

Abstract
Human lipoxygenase (hLO) isozymes have been implicated in a number of disease states and have attracted much attention with respect to their inhibition. One class of inhibitors, the flavonoids, have been shown to be potent lipoxygenase inhibitors but their study has been restricted to those compounds found in nature, which have limited structural variability. We have therefore carried out a comprehensive study to determine the structural requirements for flavonoid potency and selectivity against platelet 12-hLO, reticulocyte 15-hLO-1 and prostate epithelial 15-hLO-2. We conclude from this study that catechols are essential for high potency, that isoflavones and isoflavanones tend to select against 12-hLO, that isoflavans tend to select against 15-hLO-1, but few flavonoids target 15-hLO-2.
Keywords: lipoxygenase, flavonoids, reductive inhibition, IC50
Introduction
Lipoxygenases (LO) catalyze the first step in the conversion of polyunsaturated fatty acids to hydroxylated fatty acids.1–3 They contain a non-heme iron (Fe+3) that promotes hydrogen atom abstraction, dioxygenation of the 1,4-diene moiety and reduction of the peroxyl radical to form mono-peroxyl fatty acids.4 A wide variety of human lipoxygenase (hLO) isozymes are found in nature, which are named after the oxygenated carbon atom of the fatty acid product.5 The three human lipoxygenase isozymes of this investigation, platelet 12-human lipoxygenase (12-hLO), reticulocyte 15-human lipoxygenase-1 (15-hLO-1) and prostate epithelial 15-human lipoxygenase-2 (15-hLO-2), are involved in a number of diseases. Platelet 12-hLO6,7 is implicated as a critical signaling molecule in tumor metastasis,8 skin disease,9 and neuronal degeneration.10 Reticulocyte 15-hLO-111 has been implicated in atherogenic processes,12,13 and several pro-neoplastic effects, which contribute to prostatic adenocarcinoma by promoting cell proliferation and angiogenesis.14,15,16 Prostate epithelial 15-hLO-2,17 however, is thought to inhibit the growth of prostatic adenocarcinoma16 because its loss contributes to increased proliferation and reduced differentiation of cancerous cells.18,19 For these reasons, there is considerable interest in the discovery and development of selective inhibitors of 12-hLO and 15-hLO-1, which do not inhibit 15-hLO-2.
The search for selective lipoxygenase inhibitors has uncovered several classes of natural products with inhibitory activity. Known inhibitors against 12-hLO include, the alkaloids, such as chelerythrine and sanguinarine,20 the tropolones, such as hinokitiol,21 the arachidonic acid analogues, such as falcarindiol and panaxynol,22 and the phenolics, such as rosmarinic acid, caffeic acid,23 and anthracenones.24 Known 15-hLO-1 inhibitors include the marine–derived brominated phenol esters,25 the marine sponge-derived terpenes,26, 27 the nordihydroguaiaretic acid (NDGA) derivatives,28 and the anthraquinones.29 Nevertheless, the most effective inhibitors against both 12-hLO and 15-hLO-1 are found in the broad class of phenolic compounds,30 especially among flavonoids such as baicalein,31 quercetin,32 luteolin,33 anadanthoflavone,34 esculetin,35 and epicatechin.36 Interestingly, members of this class of potent inhibitors have rarely been simultaneously investigated to determine their relative potency and selectivity against 12-hLO, 15-hLO-1 and 15-hLO-2. This fact, together with the limited structural variability of natural flavonoids, highlighted the need to investigate the structural requirements of this broad class of flavonoid inhibitors against each of these three hLO isozymes.
In the current investigation, we have used the basic structure of flavonoids, the benzopyran skeleton, as a structural template for the design of novel, potent and selective inhibitors of hLO. We present the synthesis of new benzopyran-4-one analogues with different alkyl and/or aryl substitution at positions C-2 and C-3, and with the scaffold decorated with hydroxyl groups at different positions. This diverse library of flavonoid derivatives was subsequently screened in vitro against 12-hLO, 15-hLO-1 and 15-hLO-2 and compared with known inhibitors in order to determine the relative structural determinants for each isozyme’s inhibition.
Results and Discussion
Synthesis
The 2-substituted 6-hydroxychromen-4-ones (7a–f) were prepared according to Nussbaumer et al.37 by acylation of 2,5-dihydroxyacetophenone (1) to introduce the desired substituent. The doubly-acylated intermediate (3a–g) underwent migration of acyl residues when treated with sodium hydride. The resulting crude products (5a–g) were then cyclized to the corresponding 6-hydroxychromen-4-ones (7a–f) with sulfuric and acetic acid. The 2-substituted 7-hydroxychromen-4-ones (8a–e) were synthesized analogously (Scheme 1), starting from 2,4- dihydroxyacetophenone (2). The 2-alkyl-substituted 6,7-methylenedioxybenzopyran-4-ones (12a–c) were obtained from the corresponding 2-hydroxy-4,5-methylenedioxyacetophenone (10) using the method previously described,37 and the 6,7-dihydroxy-2-t-butylbenzopyran-4-one (14a) was prepared from 12a as shown in Scheme 1. The deprotection of the methylenedioxy group could not be achieved with 12b and 12c, because a complex mixture was obtained that we were unable to separate and identify. The 2-substituted 3-hydroxychromen-4-ones (21a–f) were prepared according to Fourgerousse et al.38 by acylation of the 2-hydroxyacetophenone (15) in pyridine solution at room temperature for 2 h. Selective bromination at the alpha position with regard to the carbonyl group was achieved using phenyltrimethylammonium tribromide (PTT). In the next step, the functional chromanol oxygen atom was introduced with potassium benzoate, under vigorous stirring (48 h) at room temperature. The Baker-Venkataraman rearrangement was carried out using a non-nucleophilic base, sodium hydride, in DMF at 0–5 °C. The product was washed with water and used immediately in the cyclization step, which took place in a 0.5 % solution of sulfuric acid in glacial acetic acid. Finally, the hydroxyl groups were deprotected by saponification in a 5 % alcoholic solution of sodium hydroxide, for 2 h at 60 °C (Scheme 2). The isoflavonoids (25a–j) were obtained by electrophilic substitution of appropriate phenols with benzyl cyanides (Houben Hoesch reaction). The resulting hydroxylketones were cyclized to the isoflavones using DMF/MeSO2Cl as a carbon atom donor in the presence of BF3Et2O (Scheme 3).39 The isoflavanone (26a–b) and isoflavane (7a–g) were obtained through catalytic hydrogenation with Pd/C (10%) from the corresponding isoflavones. The reduction of isoflavones to the corresponding isoflavanones (26a–b) was done by catalytic hidrogenation over Pd/C (10%) in dioxane/ethanol for 6 h. To obtain the isoflavanes (27a–g), the corresponding isoflavones were hydrogenated over Pd/C (10%) in acetic acid containing 0,1% concentrated sulfuric acid, for 14 h.
Scheme 1.

Synthesis of 6-and 7-hydroxybenzopyran-4-ones.
Scheme 2.

Synthesis of 3-hydroxy-benzopyran-4-ones.
Scheme 3.

Synthesis of isoflavonoids.
Biological Evaluation
Many flavonoids have been investigated over the years as lipoxygenase inhibitors, however, they are rarely screened simultaneously with a variety of hLO isozymes to determine specificity. We present the inhibitory potency of known flavonoid inhibitors, quercetin, baicalein and fisetin, in conjunction with a library of synthetic flavonoid compounds against the activity of three human lipoxygenases, 12-hLO, 15-hLO-1 and 15-hLO-2.
Initial HTP screening of all forty-seven flavonoid compounds was performed against 12-hLO, 15-hLO-1 and 15-hLO-2, using the Fe3+/xylenol orange screening method40 in a 384-well format and compared these results with our well-established manual screen.31, 41 The HTP screen uncovered twelve flavonoids with IC50 values lower than 50 μM for 12-hLO, thirteen for 15-hLO-1 and three for 15-hLO-2. Our manual screen of the entire library confirmed these results, with one additional inhibitor was found for 12-hLO and 3 for 15-hLO-1. These data indicate that our one-point HTP screen was able to detect hLO inhibitors exceptionally well, with over 95% predictive power for all three of the hLO isozymes, 12-hLO, 15-hLO-1 and 15-hLO-2
Full IC50 values were determined for all the potent flavonoid inhibitors found. As seen in Table 1, the known LO inhibitors, nordihydroguaiaretic acid (NDGA), quercetin (28), fisetin (29), and baicalein (30), are potent inhibitors of 12-hLO and 15-hLO-1, which is consistent with previous data.42,31,32 Nevertheless, despite the potency of these inhibitors against 12-hLO and 15-hLO-1, only NDGA had any effect on 15-hLO-2, albeit stronger than the published value of 76 μM.43 Interestingly, quercetin, fisetin and baicalein, are reductive inhibitors against 15-hLO-1, even though the positions of the alcohols are dramatically different among the three. This implies that multiple binding orientations are possible for all three compounds, affording each inhibitor’s catechol moiety access to the iron to perform an inner sphere reduction.44 The conclusion that there are multiple inhibitor binding modes is supported by the fact that both 29 and 30 are potent, reductive inhibitors but 36 is not. The catechol is therefore required for activity but it can be on either ring B or ring C, suggesting at least two spatially distinct orientations of flavonoids to achieve ferric ion binding and reduction.
Table 1.
a. Inhibitory activity (IC50 ± SD) of commercial flavonoids.
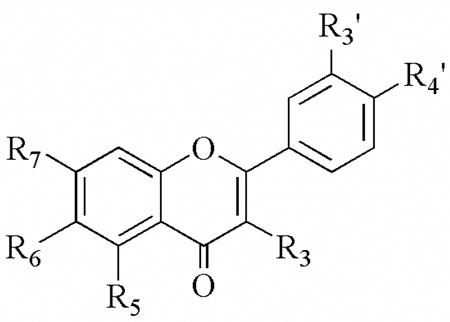 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R3 | R5 | R6 | R7 | R3′ | R4′ | 12-hLO IC50 (μM) | 15-hLO-1 IC50 (μM) | 15-hLO-1/12-hLO | 15-hLO-2 IC50 (μM) | Reduction 15-hLO-1 | |
| NDGA | --- | --- | --- | --- | --- | --- | 2.6 ± 0.3 | 0.25 ± 0.02 | 0.1 | 11 ± 0.7 | Yes |
| 28 | OH | OH | H | OH | OH | OH | 0.44 ± 0.08 | 2.2 ± 0.3 | 5 | > 100 | Yes |
| 29 | OH | H | H | OH | OH | OH | 0.95 ± 0.2 | 1.4 ± 0.2 | 1.5 | > 100 | Yes |
| 30 | H | OH | OH | OH | H | H | 0.86 ± 0.3 | 9.1 ± 0.8 | 12 | >100 | Yes |
| 31 | OH | H | H | H | H | H | > 100 | 73 ± 18 | < 0.73 | > 100 | No |
| 32 | H | H | OH | H | H | H | > 100 | > 100 | -- | > 100 | No |
| 33 | H | H | H | OH | H | H | > 100 | > 100 | -- | > 100 | - |
| 34 | OH | OH | H | H | H | H | > 100 | 3.3± 1.6 | < 0.03 | > 100 | No |
| 35 | OH | H | OH | H | H | H | > 100 | > 100 | -- | > 100 | - |
| 36 | OH | H | H | OH | H | H | > 100 | > 100 | -- | > 100 | - |
All IC50 values are expressed in micromolar units.
We also investigated commercial flavonoids, 31–36, to examine the influence of hydroxyl groups on the inhibitory activity of the flavone skeleton (Table 1). We found that none of the monohydroxylated and dihydroxylated flavonoids inhibited 12-hLO or 15-hLO-2. Compound 31, however, with a single hydroxyl group at the C-3 position, was a weak inhibitor of 15-hLO-1, and the introduction of a second hydroxyl group at C-5 (34), led to a 20-fold improvement of the IC50, with an excellent selectivity for 15-hLO-1 (IC50 15-hLO-1/12-hLO < 0.03).
The next class of compounds investigated were the 2-alkylbenzopyran-4-ones, 7a–7f, 8a–8e, 12a–12c, and 14 (Table 2). The first conclusion derived from these data is that the catechol moiety is important for activity. This is clearly seen when comparing 7e, 8d, 12a and 14, because all have the same substituent attached to the pyranone ring, but only 14 has a catechol moiety and only 14 is a lipoxygenase inhibitor. This is consistent with our previous results regarding NDGA derivatives, in which the catechol moiety was critical for inhibition due to its ability to reduce the active site ferric species of soybean lipoxygenase to its inactive ferrous form, thus interrupting the catalytic cycle.45–46 The second conclusion is that this class of compounds, along with the 3-hydroxybenzopyran-4-ones (Table 3), are in general poor inhibitors. The only exception, aside from the catechol containing 14, is 7f, which selectively inhibits 15-hLO-1 (IC50 = 38 +/− 10 μM). Interestingly, the two features that are critical for the inhibitory potency of 7f are the position of the alcohol (8e is inactive) and the length and bulk of the alkane side group (7b and 7d are inactive), indicating a specific binding mode for 7f. Compounds 21a–21f (Table 3) are all poor inhibitors, consistent with the conclusion that a catechol moiety is important for inhibitor potency.
