci U S A. 1998;95:15542–6. doi: 10.1073/pnas.95.26.15542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reddel SW, Wang YX, Sheng YH, Krilis SA. Epitope studies with anti-β2-glycoprotein I antibodies from autoantibody and immunized sources. J Autoimmun. 2000;15:91–6. doi: 10.1006/jaut.2000.0427. [DOI] [PubMed] [Google Scholar]
- 15.McNeeley PA, Dlott JS, Furie RA, Jack RM, Ortel TL, Triplett DA, et al. β2-glycoprotein I-dependent anticardiolipin antibodies preferentially bind the amino terminal domain of β2-glycoprotein I. Thromb Haemost. 2001;86:590–5. [PubMed] [Google Scholar]
- 16.De Laat B, Derksen RH, Urbanus RT, de Groot PG. IgG antibodies that recognize epitope Gly40-Arg43 in domain I of β2-glycoprotein I cause LAC, and their presence correlates strongly with thrombosis. Blood. 2005;105:1540–5. doi: 10.1182/blood-2004-09-3387. [DOI] [PubMed] [Google Scholar]
- 17.Iverson GM, Reddel SW, Victoria EJ, Cockerill KA, Wang YX, Marti-Renom MA, et al. Use of single point mutations in domain I of β2-glycoprotein I to determine fine antigenic specificity of antiphospholipid autoantibodies. J Immunol. 2002;169:7097–103. doi: 10.4049/jimmunol.169.12.7097. [DOI] [PubMed] [Google Scholar]
- 18.Steinkasserer A, Estaller C, Weiss EH, Sim RB, Day AJ. Complete nucleotide and deduced amino acid sequence of human β2-glycoprotein I. Biochem J. 1991;277(Pt 2):387–91. doi: 10.1042/bj2770387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwarzenbacher R, Zeth K, Diederichs K, Gries A, Kostner GM, Laggner P, et al. Crystal structure of human β2-glycoprotein I: implications for phospholipid binding and the antiphospholipid syndrome. Embo J. 1999;18:6228–39. doi: 10.1093/emboj/18.22.6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouma B, de Groot PG, van den Elsen JM, Ravelli RB, Schouten A, Simmelink MJ, et al. Adhesion mechanism of human β2-glycoprotein I to phospholipids based on its crystal structure. Embo J. 1999;18:5166–74. doi: 10.1093/emboj/18.19.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shoenfeld Y, Krause I, Kvapil F, Sulkes J, Lev S, von Landenberg P, et al. Prevalence and clinical correlations of antibodies against six β2-glycoprotein-I-related peptides in the antiphospholipid syndrome. J Clin Immunol. 2003;23:377–83. doi: 10.1023/a:1025321617304. [DOI] [PubMed] [Google Scholar]
- 22.Pierangeli SS, Blank M, Liu X, Espinola R, Fridkin M, Ostertag MV, et al. A peptide that shares similarity with bacterial antigens reverses thrombogenic properties of antiphospholipid antibodies in vivo. J Autoimmun. 2004;22:217–25. doi: 10.1016/j.jaut.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 23.Vlachoyiannopoulos PG, Mavragani CP, Bourazopoulou E, Balitsari AV, Routsias JG. Anti-CD40 antibodies in antiphospholipid syndrome and systemic lupus erythematosus. Thromb Haemost. 2004;92:1303–11. doi: 10.1160/TH04-02-0135. [DOI] [PubMed] [Google Scholar]
- 24.Sheng Y, Reddel SW, Herzog H, Wang YX, Brighton T, France MP, et al. Impaired thrombin generation in β2-glycoprotein I null mice. J Biol Chem. 2001;276:13817–21. doi: 10.1074/jbc.M010990200. [DOI] [PubMed] [Google Scholar]
- 25.Giles I, Lambrianides N, Latchman D, Chen P, Chukwuocha R, Isenberg D, et al. The critical role of arginine residues in the binding of human monoclonal antibodies to cardiolipin. Arthritis Res Ther. 2005;7:R47–56. doi: 10.1186/ar1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giles I, Lambrianides N, Pattni N, Faulkes D, Latchman D, Chen P, et al. Arginine residues are important in determining the binding of human monoclonal antiphospholipid antibodies to clinically relevant antigens. J Immunol. 2006;177:1729–36. doi: 10.4049/jimmunol.177.3.1729. [DOI] [PubMed] [Google Scholar]