Table 2.
a. Inhibitory activity (IC50 ± SD) of 2-alkylbenzopyran-4-ones.
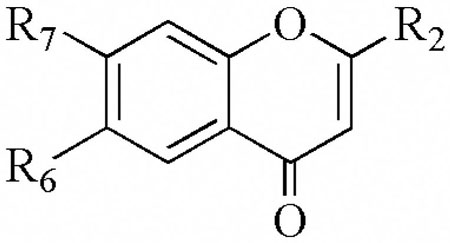 | ||||||||
|---|---|---|---|---|---|---|---|---|
| R2 | R6 | R7 | 12-hLO IC50(μM) | 15-hLO-1 IC50(μM) | 15-hLO-1/12hLO | 15-hLO-2 IC50(μM) | Reduction 15-hLO-1 | |
| 7a | CH2CH3 | OH | H | > 100 | > 100 | - | > 100 | - |
| 7b | CH2CH2CH3 | OH | H | >100 | > 100 | - | >100 | - |
| 7c | CH(CH3)2 | OH | H | > 100 | > 100 | - | > 100 | - |
| 7d | CH2CH(CH3)2 | OH | H | > 100 | > 100 | - | > 100 | - |
| 7e | C(CH3)3 | OH | H | > 100 | > 100 | - | > 100 | - |
| 7f | CH2(CH2)3CH3 | OH | H | > 100 | 38 ± 10 | < 0.38 | > 100 | no |
| 8a | CH2CH2CH3 | H | OH | > 100 | > 100 | - | > 100 | - |
| 8b | CH(CH3)2 | H | OH | > 100 | > 100 | - | > 100 | - |
| 8c | CH2CH(CH3)2 | H | OH | > 100 | > 100 | - | > 100 | - |
| 8d | C(CH3)3 | H | OH | > 100 | > 100 | - | > 100 | - |
| 8e | CH2(CH2)3CH3 | H | OH | > 100 | > 100 | - | > 100 | - |
| 12a | C(CH3)3 | O-CH2-O | > 100 | > 100 | - | > 100 | - | |
| 12b | CH2CH(CH3)2 | O-CH2-O | > 100 | > 100 | - | > 100 | - | |
| 12c | Ph | O-CH2-O | > 100 | > 100 | - | > 100 | - | |
| 14 | C(CH3)3 | OH | OH | > 100 | 18 ± 3.5 | < 0.18 | > 100 | no |
All IC50 values are expressed in micromolar units.
Table 3.
a. Inhibitory activity (IC50 ± SD) of 3-hydroxybenzopyran-4-ones.
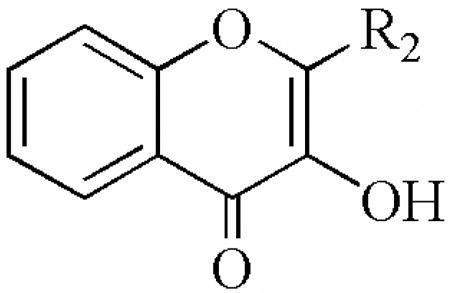 | ||||
|---|---|---|---|---|
| R2 | 12-hLO IC50(μM) | 15-hLO-1 IC50(μM) | 15-hLO-2 IC50(μM) | |
| 21a | CH2CH3 | > 100 | > 100 | > 100 |
| 21b | CH2CH2CH3 | >100 | > 100 | > 100 |
| 21c | CH(CH3)2 | > 100 | > 100 | > 100 |
| 21d | CH2CH(CH3)2 | > 100 | > 100 | > 100 |
| 21e | C(CH3)3 | > 100 | > 100 | > 100 |
| 21f | CH2(CH2)3CH3 | > 100 | > 100 | > 100 |
All IC50 values are expressed in micromolar units.
Another critical finding, beyond the presence of a catechols being present, is that the aromaticity and oxidation state of ring C are important for the potency and selectivity of these compounds against LO (Table 4a–c). Among the 6,7-catechol compounds, the parent structure (25a) is not potent against either 12-hLO or 15-hLO-1 (Table 4a). However, if the double bond is removed from ring C, 26a, the potency and selectivity against 12-hLO increase dramatically (IC50 15-hLO-1/12-hLO = 12) (Table 4b). If the ketone is also removed, i.e. 27a, the compound remains as a potent inhibitor, but its selectivity is reversed (IC50 15-hLO/12-hLO-1 = 0.019) (Table 4c).
Table 4.
| Table 4aa. Inhibitory activity (IC50 ± SD) of 6,7-dihydroxyisoflavones. | |||||||
|---|---|---|---|---|---|---|---|
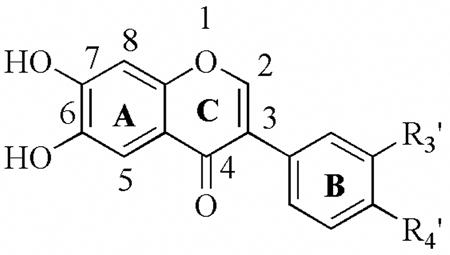 | |||||||
| R3′ | R4′ | 12-hLO IC50(μM) | 15-hLO-1 IC50(μM) | 15-hLO-1/12-hLO | 15-hLO-2 IC50(μM) | Reduction 15-hLO-1 | |
| 25a | H | OCH3 | > 100 | > 100 | - | > 100 | NA |
| 25b | H | OH | 8.7± 0.95 | 49± 7.8 | 6 | > 100 | No |
| 25c | OH | NO2 | 5.8 ± 0.54 | > 100 | > 17 | > 100 | NA |
| 25d | O-CH2-O | > 100 | > 100 | - | > 100 | NA | |
| Table 4ba. Inhibitory activity (IC50 ± SD) of 6,7-dihydroxyisoflavanones. | |||||||
|---|---|---|---|---|---|---|---|
 | |||||||
| R3′ | R4′ | 12-hLO IC50(μM) | 15-hLO-1 IC50(μM) | 15-hLO-1/12-hLO | 15-hLO-2 IC50(μM) | Reduction 15-hLO-1 | |
| 26a | H | OCH3 | 1.6± 0.1 | 19± 1.9 | 12 | > 100 | No |
| 26b | H | OH | 3.8± 0.29 | > 100 | > 26 | > 100 | NA |
| 26c | CH3 | H | 14 ± 1.4 | 0.21± 0.02 | 0.014 | > 100 | Yes |
| Table 4ca. Inhibitory activity (IC50 ± SD) of 6,7-dihydroxyisoflavans. | |||||||
|---|---|---|---|---|---|---|---|
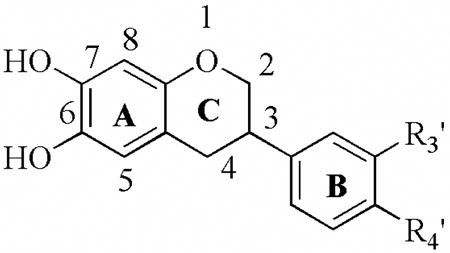 | |||||||
| R3′ | R4′ | 12-hLO IC50(μM) | 15-hLO-1 IC50(μM) | 15-hLO-1/12-hLO | 15-hLO-2 IC50(μM) | Reduction 15-hLO-1 | |
| 27a | H | OCH3 | 7.6 ± 0.6 | 0.15± 0.01 | 0.019 | > 100 | Yes |
| 27b | H | OH | 17 ± 1.7 | 0.51± 0.12 | 0.031 | 71± 28 | Yes |
| 27c | O-CH2-O | 11 ± 1.2 | 0.35± 0.06 | 0.032 | 16± 2.2 | Yes | |
| 27d | CH3 | H | 15 ± 1.4 | 0.21± 0.02 | 0.014 | 8.3± 0.92 | Yes |
Compound 25b is a potent inhibitor for 12-hLO, but not as potent against 15-hLO-1. Removal of the double bond from ring C, i.e. 26b, increases the potency towards 12-hLO, while inhibition of 15-hLO-1 is lost (IC50 15-hLO-1/12-hLO > 26) (Table 4b). If the ketone is also removed, i.e. 27b, then the potency against 15-hLO-1 increases dramatically, while the potency against 12-hLO decreases, shifting the IC50 15-hLO-1/12-hLO ratio from 26 for 26b to 0.03 for 27b. Compound 25d is not potent against either 12-hLO or 15-hLO-1, however, removal of the double bond and ketone from ring C, i.e. 27c, improves inhibition of both 12-hLO and 15-hLO-1 (IC50 15-hLO/12-hLO-1 = 0.032) (Table 4c).
These observations indicate that the selectivity is affected by the planarity of the flavonoid, with 27a, 27b, 27c and 27d being selective against 15-hLO-1, while 25b, 25c, 26a and 26b are selective against 12-hLO. These trends suggest that 6,7-dihydroxyisoflavones and 6,7-dihydroxyisoflavanones tend to be selective against 12-hLO, while 6,7-dihydroxyisoflavans tend to be selective against 15-hLO-1. These inhibitor results for 12-hLO and 15-hLO-1 are consistent with previous inhibition data against porcine 5-lipoxygenase, which indicates that some isoflavans are better inhibitors of the three LO isozymes, 5, 12 and 15-hLO-1 than the corresponding isoflavones.47 An exception to this trend is 26c, which is selective against 15-hLO, even though it is an isoflavanone, indicating that certain substituents on ring B can counter the effect of the ring C structure. Finally, the role of ring C also affects the mechanism of inhibition, with 27a being a reductive inhibitor, while 26a is not. These data suggest that the non-planarity of ring C in 27a allows better access to the iron for an inner sphere reduction.
Modifications on ring B also have effects on the inhibition potency of both isoflavones and isoflavanones (Table 4a and 4b). This is seen by inspecting the structures of compounds 25a and 25d, which do not inhibit either 12-hLO or 15-hLO-1, while 25b and 25c are both potent and selective inhibitors of 12-hLO (IC50 15-hLO-1/12-hLO = 6 and 17, respectively). Among the isoflavanones, 26a and 26b are selective against 12-hLO, while 26c is selective against 15-hLO-1 (vide supra). However, inspection of the four isoflavan compounds (27a–d), whose ring C lacks both the double bond and ketone (Table 4c), demonstrates that their potency and selectivity are relatively unaffected by R3′ and R4′ modifications. This structure/activity relationship difference between the three classes of inhibitors, isoflavones, isoflavanones and isoflavans, suggests different binding modes, where the ring B substituent’s effects are nullified by the structural flexibility of the isoflavan but not by the more rigid isoflavones and isoflavanones.
The data in Tables 5a and 5b indicate that the 7,8-catechol compounds are potent inhibitors of both 12-hLO and 15-hLO-1 and that in general, shifting the catechol does not greatly affect the inhibitor’s ability to reduce the iron. One exception to this trend is the 10-fold decrease in potency of 27e compared to 27a. Another observation is that the 7,8-isoflavones are more selective against 12-hLO, while the 7,8- dihydroxyisoflavans are more selective against 15-hLO-1. This is seen by comparing 25e with 27g, whose potencies are comparable but with reversed selectivity. These trends are similar to those observed for 6,7-dihydroxyflavonoids (Tables 4a–c) and supports our hypothesis that the structure of ring C is important for the selectivity for both 6,7-and 7,8-dihydroxyflavonoids.
Table 5.
| Table 5aa. Inhibitory activity (IC50 ± SD) of 7,8-dihydroxyisoflavones. | |||||||
|---|---|---|---|---|---|---|---|
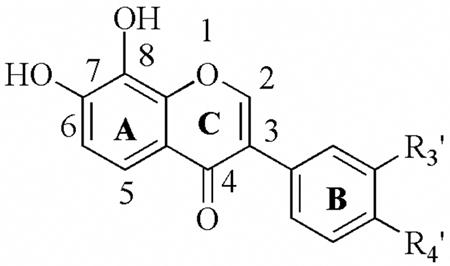 | |||||||
| R3′ | R4′ | 12-hLO IC50 (μM) | 15-hLO-1 IC50 (μM) | 15-hLO-1/12-hLO | 15-hLO-2 IC50 (μM) | Reduction 15-hLO-1 | |
| 25e | H | CH3 | 1.6 ± 0.3 | 7.8± 0.8 | 5 | > 100 | Yes |
| 25f | H | H | 6.4 ± 0.6 | 13.0± 1.6 | 2 | > 100 | Yes |
| 25g | H | Cl | 0.48± 0.09 | 9.0± 1.0 | 10 | > 100 | Yes |
| 25h | Cl | H | 0.78± 0.08 | 6.2± 0.7 | 8 | > 100 | Yes |
| 25i | CH3 | H | 3.6± 0.3 | 11.2± 0.7 | 3 | > 100 | Yes |
| 25j | CF3 | H | 0.62± 0.06 | 8.3± 0.8 | 13 | > 100 | Yes |
| Table 5ba. Inhibitory activity (IC50 ± SD) of 7,8-dihydroxyisoflavans. | |||||||
|---|---|---|---|---|---|---|---|
| R3′ | R4′ | 12-hLO IC50 (μM) | 15-hLO-1 IC50 (μM) | 15-hLO-1/12-hLO | 15-hLO-2 IC50 (μM) | Reduction 15-hLO-1 | |
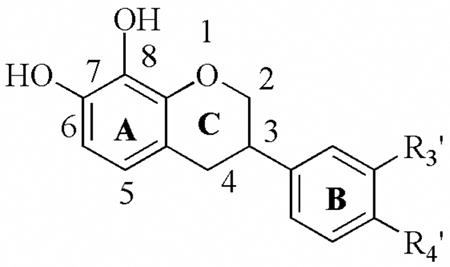 | |||||||
| 27e | H | OCH3 | 70 ± 8.7 | 5.7± 1.0 | 0.08 | > 100 | Yes |
| 27f | OCH3 | OCH3 | > 100 | > 100 | - | > 100 | Yes |
| 27g | H | CH3 | 26 ± 4.5 | 3.7± 0.66 | 0.14 | > 100 | Yes |
All IC50 values are expressed in micromolar units.
All IC50 values are expressed in micromolar units.
In the preceding discussion, we have not mentioned reduction potential as a variable because it is exceedingly hard to establish a trend for two reasons. First, it is difficult to estimate the effect on the reduction potential for substituents that are on ring B because any effect would be weak due to their relative distance from the catechols. This assertion is supported by the fact that 25g and 25h have comparable inhibitory potencies, even though the electron withdrawing chlorine is on the electronically distinct para- and meta- positions, respectively. Second, it was previously shown that the potency of very simple catechols does not track with their reduction potential,44 indicating that it would be even harder to differentiate between steric and reduction potential effects with the complex compounds presented in this investigation.
One of the most intriguing aspects of this study is the weak potency of most of these compounds against 15-hLO-2, even though many are potent against either 12-hLO or 15-hLO-1. The only inhibitors that are potent against 15-hLO-2, are those that have a 6,7-catechol but lack the double bond and ketone group in ring C (27b, 27c, and 27d). Interestingly, modifications of ring B affect the potency of the isoflavans against 15-hLO-2, with 27d being more than 10-fold more potent than 27a. This trend is opposite to that of the 7,8-isoflavans, whose changes in ring B do not affect potency, illustrating a difference in their structure/activity relationship. In addition, the 15-hLO-2 inhibitors are more potent against 15-hLO-1 than 12-hLO, which may suggest a greater similarity in the active sites between 15-hLO-2 and 15-hLO-1, however, differences in the access or exit channels can not be completely ruled out. This is reasonable given the similar regio-specificity of product generation between 15-hLO-2 and 15-hLO-1. This explanation, however, does not account for the fact that the majority of the 15-hLO-1 and 12-hLO inhibitors do not affect 15-hLO-2, possibly due to the fact that the protein sequence of 15-hLO-2 has only marginal similarity to either 12-hLO (55 %) or 15-hLO-1 (55 %).
Molecular Modeling Analysis
As discussed above, our data suggests that there are multiple modes in which flavonoids can bind to the active site of the hLO isozymes, depending on the position of the catechol moiety with respect to the flavonoid scaffold. To probe this possibility further, we flexibly docked four distinct catechol inhibitors into the active site of the 15-hLO-1 homology model to visualize their mode of binding.41 Given that these particular catechols are known to directly bind and reduce the ferric center of LO,44 we analyzed the docking behavior of compounds 25b, 25e, 29 and 30, each representing one of the four classes of catechols studied. The docking poses of these four inhibitors (Figure 1) demonstrate that even if we constrain the close proximity of the catechol to the iron (i.e. to act as a reductive inhibitor), the active site of 15-hLO-1 is large enough to accommodate four distinct binding modes of the flavonoid scaffold. This modeling result supports the experimental conclusion that for small catechols, no single binding mode predominates. It should be noted that we were unable to relate the GLIDE score of these inhibitors to their inhibitory potency, most likely due to their small size relative to the 15-hLO-1 active site and the poor estimation of the metal-catechol bond by the docking program.
Figure 1.

Representative poses of compound docking simulations for 15-hLO-1 with the closest phenolic distance to the iron, for inner sphere reduction, indicated with a dashed line. 25b, 3.2 Å (A), 25e, 3.2 Å (B), 29, 4.2 Å (C) and 30, 3.1 Å (D). The first coordination sphere ligands that surround the active site iron are also shown, with Thr# labeled.
In summary, the data presented here outlines a number of important discoveries. First, the aromaticity and oxidation state of ring C in our flavonoid compounds appears to be important for both the inhibitory potency and selectivity against hLO, with isoflavones and isoflavanones preferentially inhibiting 12-hLO and isoflavanes preferentially inhibiting 15-hLO-1. Second, modification of the basic flavonoid structure has produced a number of selective inhibitors of both 12-hLO (25c, 26b and 25j) and 15-hLO-1 (26c, 27a and 27d), indicating that the flavonoid skeleton is a viable scaffold for selective inhibitor development. Third, a variety of binding modes are possible for flavonoids in lipoxygenases as seen by the fact that compounds 25b, 25e, 29 and 30 all are potent, reductive inhibitors but their relative positioning of ring B to their catechol moiety is different. Finally, it appears that the structural requirements for 15-hLO-2 inhibition are dramatically different from those of 15-hLO-1 and 12-hLO, which indicates that if 15-hLO-2 has a beneficial role in cancer prevention, inhibitors can be developed that do not target its activity.
Experimental Section
Chemical synthesis
Melting points were determined on an Electrothermal apparatus and were not corrected. UV spectra were recorded on a Spectronic Genesys 5 instrument. 1H and 13C NMR spectra were recorded at 300 MHz or 400 MHz (1H) and 75 or 100 MHz (13C), respectively (Bruker AMX 300 and Bruker AMX 400 spectrometers) with (CH3)4Si as internal standard. Chemical shifts are reported in parts per million, using tetramethylsilane as an internal reference. High-resolution mass spectra were recorded with an EI MS-50 AEI instrument at Bonn University, Germany and with a MAT 95XP, Thermo-Finnigan spectrometer at the University of Chile, Santiago. All starting materials were commercially available, > 98% purity, and used without further purification. Quercetin, baicalein, fisetin, 3-hydroxyflavone, 6-hydroxyflavone, 7-hydroxyflavone, 3,5-dihydroxyflavone, 3,6-dihydroxyflavone and 3,7-dihydroxyflavone were purchased from Aldrich.
The HPLC analyses of the compounds were performed using a Merck-Hitachi Intelligent L-6200A Pump, an L-4250 UV-Vis Detector and a D-7000 HSM System Manager Report, a C18 reverse phase column (Hypersil ODS-5, 250 mm × 4 mm) and a flow rate of 1 mL/min. Compounds 7a to 14 were detected at 260 nm, 21a to 21f at 319 nm, 25a to 25j, 26a, 26b, 27b, 27f and 27g at 255 nm, and 27a and 27c to 27e at 295 nm. Two different solvent systems were used: system 1: (A) acetonitrile and (B) 1% acetic acid and system 2: (A) acetonitrile and (B) a 1:1 mixture of 1% acetonitrile/methanol. A gradient of 30 minutes of duration was used in both cases, beginning with 30% of (A), reaching 99% at 30 minutes, and (B) starting with 70% and ending with 1% in the same period. In both systems all the compounds used were found to be more than 95 % pure.
General procedure for the Synthesis of 2-alkyl-6-hydroxy-4-H-benzopyran-4-one (7a–f)
Synthesis of intermediates compounds 3a–g, 4a–f
A solution of 1 (5 g, 32.8 mmol) in dry pyridine (35 mL) was treated with propionyl chloride (10 g, 83 mmol) under cooling in an ice-bath to keep the reaction temperature at about 20 °C. The mixture was stirred for 18 h at room temperature; and then the solvent was partially removed under reduced pressure. The residue was poured onto ice and hydrochloric acid (32 %, 30 mL) and extracted with diethyl ether three times. The combined organic layers were washed with aqueous sodium carbonate solution and water, dried (MgSO4) and concentrated to obtain the crude title compound 3a, which was purified by chromatography on silica gel eluting with dichloromethane to give 3a.
Compounds 3b–g and 4a–f were prepared in a similar fashion using 1 or 2 and 2.5 eq of the requisite acyl chloride.
2′,5′-Dicyloxypropanoyloxyacetophenone (3a) (77%).1H NMR (CDCl3) δH 7.50 (1H, d, J = 2.8 Hz), 7.24 (1H, dd, J = 8.6 Hz, J = 2.8 Hz), 7.09 (1H, d, J = 8.6 Hz), 2.60 (4H, m), 2.51 (3H, s), 1.25 (3H, t, J = 7.4 Hz), 1.25 (3H, t, J = 7.5 Hz).
2′,5′-Dibutanoyloxyacetophenone (3b) (91%). 1H NMR (CDCl3) δH 7.49 (1H, d, J = 2.8 Hz), 7.23 (1H, dd, J = 8.6 Hz; J = 2.8 Hz), 7.09 (1H, d, J = 8.6 Hz), 2.55 (4H, m), 2.50 (3H, s), 1.77 (4H, m), 1.02 (6H, t, J = 7.4 Hz).
2′,5′-Di-(2-methylpropanoyloxy)-acetophenone (3c) (85%). 1H NMR (CDCl3) δH 7.46 (1H, d, J = 2.6 Hz), 7.22 (1H, dd, J = 8.9 Hz, J = 2.8 Hz), 7.06 (1H, d, J = 8.7 Hz), 2.80 (2H, m), 2.50 (3H, s), 1.31 (6H, d, J = 6.9 Hz), 1.29 (6H, d, J = 6.9 Hz).
2′5′-Di-(3-methylbutanoyloxy)-acetophenone (3d) (91%). 1H NMR (CDCl3) δH 7.47 (1H, d, J = 2.8 Hz), 7.23 (1H, dd, J = 8.6 Hz, J = 2.8 Hz), 7.09 (1H, d, J = 8.8 Hz), 2.51 (3H, s), 2.47 (2H, d, J = 7.1 Hz), 2.42 (2H, d, J = 7.1 Hz), 2.22 (2H, m), 1.04 (12H, d, J = 6.6 Hz).
2′,5′-Di-(2,2-dimethylpropanoyloxy)-acetophenone (3e) (95%). 1H NMR (CDCl3) δH 7.42 (1H, d, J = 2.6 Hz), 7.19 (1H, dd, J = 8.7 Hz, J = 2.6 Hz), 7.01 (1H, d, J = 8.7 Hz), 2.50 (3H, s), 1.35 (9H, s), 1.33 (9H, s).
2′,5′-Dihexanoyloxyacetophenone (3f) (82%). 1H NMR (CDCl3) δH 7.49 (1H, d, J = 2.8 Hz), 7.24 (1H, dd, J = 8.6 Hz, J = 2.8 Hz), 7.09 (1H, d, J = 8.6 Hz) 2.58 (4H, m), 2.54 (3H, s), 1.74 (4H, m), 1.37 (8H, m), 0.90 (6H, m).
2′,5′-Dicinnamoyloxyacetophenone (3g) (87%). 1H NMR (CDCl3) δH 7.83 (1H, d, J = 15.9 Hz), 7.82 (1H, d, J = 15.9 Hz), 7.57 (1H, d, J = 2.8 Hz), 7.51 (4H, m), 7.40 (6H, m), 7.30 (1H, dd, J = 8.8 Hz, J = 3.0 Hz), 7.15 (1H, d, J = 8.8 Hz), 6.59 (1H, d, J = 16.1 Hz), 6.56 (1H, d, J = 16.1 Hz), 2.49 (1H, s).
2′,4′-Dipropanoyloxyacetophenone (4a) (88%). 1H NMR (CDCl3)δH 7.82 (1H, d, J = 8.6 Hz), 7.08 (1H, dd, J = 8.6 Hz, J = 2.3 Hz), 6.94 (1H, d, J = 2.3 Hz), 2.52 (3H, s), 2.32 (3H, s), 2.28 (3H, s).