- 27.Ioannou Y, Giles I, Lambrianides A, Richardson C, Pearl LH, Latchman DS, et al. A novel expression system of domain I of human β2 glycoprotein I in Escherichia coli. BMC Biotechnol. 2006;6:8. doi: 10.1186/1472-6750-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson WA, Gharavi AE, Koike T, Lockshin MD, Branch DW, Piette JC, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42:1309–11. doi: 10.1002/1529-0131(199907)42:7<1309::AID-ANR1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 29.Hochberg MC, the Diagnostic and Therapeutic Criteria Committee of the American College of Rheumatology Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter] Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 30.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 31.Giles IP, Haley J, Nagl S, Latchman DS, Chen PP, Chukwuocha RU, et al. Relative importance of different human aPL derived heavy and light chains in the binding of aPL to cardiolipin. Mol Immunol. 2003;40:49–60. doi: 10.1016/s0161-5890(03)00100-7. [DOI] [PubMed] [Google Scholar]
- 32.Rahman MA, Kettleborough CA, Latchman DS, Isenberg DA. Properties of whole human IgG molecules produced by the expression of cloned anti-DNA antibody cDNA in mammalian cells. J Autoimmun. 1998;11:661–9. doi: 10.1006/jaut.1998.0241. [DOI] [PubMed] [Google Scholar]
- 33.Peitsch MC. ProMod and Swiss-Model: Internet-based tools for automated comparative protein modelling. Biochem Soc Trans. 1996;24:274–9. doi: 10.1042/bst0240274. [DOI] [PubMed] [Google Scholar]
- 34.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modelling. Electrophoresis. 1997;18:2714–23. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 35.Cervera R, Piette JC, Font J, Khamashta MA, Shoenfeld Y, Camps MT, et al. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum. 2002;46:1019–27. doi: 10.1002/art.10187. [DOI] [PubMed] [Google Scholar]
- 36.Petri M. Epidemiology of the antiphospholipid antibody syndrome. J Autoimmun. 2000;15:145–51. doi: 10.1006/jaut.2000.0409. [DOI] [PubMed] [Google Scholar]
- 37.De Laat B, Derksen RH, van Lummel M, Pennings MT, de Groot PG. Pathogenic anti-β2-glycoprotein I antibodies recognize domain I of β2-glycoprotein I only after a conformational change. Blood. 2005;107:1916–24. doi: 10.1182/blood-2005-05-1943. [DOI] [PubMed] [Google Scholar]
- 38.Giles IP, Isenberg DA, Latchman DS, Rahman A. How do antiphospholipid antibodies bind β2-glycoprotein I? [review] Arthritis Rheum. 2003;48:2111–21. doi: 10.1002/art.11101. [DOI] [PubMed] [Google Scholar]
- 39.Iverson GM, Matsuura E, Victoria EJ, Cockerill KA, Linnik MD. The orientation of β2GPI on the plate is important for the binding of anti-β2GPI autoantibodies by ELISA. J Autoimmun. 2002;18:289–97. doi: 10.1006/jaut.2002.0590. [DOI] [PubMed] [Google Scholar]
- 40.Igarashi M, Matsuura E, Igarashi Y, Nagae H, Ichikawa K, Triplett DA, et al. Human β2-glycoprotein I as an anticardiolipin cofactor determined using mutants expressed by a baculovirus system. Blood. 1996;87:3262–70. [PubMed] [Google Scholar]
- 41.George J, Gilburd B, Hojnik M, Levy Y, Langevitz P, Matsuura E, et al. Target recognition of β2-glycoprotein I (β2GPI)-dependent anticardiolipin antibodies: evidence for involvement of the fourth domain of β2GPI in antibody binding. J Immunol. 1998;160:3917–23. [PubMed] [Google Scholar]
- 42.Horizon A, Weisman MH, Wallace DJ, Merrill JT, Linnik MD, Cockerill KA, et al. Results of a randomized, placebo controlled, double blind phase 1/2 clinical trial to assess the safety and tolerability of LJP 1082 in patients with antiphospholipid syndrome [abstract] Arthritis Rheum. 2003;48(Supp 9):S364–5. [Google Scholar]
- 43.Alarcon-Segovia D, Tumlin JA, Furie RA, McKay JD, Cardiel MH, Strand V, et al. LJP 394 for the prevention of renal flare in patients with systemic lupus erythematosus: results from a randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2003;48:442–54. doi: 10.1002/art.10763. [DOI] [PubMed] [Google Scholar]
- 44.Linnik MD, Hu JZ, Heilbrunn KR, Strand V, Hurley FL, Joh T, the LJP 394 Investigator Consortium Relationship between anti–double-stranded DNA antibodies and exacerbation of renal disease in patients with systemic lupus erythematosus. Arthritis Rheum. 2005;52:1129–37. doi: 10.1002/art.20980. [DOI] [PubMed] [Google Scholar]