2′,4′-Dibutanoyloxyacetophenone (4b) (72%). 1H NMR (CDCl3) δH 7.81 (1H, d, J = 8.6 Hz), 7.06 (1H, dd, J = 8.6 Hz, J = 2.3 Hz), 6.92 (1H, d, J = 2.3 Hz), 2.57 (2H, t, J = 7.4 Hz), 2.52 (2H, t, J = 7.3 Hz), 2.51 (3H, s), 1.77 (4H, m), 1.03 (3H, t, J = 7.4 Hz), 1.02 (3H, t, J = 7.4 Hz).
2′,4′-Di-(2-methylpropanoyloxy)-acetophenone (4c) (97%). 1H NMR (CDCl3) δH 7.80 (1H, d, J = 8.6 Hz), 7.06 (1H, d, J = 8.6 Hz, J = 2.3 Hz), 6.89 (1H, d, J = 2.3 Hz), 2.80 (3H, s), 2.51 (2H, m), 1.32 (6H, d, J = 7.1 Hz), 1.21 (6H, d, J = 7.1 Hz).
2′4′-Di-(3-methylbutanoyloxy)-acetophenone (4d) (93%). δH 1H NMR (CDCl3) 7.78 (1H, d, J = 8.6 Hz), 7.04 (1H, dd, J = 8.6 Hz, J = 2.3 Hz), 6.89 (1H, d, J = 2.3 Hz), 2.49 (3H, s), 2.46 (2H, d), 2.41 (2H, d), 2.198 (2H, m), 1.03 (6H, s), 1.02 (6H, s).
2′,4′-Di-(2,2-dimethylpropanoyloxy)-acetophenone (4e) (89%). 1H NMR (CDCl3) δH 7.77 (1H, d, J = 8.6 Hz), 7.02 (1H, dd, J = 8.6 Hz, J = 2.3 Hz), 6.81 (1H, d, J = 2.3 Hz), 2.50 (3H, s), 1.35 (9H, s), 1.32 (9H, s).
2′,4′-Dihexanoyloxyacetophenone (4f) (76%). 1H NMR (CDCl3) δH 7.81 (1H, d, J = 8.6 Hz), 7.06 (1H, dd, J = 8.6 Hz, J = 2.3 Hz), 6.92 (1H, d, J = 2.3 Hz), 2.58 (4H, m), 2.51 (3H, s), 1.72 (4H, m), 1.36 (8H, m), 0.91 (6H, t, J = 7.1 Hz).
Synthesis of the intermediate 3-(1,3-dioxopentyl)-4-hydroxyphenyl propanoate (5a–g, 6a–f)
Crude 3a (5.2 g, 20 mmol) was dissolved in dry DMF (30 mL) and added slowly at 0 °C under argon to a suspension of sodium hydride (95% pure, 870 mg, 34.4 mmol) in dry DMF (30 mL). Acetic acid was added to the mixture with caution and then, it was poured into water (300 mL) and extracted with ethyl acetate (3 × 70 mL). The combined organic layers were washed with saturated aqueous sodium chloride solution, dried (MgSO4), and concentrated to yield the crude title compound, which was used in the next step without further purification.
Synthesis of the 2-alkyl-6-hydroxy-4H-benzopyran-4-ones (7a–f)
A solution of 4.3 mmol of crude 5a in methanol (30 mL) was treated with 15 mL of concentrated aqueous hydrochloric acid in 20 mL of dioxane. The mixture was heated to 60 °C and then poured into water, and extracted with ethyl acetate. Subsequent drying over magnesium sulfate and evaporation yielded (7a), which was cristallized in methanol.
2-Ethyl-6-hydroxy-4H-benzopyran-4one (7a) (60%). 1H NMR (DMSO-d6) δH 9.90 (1H, s, OH-6), 7.46 (1H, d, J = 9.0 Hz, H-5), 7.27 (1H, d, J = 2.8 Hz, H-8), 7.18 (1H, dd, J = 9.0 Hz, J = 3.0 Hz, H-7), 6.11 (1H, s, H-3), 2.63 (2H, c, J = 7.5 Hz, Ar-CH2-), 1.21 (3H, t, J = 7.5 Hz, Ar-CH2CH3). 13C NMR (DMSO-d6) δC 177.3, 171.2, 154.9, 150.1, 124.3, 123.1, 119.7, 107.8, 27.0, 11.2. UV: 328nm, 226nm. HRMS (M+H)+ calcd. for C11H10O3, 190.0629; found, 190.0622, Mp. 164–165 °C, lit.,48 mp. 165 °C.
6-Hydroxy-2-propyl-4H-benzopyran-4one (7b) (58%). 1H NMR (DMSO-d6) δH 9.89 (1H, s, OH-6), 7.44 (1H, d, J = 8.8 Hz, H-5), 7.27 (1H, d, J = 3.0 Hz, H-8), 7.18 (1H, dd, J = 8.8 Hz; J = 3.0 Hz, H-7), 6.12 (1H, s, H-3), 2.57 (2H, t, J = 7.4 Hz, Ar-CH2-), 1.66 (2H, m, Ar-CH2CH2-), 0.93 (3H, t, J = 7.3 Hz, Ar-CH2CH2CH3). 13C NMR (DMSO-d6) δC 177.2, 169.5, 155.0, 150.1, 124.4, 123.1, 119.8, 108.8, 108.0, 35.6, 20.2, 13.7. UV: 328 nm, 226 nm. HRMS (M+H)+ calcd. for C12H12O3, 204.0786; found, 204.0792, Mp. 155–156 °C, lit.,37 mp. 150 °C.
6-Hydroxy-2-isopropyl-4H-benzopyran-4one (7c) (56%) 1H NMR (DMSO-d6) δH 9.89 (1H, s, 6-OH), 7.46 (1H, d, J = 9.0 Hz, H-5), 7.27 (1H, d, J = 2.8 Hz, H-8), 7.18 (1H, dd, J = 9.0 Hz, J = 2.8 Hz, H-7), 6.10 (1H, s, H-3), 2.87 (1H, m, Ar-CH-), 1.24 (6H, d, J = 7.0 Hz, Ar-CH(CH3)2). 13C NMR (DMSO-d6) δC 177.4, 174.0, 155.0, 150.0, 124.4, 123.2, 119.8, 107.9, 106.4, 32.9, 20.3. UV: 328 nm, 226 nm. HRMS (M+H)+ calcd for C12H12O3, 204.0786; found, 204.0790. Mp. 182–183 °C.
6-Hydroxy-2-isobutyl-4H-benzopyran-4one (7d) (50%) 1H NMR (DMSO-d6) δH 9.90 (1H, s, OH-6), 7.44 (1H, d, J = 8.8 Hz, H-5), 7.27 (1H, d, J = 3.0 Hz, H-8), 7.17 (1H, dd, J = 8.8 Hz; J = 3.0 Hz, H-7), 6.12 (1H, s, H-3), 2.48 (2H, d, J = 7.3 Hz, Ar-CH2), 2.06 (1H, m, Ar-CH2CH-), 0.93 (6H, d, J = 6.6 Hz, Ar-CH2CH(CH3)2). 13C NMR (DMSO-d6) δC 177.1, 168.7, 155.0, 150.1, 124.4, 123.1, 119.8, 109.7, 108.0, 42.7, 27.0, 22.4. UV: 328 nm, 229 nm. HRMS (M+H)+ calcd for C13H14O3, 218.0942; found, 218.0942. Mp. 145–146 °C.
6-Hydroxy-2-t-butyl-4H-benzopyran-4one (7e) (50%) 1H NMR (DMSO-d6) δH 9.90 (1H, s, OH-6), 7.48 (1H, d, J = 8.8 Hz, H-5), 7.26 (1H, d, J = 2.8 Hz, H-8), 7.19 (1H, dd, J = 8.8 Hz; J = 2.8 Hz, H-7), 6.13 (1H, s, H-3), 1.30 (9H, s, C(CH3)3). 13C NMR (DMSO-d6) δC 177.6, 175.8, 162.3, 154.9, 150.1, 123.3, 119.7, 107.8, 105.4, 36.5, 28.1. UV: 328 nm, 226 nm. HRMS (M+H)+ calcd for C13H14O3, 218.0942; found, 218.0937, Mp. 179–180 °C, lit.,37 mp. 170 °C.
6-Hydroxy-2-pentyl-4H-benzopyran-4one (7f) (40%) 1H NMR (DMSO-d6) δH 9.89 (1H, s, OH-6), 7.44 (1H, d, J = 9.1 Hz, H-5), 7.27 (1H, d, J = 3.0 Hz, H-8), 7.16 (1H, dd, J = 9.1 Hz, J = 3.0 Hz, H-7), 6.13 (1H, s, H-3), 3.31 (2H, t, J = 7.6 Hz, Ar-CH2-), 1.64 (4H, m, Ar-CH2CH2CH2CH2-), 1.31 (2H, m, Ar-CH2CH2CH2-), 0.85 (3H, t, J = 6.8 Hz, Ar-(CH2)4CH3). 13C NMR (DMSO-d6) δC 177.1, 169.7, 155.0, 150.1, 124.4, 123.1, 119.8, 108.7, 108.0, 33.7, 31.0, 26.4, 22.2, 14.2. UV: 328 nm, 226 nm. HRMS (M+H)+ calcd for C14H16O3, 232.1099; found, 232.1093, Mp. 118–120 °C.
7-Hydroxy-2-propyl-4H-benzopyran-4one (8a) (42%) 1H NMR (DMSO-d6) δH 10.68 (1H, s, OH-7), 7.82 (1H, d, J = 8.5 Hz, H-5), 6.87 (1H, dd, J = 8.5 Hz; J = 2.5 Hz, H-6), 6.79 (1H, d, J = 2.5 Hz, H-8), 6.06 (1H, s, H-3), 2.56 (2H, t, J = 7.4 Hz, Ar-CH2-), 1.67 (2H, m, Ar-CH2CH2-), 0.93 (3H, t, J = 7.4 Hz, Ar-CH2CH2CH3). 13C NMR (DMSO-d6) δC 176.6, 169.0, 162.9, 158.2, 126.9, 116.3, 115.1, 109.3, 102.6, 35.5, 20.1, 13.7. UV: 292 nm, 247 nm, 214 nm. HRMS (M+H)+ calcd for C12H12O3, 204.0786; found, 204.0778. Mp. 150–152 °C, lit.,37 mp. 143 °C.
7-Hydroxy-2-isopropyl-4H-benzopyran-4one (8b) (45%) 1H NMR (DMSO-d6) δH 10.68 (1H, s, OH-7), 7.82 (1H, d, J = 8.6 Hz, H-5), 6.87 (1H, dd, J = 8.8 Hz, J = 2.3 Hz, H-6), 6.80 (1H, d, J = 2.0 Hz, H-8), 6.04 (1H, s, H-3), 2.83 (1H, m, Ar-CH-) 1.23 (6H, d, J = 7.1 Hz, Ar-CH(CH3)2). 13C NMR (DMSO-d6) δC 176.9, 173.6, 163.1, 157.8, 126.9, 116.5, 114.9, 107.0, 102.7, 32.8, 20.5. UV: 295 nm, 247 nm, 214 nm. HRMS (M+H)+ calcd. for C12H12O3, 204.0786; found, 204.0791. Mp. 156–158 °C.
7-Hydroxy-2-isobutyl-4H-benzopyran-4one (8c) (52%) 1H NMR (DMSO-d6) δH 10.68 (1H, s, OH-7), 7.83 (1H, d, J = 8.6 Hz, H-5), 6.87 (1H, dd, J = 8.8 Hz, J = 2.3 Hz, H-6), 6.80 (1H, d, J = 2.0 Hz, H-8), 6.06 (1H, s, H-3), 2.46 (2H, d, J = 7.3 Hz, Ar-CH2), 2.05 (1H, m, Ar-CH2CH-), 0.93 (6H, d, J = 6.8 Hz, ArCH2CH(CH3)2). 13C NMR (DMSO-d6) δC 176.6, 168.2, 162.9, 158.3, 126.9, 116.5, 115.2, 110.3, 102.7, 42.5, 26.9, 22.1. UV: 292 nm, 247 nm, 214 nm. HRMS (M+H)+ calcd for C13H14O3, 218.0942; found, 218.0936. Mp. 152–153 °C.
7-Hydroxy-2-t-butyl-4H-benzopyran-4one (8d) (40%) 1H NMR (DMSO-d6) δH 10.69 (1H, s, OH-7), 7.82 (1H, d, J = 8.9 Hz, H-5), 6.87 (1H, dd, J = 8.7 Hz, J = 2.3 Hz, H-6), 6.82 (1H, d, J = 2.0 Hz, H-8), 6.07 (1H, s, H-3), 1.28 (9H, s, -C(CH3)3). 13C NMR (DMSO-d6) δC 177.0, 175.2, 163.0, 158.1, 126.8, 116.0, 115.2, 106.0, 102.6, 36.4, 27.9. UV: 295 nm, 247 nm, 214 nm. HRMS (M+H)+ calcd for C13H14O3, 218.0942; found, 218.0943. Mp 222–223 °C.
7-Hydroxy-2-pentyl-4H-benzopyran-4one (8e) (45%) 1H NMR (DMSO-d6) δH 10.65 (1H, s, OH-7), 7.82 (1H, d, J = 8.7 Hz, H-5), 6.87 (1H, dd, J = 8.9 Hz, J = 2.3 Hz, H-6), 6.80 (1H, d, J = 2.3 Hz, H-8), 6.07 (1H, s, H-3), 2.58 (2H, t, J = 7.5 Hz, Ar-CH2), 1.64 (2H, m, Ar-CH2CH2CH2-), 1.31 (4H, m, Ar-CH2CH2CH2CH2-), 0.86 (3H, m, Ar-(CH2)4CH3). 13C NMR (DMSO-d6) δC 176.6, 169.3, 162.9, 158.2, 126.9, 116.3, 115.1, 109.2, 102.6, 33.6, 31.0, 26.3, 22.2, 14.2. UV: 247 nm, 214 nm. HRMS (M+H)+ calcd for C14H16O3, 232.1099; found: 232.1094, Mp 135–136 °C.
General procedure for the synthesis of 2-alkyl-6,7-methylenedioxybenzopyran-4-ones (12a–c)
Synthesis of 2-hydroxy-4,5-methylenedioxyacetophenone (10) (77%)
To a solution of sesamol (2.76 g, 0.02 mol) in Ac2O (10 mL) was added 5 mL of the boron trifluoride etherate complex at 0°C, under argon. The resulting solution was heated to 80–90 °C for 1h and poured into a saturated NaOAc solution (20mL). After extracting with Et2O, the Et2O solution was washed with 10% aqueous NaHCO3 solution and H2O, dried (Na2SO4) and concentrated under reduced pressure. The residue was recrystallized from absolute EtOH to obtain the acetophenone 10. 1H NMR (CDCl3) δH 12.99 (1H, s), 7.03 (1H, s), 6.43 (1H,s), 5.95 (2H,s), 2.50 (3H,s). 13C NMR (CDCl3) δC 201.9. 162.1, 1154.4, 140.5, 112.3, 107.2, 101.9, 98.7, 26.4.
Synthesis of 2-alkyl benzopyran-4-ones (12a–c)
A solution of 10 (5 g, 32.8 mmol) in dry pyridine (35 mL) was treated with trimethylacetyl chloride (10 g, 83 mmol) under cooling with an ice-bath to keep the reaction temperature at about 20 °C. The mixture was stirred for 18 h at room temperature; and the solvent was partially distilled off under reduced pressure. The residue was poured onto ice and hydrochloric acid (32 %, 30 mL) and extracted with diethyl ether. The combined organic layers were washed with aqueous sodium carbonate solution and water, dried (MgSO4) and concentrated to give the crude title compound 11a.
Crude 11a (5.2 g, 19 mmol) was dissolved in dry DMF (30 mL) and added slowly to a suspension of sodium hydride (95% pure, 870 mg, 34.4 mmol) in dry DMF (30 mL), at 0 °C, under argon. Acetic acid was added to the mixture with caution and then, it was poured into water (300 mL) and extracted with ethyl acetate (3 × 70 mL). The combined organic layers were washed with saturated aqueous sodium chloride solution, dried (MgSO4), and evaporated to yield the crude title compound. A solution of this crude compound in methanol (30 mL) was treated with concentrated aqueous hydrochloric acid in dioxane. The mixture was heated to 60 °C and then poured into water, and washed with ethyl acetate. Subsequent drying over magnesium sulfate and evaporation yielded (12a).
6,7-Methylenedioxy-2-t-butylbenzopyran-4-one (12a). 1H NMR (CDCl3) δH 7.46 (1H,s, H-5), 6.84 (1H, s, H-8), 6.27 (1H,s, H-3), 6.06 (2H, s, O-CH2-O), 1.31 (9H, s, C(CH3)3). 13C NMR (CDCl3) δC 177.9, 175.5, 153.9, 152.8, 146.1, 118.1, 106.2, 102.4, 102.2, 97.9, 36.4, 28.0. UV: 319 nm, 277 nm, 235 nm. HRMS (M+H)+ calcd for C14H14O4, 246.0892; found: 246.0897, Mp 214–215 °C.
2-Isobutyl-6,7-methylenedioxybenzopyran-4-one (12b). 1H NMR (CDCl3) δH 7.48 (1H,s, H-5), 6.82 (1H, s, H-8), 6.19 (1H, s, H-3), 6.07 (2H, s, O-CH2-O), 2.44 (2H, d, J = 7.1 Hz, Ar-CH2-), 2.10 (1H, m, Ar-CH2CH-), 0.98 (6H, d, J = 6.6 Hz, Ar-CH2CH(CH3)2). 13C NMR (CDCl3) δC 177.3, 168.5, 154.0, 152.8, 146.1, 118.4, 110.0, 102.3, 97.9, 43.3, 27.2, 22.3. UV: 319 nm, 277 nm, 235 nm. HRMS (M+H)+ calcd for C14H14O4, 246.0892; found: 246.0890, Mp 90–92 °C.
6,7-Methylenedioxy-2-phenylbenzopyran-4-one (12c). 1H NMR (CDCl3) δH 8.03, (2H, dd, J = 8.0 Hz, J = 2.1 Hz, H-2′, H-6′), 7.56 (3H, m, H-3′, H-4′, H-5′), 7.37 (1H, s, H-5), 7.31 (1H, s, H-8), 6.94 (1H, s, H-3), 6.21 (2H, s, O-CH2-O). 13C NMR (CDCl3) δC 176.6, 162.5, 153.6, 153.3, 146.8, 132.2, 131.8, 129.8, 118.9, 106.9, 103.5, 101.5, 99.2. HRMS (M+H)+ calcd for C16H10O4, 266.5792; found 265.99224 (The molecular ion does not appear with sufficient intensity) Mp 210–212 °C.
Synthesis of 6,7-dihydroxy-2-t-butylbenzopyran-4-one (14). A solution of 12c (0.7 g, 0.65 mmol) in chloroform (8 mL) was treated with PCl5 (0.3 g, 1.3 mmol). The mixture was refluxed for 7 h and after cooling the solvent was removed under reduced pressure. The residue was hydrolyzed in water refluxing for 2 h, and after cooling was extracted twice with ethyl acetate. The solvent was removed and the product was purified through a silica gel column, eluting with a 2:1 CH2Cl2/EtOAc in a 2:1 proportion as eluent to give (14) (53%). 1H NMR (CDCl3) δH 8.06 (1H, s, OH-7), 7.98 (1H, s, OH-6), 7.55 (1H, s, H-5), 7.26 (1H, s, H-8), 6.29 (1H, s, H-3), 1.35 (9H, s, -C(CH3)3). 13C NMR (CDCl3) δC 179.1, 177.0, 150.5, 150.3, 128.2, 123.1, 119.1, 110.6, 106.1, 37.8, 28.1. HRMS (M+H)+ calcd for C13H14O4, 234.0892; found: 234.0894, Mp 148–149 °C.
General procedure for the synthesis of the intermediate compounds (16a–f). Synthesis of 2′-propanoyloxy acetophenone (16a)
A mixture of (15) (1.5 g, 11 mmol) and propionyl chloride (2.62 mL, 22 mmol) was stirred in dry pyridine (5 mL) at room temperature for 2 h. The mixture was then poured into a mixture of crushed ice (25 mL) and concentrated HCl (1.5 mL), extracted twice with dichloromethane and the combined organics were washed three times with water. The solvent was removed under reduced pressure. The residue was purified through a silica gel column using CH2Cl2 as eluent to give (16a) (2.3g, 97%). 1H NMR (CDCl3) δH 7.78 (1H, dd, J = 8.1 Hz, J = 2.1 Hz), 7.50 (1H, ddd, J = 7.7 Hz, J = 8.1 Hz, J = 1.7 Hz), 7.29 (1H, ddd, J = 7.7 Hz, J = 8.1 Hz, J = 1.7 Hz), 7.08 (1H, dd, J = 8.5 Hz, J = 1.1 Hz), 2.63 (2H, q), 2.52 (3H, s), 1.26 (3H, t, J = 7.5 Hz).
2′-Butanoyloxyacetophenone (16b) (90%). 1H NMR (CDCl3) δH 7.76 (1H, dd, J = 7.8 Hz, J = 1.5 Hz), 7.50 (1H, ddd, J = 7.8 Hz, J = 7.6 Hz, J = 8.0 Hz, J = 1.8 Hz), 7.29 (1H, ddd, J = 7.8 Hz, J = 7.6 Hz, J = 7.6 Hz, J =1.0 Hz), 7.06 (1H, dd, J = 8,1 Hz, J = 1.3 Hz), 2.84 (1H, m), 2.53 (3H, s), 1.33 (6H, d, J = 6.8 Hz).
2′-(2-Methylpropanoyloxy)-acetophenone (16c) (98%). 1H NMR (CDCl3) δH 7.77 (1H, dd, J = 7.8 Hz, J = 1.8 Hz), 7.50 (1H, ddd, J = 7.5 Hz, J = 8.1 Hz, J = 1.8 Hz, J = 1.5 Hz), 7.29 (1H, ddd, J = 7.6 Hz, J = 7.5 Hz, J = 7.7 Hz, J = 1.3 Hz), 7.08 (1H, dd, J = 8.1 Hz, J = 1.0 Hz), 2.58 (2H, t, J = 7.4 Hz), 2.53 (3H, s), 1.79 (2H, m), 1.04 (3H, t, J = 7.3 Hz).
2′-(3-Methylbutanoyloxy)-acetophenone (16d) (80%). 1H NMR (CDCl3) δH 7.76 (1H, dd, J = 7.8 Hz, J = 1.8 Hz), 7.49 (1H, ddd, J = 7.5 Hz, J = 8.0 Hz, J = 8.2 Hz, J = 1.8 Hz), 7.28 (1H, ddd, J = 7.6 Hz, J = 7.5 Hz, J = 7.8 Hz, J = 1.3 Hz), 7.08 (1H, dd, J = 8.1 Hz, J = 1.5 Hz), 2.52 (3H, s), 2.48 (2H, d, J = 7.1 Hz), 2.24 (1H, m), 1.05 (6H, d, J = 6.6 Hz).
2′-(2,2-Dimethylpropanoyloxy)-acetophenone (16e) (98%). 1H NMR (CDCl3) δH 7.74 (1H, dd, J = 7.8 Hz, J = 1.8 Hz), 7.49 (1H, ddd, J = 7.3 Hz, J = 8.1 Hz, J = 8.1 Hz, J =1.8 Hz), 7.28 (1H, ddd, J = 7.6 Hz, J = 7.6 Hz, J = 7.6 Hz, J = 1.3 Hz), 7.02 (1H, dd, J = 8.1 Hz, J = 1.0 Hz), 2.5 (3H, s), 1.4 (9H, s).
2′-Hexanoyloxyacetophenone (16f) (49%). 1H NMR (CDCl3) δH 7.77 (1H, dd, J = 7.8 Hz, J = 1.8 Hz), 7.50 (1H, ddd, J = 7.4 Hz, J = 8.0 Hz, J = 8.0 Hz, J = 1.8 Hz), 7.28 (1H, ddd, J = 7.6 Hz, J = 7.6 Hz, J = 7.6 Hz, J = 1.3 Hz), 7.08 (1H, dd, J = 8.1 Hz, J = 1.3 Hz), 2.59 (2H, t, J = 7.6 Hz), 2.52 (3H, s), 1.75 (2H, m), 1.38 (4H, m), 0.91 (3H, t, J = 7.1 Hz).
General procedure for the synthesis of 17a–f. Synthesis of 2′-propanoyloxy-2-bromoacetophenone (17a)
To a solution of (16a) (2.2 g, 11.5 mmol) in anhydrous tetrahydrofuran (10 mL), PTT (4.3 g, 11.5 mmol) was added in portions over a period of 10 min. The reaction mixture was stirred until a colorless precipitate was formed. The residue was recrystallized from ethanol to give (17a) (3.0 g, 97 %), 1H NMR (CDCl3) δH 7.80 (1H, dd, J = 7.7 Hz, J = 1.5 Hz), 7.56 (1H, ddd, J = 7.8 Hz, J = 7.9 Hz, J = 7.7 Hz J = 1.5 Hz), 7.32 (1H, ddd, J = 7.6 Hz, J = 7.7 Hz, J = 7.5 Hz, J = 1.1 Hz), 7.18 (1H, dd, J = 8.1 Hz, J = 0.8 Hz), 4.39 (2H, s), 2.65 (2H, c, J = 7.5 Hz), 1.27 (3H, t, J = 7.5 Hz).
2′-Butanoyloxy-2-bromoacetophenone (17b) (98%). 1H NMR (CDCl3) δH 7.77 (1H, dd, J = 7.9 Hz, J = 1.7 Hz), 7.55 (1H, ddd, J = 7.5 Hz, J = 8.2 Hz, J = 8.1, Hz J = 1.7 Hz), 7.31 (1H, ddd, J = 7.6 Hz, J = 7.5 Hz, J = 7.7 Hz, J = 1.1 Hz), 7.13 (1H, dd, J = 8.1 Hz, J = 1.1 Hz), 4.38 (2H, s), 2.84 (1H, m), 1.33 (6H, d, J = 7.2 Hz).
2′-(2-Methylpropanoyloxy)-2-bromoacetophenone (17c) (98%). 1H NMR (CDCl3) 7.79 (1H, dd, J = 7.7 Hz, J = 1.7 Hz), 7.55 (1H, ddd, J = 7.5 Hz, J = 8.2 Hz, J = 7.9 Hz, J =1.5 Hz), 7.31(1H, ddd, J = 7.7 Hz, J = 7.6 Hz, J = 8.1 Hz, J = 1.1Hz), 7.16 (1H, dd, J = 8.3 Hz, J = 1.3 Hz), 4.39 (2H, s), 2.59 (2H, t, J = 7.4 Hz), 1.78 (2H, m), 1.03 (3H, t, J = 7.3 Hz).
2′-(3-Methylbutanoyloxy)-2-bromoacetophenone (17d) (98%). 1H NMR (CDCl3) δH 7.78 (1H, dd, J = 7.9 Hz, J = 1.7 Hz), 7.55 (1H, ddd, J = 7.5 Hz, J = 8.1 Hz, J = 7.7 Hz, J = 1.7 Hz), 7.31(1H, ddd, J = 7.7 Hz, J =7.8 Hz, J =7.5 Hz, J = 1.1 Hz), 7.15 (1H, dd, J =8.1 Hz, J = 1.1 Hz), 4.39 (2H, s), 2.50 (2H, d, J = 7.0 Hz), 2.25 (1H, m), 1.05 (6H, s).
2′-(2,2-Dimethylpropanoyloxy)-2-bromoacetophenone (17e) (96%). 1H NMR (CDCl3) δH 7.75 (1H, dd, J = 7.7 Hz, J = 1.7 Hz), 7.54 (1H, ddd, J = 7.5 Hz, J = 8.1 Hz, J = 8.1 Hz, J =1.7 Hz), 7.33 (1H, ddd, J = 7.0 Hz, J = 7.4 Hz, J = 7.6 Hz, J = 1.1 Hz), 7.07 (1H, dd, J = 8.1 Hz, J = 1.1 Hz), 4.37 (2H, s), 1.37 (9H, s).
2′-Hexanoyloxy-2-bromoacetophenone (17f) (97%). 1H NMR (CDCl3) δH 7.79 (1H, dd, J = 7.9 Hz, J = 1.7 Hz), 7.56 (1H, ddd, J = 7.4Hz, J = 8.3 Hz, J = 8.1 Hz, J =1.7 Hz), 7.32 (1H, ddd, J = 7.6 Hz, J = 7.9 Hz, J = 7.4 Hz, J = 1.1 Hz), 7.16 (1H, dd, J = 8.1 Hz, J = 1.1 Hz), 4.39 (2H, s), 2.61 (2H, m), 1.75 (2H, m), 1.37 (4H, m), 0.91 (3H,t, J = 7.0 Hz).
General procedure for the synthesis of 18a–f. Synthesis of 2-(2-phenoxyacetyl) phenyl propionate (18a)
A mixture of (18a) (3.0 g, 11 mmol) and potassium benzoate (2.7 g, 17 mmol) was stirred in acetonitrile (48 mL) at room temperature for 48 h. The salts were filtered off, and the solvent was removed under reduced pressure. The residue was taken up in dichloromethane, washed with aqueous sodium carbonate, and then washed with water. The solvent was removed under reduced pressure, and the residue was recrystallized from ethanol to give 18a (53%).
General procedure for the synthesis of (20a–f). Synthesis of 2-ethyl-4-oxo-4H-chromen-3-yl benzoate (20a)
To a suspension of sodium hydride (148 mg, 6.2 mmol) in dry DMF (2.5 mL), was added 18a (1.8 g, 5.6 mmol) in DMF (2.5 mL). The reaction mixture was refluxed for 90 min with stirring, and then the cooled mixture was poured into a mixture of ice (120 g) and concentrated HCl (2 mL). The crude β-diketone (19a) precipitated subsequently, was washed with water, and was used in the following ring closure step without further purification.
To a solution of crude β-diketone (19a) in acetic acid (10 mL), concentrated sulfuric acid (0.25 mL) was added dropwise. The reaction mixture was heated at 60 °C for 90 min with stirring, and the solution was poured over ice (60 g). The precipitate was filtered, washed with water and recrystallized from ethanol to yield the pure (20a) (556 mg, 60 %) as colorless needles. 1H NMR (CDCl3) δH 8.17 (1H, dd, J = 7.6 Hz, J = 1.8 Hz), 8.16 (1H, dd, J = 8.3 Hz, J = 1.3 Hz), 7.61 (1H, ddd, J = 7.8 Hz, J = 7.8 Hz, J = 7.8 Hz, J = 1.5 Hz), 7.56 (1H, dd), 7.43 (3H, m), 7.33 (1H, ddd, J = 7.6 Hz, J = 7.6 Hz, J = 7.6 Hz, J = 1.0 Hz), 2.70 (2H, c, J = 7,6 Hz), 1.27 (3H, t, J = 7,6 Hz).
2-Isopropyl-4-oxo-4H-chromen-3-yl benzoate (20b) (80%). 1H NMR (CDCl3) δH 8.15 (3H, m), 7.59 (2H, m), 7.45 (3H, m), 7.32 (1H, ddd, J = 7.5 Hz, J = 7.5 Hz, J = 7.6 Hz, J = 1.0 Hz), 3.21 (1H, q, J = 6.8 Hz), 1.3 (6H, d, J = 6.8 Hz).
4-Oxo-2-propyl-4H-chromen-3-yl benzoate (20c) (55%). 1H NMR (CDCl3) δH 8.22 (3H, m), 7.65 (2H, m), 7.50 (3H, m), 7.40 (1H, ddd, J = 7.6 Hz, J = 7.5 Hz, J = 7.7 Hz, J = 1.0 Hz), 2.70 (2H, t, J = 7.3 Hz), 1.81 (2H, m), 1.01 (3H, t, J = 7.3 Hz).
2-Isobutyl-4-oxo-4H-chromen-3-yl benzoate (20d) (92%). 1H NMR (CDCl3) δH 8.17 (3H, m), 7.59 (2H, m), 7.43 (3H, m), 7.34 (1H, ddd, J = 7.6 Hz, J = 7.5 Hz, J = 7.7 Hz, J = 1.0 Hz), 2.55 (2H, d, J = 7.3 Hz), 2.16 (1H, m), 0.96 (6H, d, J = 6.8 Hz).
2-t-Butyl-4-oxo-4H-chromen-3-yl benzoate (20e) (61%). 1H NMR (CDCl3) δH 8.12 (3H, m), 7.60 (2H, m), 7.44 (3H, m), 7.31 (1H, ddd, J = 7.6 Hz, J = 7.6 Hz, J = 7.6 Hz, J = 1.01 Hz), 1.36 (1H, s).
4-Oxo-2-pentyl-4H-chromen-3-yl benzoate (20f) (85%). 1H NMR (CDCl3) δH 8.13 (3H, m), 7.59 (2H, m), 7.43 (3H, m), 7.33 (1H, ddd, J = 7.6 Hz, J = 7.6 Hz, J = 7.6 Hz, J = 0.8 Hz), 2.55 (2H, t, J = 7.6 Hz), 1.68 (2H, t, J = 7,0 Hz), 1.29 (4H, m), (0.82, 3H, t, J = 7.1 Hz).
General procedure for the synthesis of (21a–f)
2-Ethyl-3-hydroxy-4H-benzopyran-4-one (21a)
Compound (20a) (500 mg, 1.7 mmol) was hydrolyzed with 5 % aqueous sodium hydroxide solution (5 mL) in ethanol (40 mL) at 60 °C for 2 h. The reaction mixture was poured into a mixture of crushed ice (60 g) and concentrated HCl (1 mL). It was then extracted with ethyl acetate and washed with aqueous sodium hydrogen carbonate and water. The solvent was removed under reduced pressure, and the residue was recrystallized from aqueous ethanol to give 21a (170 mg, 53 %) 1H NMR (CDCl3) δH 8.15 (1H, dd, J = 7.3 Hz, J = 1.8 Hz, H-5), 7.57 (1H, ddd, J = 8.7 Hz, J = 7.2 Hz, J = 1.5 Hz, H-7), 7.40 (1H, dd, J = 8.6 Hz, J = 1.0 Hz, H-8), 7.30 (1H, ddd, J = 8.1 Hz, J = 7.1 Hz, J = 1.0 Hz, H-6), 6.08 (1H, s, OH-3), 2.81 (2H, c, Ar-CH2), 1.28 (3H, t, J = 7.7 Hz, CH2CH3). 13C NMR (CDCl3) δC 172.5, 155.7, 153.3, 137.7, 133.0, 125.5, 124.3, 121.5, 118.1, 22.4, 10.9. HRMS (M+H)+ calcd for C11H10O3, 190.0629; found, 190.0630. Mp 99–101 °C, UV (MeOH) λmax 319 nm, 283 nm, 232 nm, 205 nm.
2-Isopropyl-3-hydroxy-4H-benzopyran-4-one (21b) (61%). 1H NMR (CDCl3) δH 8.15 (1H, dd, J = 8.1 Hz, J = 1.8 Hz, H-5), 7.57 (1H, ddd, J = 8.7 Hz, J = 7.1 Hz, J = 1.8 Hz, H-7), 7.42 (1H, dd, J = 8.8 Hz, J =1.0 Hz, H-8), 7.30 (1H, ddd, J = 8.0 Hz J = 7.0 Hz, J = 1.2 Hz, H-6), 6.27 (1H, s, OH-3), 3.41 (1H, m, Ar-CH-), 1.28 (6H, d, J = 6.8 Hz, Ar-CH(CH3)2). 13C NMR (CDCl3) δC 172.7, 156.0, 155.7, 136.9, 132.9, 125.5, 124.3, 121.4, 118.1, 28.2, 19.4. HRMS (M+H) + calcd for C12H12O3, 204.0786; found, 204.0788. Mp. 152–153 °C, UV (MeOH) λmax 319 nm, 283 nm, 232 nm, 205 nm.
3-Hydroxy-2-propyl-4H-benzopyran-4-one (21c) (63%). 1H NMR (CDCl3) δH 8.16 (1H, dd, J = 7.8 Hz, J = 1.5 Hz, H-5), 7.56 (1H, ddd, J = 8.7 Hz, J = 7.0 Hz, J = 1.8 Hz, H-7), 7.39 (1H, dd, J = 8.6 Hz, J = 1.3 Hz, H-8), 7.30 (1H, ddd, J = 8.1 Hz, J = 7.1 Hz, J = 1.1 Hz, H-6), 6.23 (1H, s, OH-3), 2.75 (2H, t, J = 7.4 Hz, Ar-CH2), 1.74 (2H, m, Ar-CH2CH2-), 0.86 (3H, t, J = 7.4 Hz, Ar-CH2CH2CH3). 13C NMR (CDCl3) δC 172.5, 155.7, 152.3, 138.3, 132.9, 125.5, 124.3, 121.4, 118.1, 30.8, 20.1, 13.7. HRMS (M+H)+ calcd for C12H12O3 204.0786; found, 204.0790. Mp. 102–104 °C. UV (MeOH) λmax 319 nm, 283 nm, 232 nm, 205 nm.
2-Isobutyl-3-hydroxy-4H-benzopyran-4-one (21d) (45%). 1H NMR (CDCl3) δH 8.21 (1H, dd, J = 8.1 Hz, J = 1.7 Hz, H-5), 7.62 (1H, ddd, J = 8.6 Hz, J = 7.0 Hz, J = 1.7 Hz, H-7), 7.44 (1H, dd, J = 8.5 Hz, J = 0.9 Hz, H-8) 7.35 (1H, ddd, J = 8.1 Hz, , J = 7.1 Hz, J = 1.0 Hz, H-6), 6.23 (1H, s, OH-3), 2.71 (2H, d, J = 7.2 Hz, Ar-CH2-, 2.21 (1H, m, Ar-(CH2CH(CH3)2), J = 1.01 (6H, d, J = 6.6 Hz, Ar- (CH2CH(CH3)2). 13C NMR (CDCl3) δC 172.4, 155.7, 151.7, 138.8, 133.0, 125.4, 124.2, 121.4, 118.1, 37.9, 27.2, 22.5. HRMS (M+H)+ calcd for C13H14O3, 218.0942; found, 218.0947. Mp. 130–131 °C, UV (MeOH) λmax 319 nm, 283 nm, 232 nm, 205 nm.
3-Hydroxy-2-t-butyl-4H-benzopyran-4-one (21e) (40%) 1H NMR (CDCl3) δH 8.13 (1H, dd, J = 8.1 Hz, J = 1.5 Hz, H-5), 7.57 (1H, ddd, J = 8.6 Hz, J = 7.0 Hz, J = 1.6 Hz, H-7), 7.41 (1H, dd, J = 8.5 Hz, J = 0.9 Hz, H-8), 7.30 (1H, ddd, J = 8.0 Hz, J = 7.0 Hz, J = 1.1 Hz, H-6), 6.45 (1H, s, OH-3), 1.41 (9H, s, C(CH3)3). 13C NMR (CDCl3) δC 173.3, 162.6, 155.5, 137.1, 133.1, 125.4, 124.4, 121.1, 118.3, 36.5, 27.6. HRMS (M+H)+ calcd for C13H14O3, 218.0942; found, 218.0948. Mp. 169–171 °C, UV (MeOH) λmax 319 nm, 283 nm, 232 nm, 205 nm.
3-Hydroxy-2-pentyl-4H-benzopyran-4-one (21f) (65%) 1H NMR (CDCl3) δH 8.15 (1H, dd, J = 8.3 Hz, J = 1.5 Hz, H-5), 7.57 (1H, ddd, J = 8.6 Hz, J = 7.0 Hz, J = 1.7 Hz, H-7), 7.40 (1H, dd, J = 8.5 Hz, J = 0.6 Hz, H-8), 7.30 (1H, ddd, J = 8.1 Hz, J = 7.0 Hz, J = 1.1 Hz, H-6), 6.18 (1H, s, OH-3), 2.77 (2H, t, J = 7.6 Hz, Ar-CH2-), 1.71 (2H, m, Ar-CH2CH2CH2-), 1.32 (4H, m, Ar-CH2CH2CH2CH2-), 0.84 (3H, m, Ar-(CH2)4CH3). 13C NMR δC 172.5, 155.6, 152.5, 138.2, 133.1, 125.5, 124.3, 121.5, 118.1, 31.4, 28.9, 26.4, 22.4, 14.0. HRMS (M+H)+ calcd for C14H16O3, 232.1099; found, 232.1105. Mp. 96–99 °C. UV (MeOH) λmax 319 nm, 283 nm, 232 nm, 205 nm.
General procedure for the synthesis of compounds 25a–j
Dry HCl (g) was passed into a cooled (0 °C), stirred mixture of the substituted phenylacetonitrile (22) (0.34 mol) and anhydrous ZnCl2 (30 g, 0.22 mol) in dry diethyl ether (200 mL). The corresponding polyhydroxybenzene (23) (0.28 mol) was added portionwise with constant bubbling of gaseous HCl for a further 4–6 hours. The reaction mixture was then stirred at room temperature for 4 to 20 hours, until the complete disappearance of the reagents, as shown by thin-layer chromatography.
The ketiminium chloride intermediate that separated as an oil after 3–4 days was washed with diethyl ether and hydrolyzed by refluxing in 5% HCl (1 L) for 4 to 5 hours. The ketone (24), which separated upon cooling, was filtered and recrystallized in the appropriate solvent.
To a solution of benzylketone (24) (0.18 mol) in dry DMF (200 mL) BF3Et2O (0.88 mol) was added dropwise. This solution was warmed to 50 °C and a solution of methanesulfonyl chloride (0.56 mol) in DMF (100 mL) was slowly added. The resulting mixture was then heated to 100 °C for 2 hours. After cooling, it was poured into water (400 mL) and left overnight to give a precipitate, which was then stirred for 2 hours in cold methanol (50 mL), filtered and crystallized in a mixture of water/ethanol.
6,7-Dihydroxy-4′-methoxyisoflavone (25a) (72%). 1H NMR (DMSO-d6) δH 10.38 (1H, s, 7-OH), 9.75 (1H, s, 6-OH), 8.27 (1H, s, 2-H), 7.49 (2H, d, J = 8.6 Hz, 2′-H, 6′-H), 7.37 (1H, s, 5-H), 6,97 (2H, d, J = 8.6 Hz, 3′-H, 5′-H), 6.89 (1H, s, H-8), 3.76 (3H, s, OCH3). 13C NMR (DMSO-d6) δC 174.9, 159.5, 153.3, 152.9, 151.5, 145.4, 130.7, 125.3, 123.1, 117.2, 114.2, 108.8, 103.4, 55.8. Mp. 251 °C.
4′,6,7-Trihydroxyisoflavone (25b) (53%). 1H NMR (DMSO-d6) δH 9.84 (1H, s, 7-OH), 8.21 (1H, s, 6-OH), 7.36 (1H, s, H-5), 7.35 (2H, d, J = 8.7 Hz, H-2′, H-6′), 6.87 (1H, s, H-8), 6.78 (2H, d, J = 8.7 Hz, H-3′, H-5′), 4.46 (1H, dd, J = 11.0 Hz, J = 13.1 Hz, H-3), 4.43 (1H, dd, J = 5.5 Hz, J = 5.9 Hz, H-2a), 3.96 (1H, dd, J = 5.3 Hz, J = 7.2 Hz, H-2e). 13C NMR (DMSO-d6) δC 187.9, 174.98, 157.8, 153.1, 151.4, 145.3, 130.7, 130.3, 123.4, 117.6, 115.6, 108.6, 103.4. Mp. 225°C
6,7-Dihydroxy-4′-nitroisoflavone (25c) (81%) 1H NMR (DMSO-d6) δH 10.11 (1H, s, 7-OH), 8.54 (1H, s, 2-H), 8.26 (2H, d, J = 8.8 Hz, H-3′, H-5′), 7.89 (2H, d, J = 8.8 Hz, H-2′, H-6′), 7.40 (1H, s, H-5), 6.93 (1H, s, H-8), [signal for 8-OH not seen]. 13C NMR (DMSO-d6) δC 174.3, 155.5, 153.3, 151.7, 147.2, 145.8, 140.4, 130.5, 123.9, 121.6, 117.1, 108.9, 103.7. Mp. 168 °C.
6,7-Dihydroxy-3′,4′-methylenedioxyisoflavone (25d) (67%). 1H NMR (DMSO-d6) δH 10.1 (1H, s, 7-OH), 9.63 (1H, s, 6-OH), 8.28 (1H, s, 2-H), 7.37 (1H, s, H-5), 7.12 (1H, d, J = 1.6 Hz, H-2′), 7.03 (1H, dd, J = 1.6 Hz, J = 8.0 Hz, H-6′), 6.94 (1H, d, J = 8.0 Hz, H-5′), 6.88 (1H, s, H-8), 6.02 (2H, s, O-CH2-O). 13C NMR (DMSO-d6) δC 174.8, 153.6, 153.0, 151.5, 147.4, 145.4, 126.8, 123.1, 123.0, 117.2, 110.1, 108.8, 108.7, 103.4, 101.6. HRMS (M+H)+ calcd for C16H10O6, 298.0478; found 298.0480. Mp. 260°C.
7,8-Dihydroxy-4′-methylisoflavone (25e) (78%). 1H NMR (DMSO-d6) δH 9.81 (1H, s, 8-OH), 8.38 (1H, s, 2-H), 7.47 (1H, d, J = 8.7 Hz, H-5), 7.45 (2H, d, J = 8.1 Hz, H-2′, H-6′), 7.21 (2H, d, J = 8.1 Hz, H-3′, H-5′), 6.95 (1H, d, J = 8.7 Hz, H-6), 2.32 (3H, s, CH3), [signal for 7-OH not seen]. 13C NMR (DMSO-d6) δC 175.6, 153.9, 150.8, 147.4, 137.6, 133.6, 129.9, 129.5, 129.3, 123.5, 118.1, 116.3, 114.9, 21.5. Mp. 241 °C.
7,8-Dihydroxyisoflavone (25f) (76%). 1H NMR (DMSO-d6) δH 9.83 (1H, s, 8-OH), 8.43 (1H, s, 2-H), 7.57 (2H, d, J = 7.0 Hz, H-3′, H-5′), 7.48 (1H, d, J = 8.6 Hz, H-5), 7.42 (2H, t, J = 7.0 Hz, H-2′, H-6′), 7.37 (1H, d, J = 8.6 Hz, H-6), [signal for 7-OH not seen]. 13C NMR (DMSO-d6) δC 175.5, 154.3, 150.8, 147.4, 133.6, 132.9, 129.7, 128.7, 128.3, 123.7, 118.1, 116.4, 114.9. Mp. 211 °C.
4′-Chloro-7,8-dihy droxyisoflavone (25g) (65%). 1H NMR (DMSO-d6) δH 10.35 (1H, s, 7-OH), 9.47 (1H, s, 8-OH), 8.47 (1H, s, H-2), 7.61 (2H, d, J = 8.6 Hz, 2′-H, 6′-H), 7.48 (1H, d, J = 8.7 Hz, H-5), 7.47 (2H, d, J = 8.59 Hz, H-3′, H-5′), 6.96 (1H, d, J = 8.7 Hz, H-8). 13C NMR (DMSO-d6) δC 175.3, 154.6, 150.9, 147.4, 133.6, 133.1, 131.8, 131.4, 128.8, 122.4, 118.0, 116.4, 115.0. HRMS (M+H)+ calcd. for C15H9O4Cl, 288.0189; found. 288.0191. Mp. 165 °C.
3′-Chloro-7,8-dihydroxyisoflavone (25h) (69%). 1H NMR (DMSO-d6) δH 9.89 (2H, s, 7-OH, 8-OH), 8.42 (1H, s, 2-H), 7.46 (1H, d, J = 9.0 Hz, 5-H), 7.42–7.68 (4H, m, 2′-H, 4′-H, 5′-H, 6′-H), 7.01 (1H, d, J = 9.0 Hz, 6-H). 13C NMR (DMSO-d6) δC 175.4, 153.3, 151.8, 137.8, 134.3, 134.0, 130.1, 128.1, 126.3, 124.4, 123.6, 118.1, 112.1. HRMS (M+H)+ calcd. for C15H9O4Cl, 288.0189; found, 288.0192. Mp. 258°C. UV (MeOH) λmax 261 nm, 302 nm.
7,8-Dihydroxy-3′-methylisoflavone (25i) (71%). 1H NMR (DMSO-d6) δH 10.28 (1H, s, 7-OH), 9.69 (1H, s, 8-OH), 8.42 (1H, s, 2-H), 7.51 (1H, d, J = 9.0 Hz, 5-H), 7.24–7.53 (4H, m, 2′-H, 4′-H, 5′-H, 6′-H) 7,0 (1H, d, J = 9.0 Hz, 6-H) 2.37 (3H, s, CH3). 13C NMR (DMSO-d6) δC 175.7, 153.8, 152.9, 151.6, 138.1, 137.9, 132.4, 128.6, 126.8, 124.7, 123.4, 118.1, 113.2, 23.7. HRMS (M+H)+ calcd. for C16H12O4, 268.0736; found, 269.0740. Mp. 268 °C. UV (MeOH) λmax 257 nm, 309 nm.
7,8-Dihydroxy-3′-trifluoromethylisoflavone (25j) (79%). 1H NMR (DMSO-d6) δH 10.00 (1H, broad, s, 8-OH), 8.64 (1H, s, 2-H), 8.04 (1H, s, 2′-H), 7.71–7.88 (3H, m, 4′-H, 5′-H, 6′-H), 7.55 (1H, d, J = 9.0 Hz, 5-H) 7.04 (1H, d, J = 9.0 Hz, 6-H) [signal for 7-OH not seen]. 13C NMR (DMSO-d6) δC 174.7, 154.5, 150.5, 146.8, 133.5, 133.1, 132.8, 129.2, 129.0, 125.6, 124.4, 124.3, 121.6, 117.4, 115.8, 114.5. HRMS (M+H)+ calcd for C16H9O4F3, 322.0453; found, 322.0454. Mp. 225°C.
General procedure for the synthesis of compounds 26a,b
Reduction of isoflavone to isoflavanone
To a stirred solution of 1.0 g of the isoflavone in 200 ml of ethanol at room temperature was added 0.5 g of Pd/C (10 %) and the mixture was hydrogenated at normal pressure for 6h. Once the hydrogenation was completed, as verified by thin layer chromatography, the solution was filtered and concentrated to dryness under reduced pressure. The resulting residue was taken up with 10 mL of water, filtered, washed with water and dried to afford the corresponding isoflavanone which was purified by chromatography over a silica gel column with a 2:1 benzene/ether mixture as eluent. The yields were 60 to 75 %.
General procedure for the synthesis of compounds 27a–g
Reduction of isoflavone to isoflavan
To a stirred solution of 5 mmol of the isoflavone in 100 ml of acetic acid containing 0.1% sulfuric acid was added 0.5 g of Pd/C (10 %) and the mixture was hydrogenated for 14 h at normal pressure and room temperature. Once the hydrogenation was completed, verified by thin layer chromatography, the solution was filtered and concentrated under reduced pressure. The resulting residue was taken up with 50 mL of water, extracted with ether (3×50 mL) and the organic lalyer dried with sodium sulfate to give the corresponding isoflavan which was purified by chromatography over a silica gel column using ether/methanol mixtures of increasing polarity as eluent. The yields were 40 to 50 %.
6,7-Dihydroxy-4′-methoxyisoflavanone (26a) (52%). 1H NMR (DMSO-d6) δH 9.80 (1H, s, 7-OH), 7.56 (1H, s, H-5), 7.14 (2H, d, J = 8.7 Hz, H-2′, H-6′), 7.09 (1H, s, H-8), 6.86 (2H, d, J = 8.6 Hz, H-3′, H-5′), 4.49 (1H, dd, J = 11.2 Hz, J = 13.9 H, H-3z), 4.47 (1H, dd, J = 5.6 Hz, J = 5.7 Hz, H-2a), 3.85 (1H, dd, J = 5.5 Hz, J = 7.6 Hz, H-2e), 3.70 (3H, s, OCH3), [signal for 6-OH not seen]. 13C NMR (DMSO-d6) δC 191.1, 159.1, 157.1, 154.9, 141.7, 130.3, 129.1, 114.5, 113.0, 111.4, 103.6, 72.0, 55.7, 50.9. Calculated for C16H14O5, C, 67.13%; H, 4.89%, found C, 66.95%; H, 4.91%. Mp. 214 °C.
4′,6,7-Trihydroxyisoflavanone (26b) (48%). 1H NMR (DMSO-d6) δH 9.84 (1H, s, 7-OH), 8.21 (1H, s, 4′-OH), 7.56 (1H, s, H-5), 6.95 (2H, d, J = 8.7 Hz, H-2′, H-6′), 6.68 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.15 (1H, s, H-8), 4.79 (1H, dd, J = 11.2 Hz, J = 13.9 H, H-3z), 4.55 (1H, dd, J = 5.6 Hz, J = 5.7 Hz, H-2a), 4.20 (1H, dd, J = 5.5 Hz, J = 7.6 Hz, H-2e), 3.70 (3H, s, OCH3), [signal for 6-OH not seen]. 13C NMR (DMSO-d6) δC 193.1, 157.4, 152.3, 150.9, 138.8, 131.1, 129.0, 116.6, 116.4, 114.4, 103.4, 70.9, 56.1. Calculated for C15H12O5, C, 66.17%; H, 4.41%, found C, 66.32%; Mp. 235 °C.
6,7-Dihydroxy-4′-methoxyisoflavan (27a) (49%). 1H NMR (DMSO-d6) δH 8.68 (1H, s, OH-7), 8.25 (1H, s, OH-6), 7.19 (2H, d, J = 8.7 Hz, H-2′, H-6′), 6.87 (2H, d, J = 8.7 Hz, H-3′, H-5′), 6.43 (1H, s, H-5), 6.19 (1H, s, H-8), 4.07 (1H, ddd, J = 3.2 Hz, J = 10.3 Hz, J = 1.6 Hz; H-2e), 3.83 (1H, dd, J = 10.4 Hz, J = 3.2 Hz, H-2a), 3.71 (3H, s, OCH3), 3.03 (1H, dddd, J = 10.0 Hz, J = 10.1 Hz, J = 5.7 Hz, J = 4.1 Hz, H-3a), 2.80 (1H, dd, J = 10.5 Hz, J = 15.8 Hz, H-4a), 2.71 (1H, ddd, J = 5.5 Hz, J = 16 Hz, J = 1.6 Hz, H-4e). 13C NMR (DMSO-d6) δC 158.7, 147.1, 144.9, 139.7, 134.4, 129.1, 116.3, 114.6, 112.2, 104.0, 70.6, 55.7, 37.9, 31.9. HRMS (M+H)+ calcd. for C16H16O4 272.10486; found, 272.10363. Mp. 144°C.
4′6,7-Trihydroxyisoflavan (27b) (55%) 1H NMR (DMSO-d6) δH 9.22 (1H, s, OH-4′), 8.66 (1H, s, OH-7), 8,24 (1H, s, OH-6), 7.07 (2H, d, J = 8.4 Hz, H-2′, H-6′), 6.69 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.41 (1H, s, H-5), 6.18 (1H, s, H-8), 4.05 (1H, ddd, J = 3.2 Hz, J = 10.5 Hz, J = 1.6 Hz; H-2e), 3.78 (1H, dd, J = 10.3 Hz, J = 10.3 Hz, H-2a), 3.06 (1H, dddd, J = 10.1 Hz, J = 10.1 Hz, J = 5.8 Hz, J = 4.1 Hz, H-3), 2.77 (1H, dd, J = 10.4 Hz, J = 15.7 Hz, H-4a), 2.68 (1H, ddd, J = 5.6 Hz, J = 16,0 Hz, J = 1.1 Hz, H-4e). 13C NMR (DMSO-d6) δC 156.7, 147.1, 144.9, 139.7, 132.1, 128.9, 116.4, 115.9, 112.4, 104.0, 70.8, 38.0, 30.1. HRMS (M+H)+ calcd. for C15H14O4 258.08920; found, 258.08590. Mp. 176–177°C.
6,7-Dihydroxy-3′,4′-methylenedioxyisoflavan (27c) (52%). 1H NMR (DMSO-d6) δH 8.71 (1H, s, OH-7), 8.26 (1H, s, OH-6), 6.91 (1H, d, J = 2.0 Hz, H-2′), 6.84 (1H, d, J = 8.0 Hz, H-5′), 6.76 (1H, dd, J = 2.0 Hz, J = 8.0 Hz, H-6′), 6.42 (1H, s, H-5), 6.19 (1H, s, H-8), 5.96 (2H, s, O-CH2-O), 4.06 (1H, ddd, J = 3.5 Hz, J = 10.5 Hz, J = 1.5 Hz, H-2e), 3.84 (1H, dd, J = 10.5 Hz, J = 10,5 Hz, H-2a), 3.02 (1H, ddd, J = 10.5 Hz, J = 10.5 Hz, J = 5.5 Hz, H-3a), 2.79 (1H, dd, J = 10.5 Hz, J = 16,0 Hz, H-4a), 2.71 (1H, ddd, J = 5.5 Hz, J = 16,0 Hz, J = 1.5 Hz, H-4e). 13C NMR (DMSO-d6) δC 147.4, 146.5, 145.9, 144.3, 139.4, 135.9, 120.5, 115.7, 111.6, 108.2, 107.9, 103.4, 100.8, 69.9, 37.9, 31.4. HRMS (M+H)+ calcd for C16H14O5, 286.0841; found, 286.0842, Mp. 162 °C.
6,7-Dihydroxy-3′-methylisoflavan (27d) (90%). 1H NMR (DMSO-d6) δH 8.75 (1H, s, OH-7), 8.25 (1H, s, OH-6), 7.19 (1H, dd, J = 8.0 Hz, H-5′), 7.00–7.10 (3H, m, H-2′, H-4′, H-6′), 6.46 (1H, s, H-5), 6.24 (1H, s, H-8), 4.12 (1H, ddd, J = 3.0 Hz, J = 10.5, Hz J = 1.6 Hz, H-2e), 3.86 (1H, dd, J = 16 Hz; J = 16 Hz, H-2a), 3.05 (1H, dddd, J = 10.5 Hz, J = 16.0 Hz, J = 5.5 Hz, J = 4.6 Hz H-3a), 2.86 (1H, dd, J = 10.5 Hz, J = 16.0 Hz, H-4a), 2.26 (1H, ddd, J = 16.0 Hz, J = 16,0 Hz, J = 1.5 Hz, H-4e). 13C NMR (DMSO-d6) δC 146.5, 144.3, 141.8, 139.1, 137.5, 128.6, 128.1, 127.3, 124.6, 115.7, 111.5, 103.4, 69.8, 37.3, 31.2, 21.1. HRMS (M+H)+ calcd for C16H16O3, 256.1099; found, 256.1101, Mp. 164 °C.
7,8-Dihydroxy-4′-methylisoflavan (27e) (68%). 1H NMR (DMSO-d6) δH 8.54 (1H, s, OH-7), 8.09 (1H, s, OH-8), 7.19 (2H, d, J = 8.1 Hz, H-2′, H-6′), 7.12 (2H, d, J = 8.1 Hz, H-3′, H-5′), 6.36 (1H, d, J = 8.2 Hz, H-5), 6.28 (1H, d, J = 8.2 Hz, H-6), 4.23 (1H, ddd, J = 3.4 Hz, J = 10.4 Hz, J = 1.7 Hz, H-2e), 3.97 (1H, dd, J = 10.2 Hz; J = 10.2 Hz, H-2a), 3.09 (1H, ddd, J = 4.0 Hz, J = 5.7 Hz, J = 10.0 Hz, J = 8.9 Hz, H-3), 2.89 (1H, dd, J = 10.4 Hz, J = 16.1 Hz, H-4a), 2.80 (1H, ddd, J = 5.4 Hz, J = 15.6 Hz, J = 1.2 Hz, H-4e). 13C NMR (DMSO-d6) δC 144.7, 143.6, 139.5, 136.5, 133.7, 129.7, 127.9, 119.1, 113.9, 108.7, 70.5, 38.2, 32.0, 21.3. HRMS (M+H)+ calcd for C16H16O3, 256.1099; found, 256.1100, Mp. 125–126. °C.
7,8-Dihydroxy-3′,4′-dimethoxyisoflavan (27f) (49 %). 1H NMR (DMSO-d6) δH 8.53 (1H, s, OH-7), 8.09 (1H, s, OH-6), 6.94 (1H, d, J = 1.8 Hz, H-2′), 6.88 (1H, d, J = 8.4 Hz, H-5′), 6.81 (1H, dd, J = 8.4 Hz, J = 1.8 Hz, H-6′), 6.35 (1H, d, J = 8.2 Hz, H-5), 6.27 (1H, d, J = 8.2 Hz, H-6), 4.24 (1H, ddd, J = 3.2 Hz, J = 10.2 Hz, J = 1.6 Hz, H-2e), 3.96 (1H, dd, J = 10.2 Hz, J = 10.2 Hz, H-2a), 3.73 (3H, s, OCH3-4′), 3.71 (3H, s, OCH3-3′), 3.06 (1H, dddd, J = 10.0 Hz, J = 16.0 Hz, J = 5.4 Hz, J = 4.6 Hz, H-3), 2.90 (1H, dd, J = 10.7 Hz, J = 15.7 Hz, H-4a), 2.79 (1H, ddd, J = 5.3 Hz, J = 15.7 Hz, J = 1.1 Hz, H-4e). 13C NMR (DMSO-d6) δC 149.7, 148.4, 144,7, 143.7, 134.9, 133.8, 119.9, 118.9, 114.0, 112.8, 108.7, 70.8, 56.3, 56.2, 38.2, 32.2. HRMS (M+H)+ calcd for C17H18O5, 302.1154; found, 302.1156. Mp. 159–160 °C.
7,8-Dihydroxy-4′-methoxyisoflavan (27g) (56 %). 1H NMR (DMSO-d6) δH 8.54 (1H, s, OH-7), 8.09 (1H, s, OH-8), 7.22 (2H, d, J = 8.8 Hz, H-2′, H-6′), 6.89 (2H, d, J = 8.8 Hz, H-3′, H-5′), 6.35 (1H, d, J = 8.2 Hz, H-5), 6.29 (1H, d, J = 8.2 Hz, H-6), 4.22 (1H, ddd, J = 3.3 Hz, J = 16 Hz, J = 1.5 Hz, H-2e), 3.92 (1H, dd, J = 10.2 Hz, J = 10.2 Hz, H-2a), 3.72 (3H, s, OCH3), 3.07 (1H, dddd, J = 10.0 Hz, J = 9.9 Hz, J = 5.7 Hz; J = 3.9 Hz, H-3), 2.88 (1H, dd, J = 10.1 Hz, J = 15.6 Hz, H-4a), 2.79 (1H, ddd, J = 5.5 Hz, J = 15.7 Hz, J = 1.8 Hz, H-4e). 13C NMR (DMSO-d6) δC 158.8, 144.8, 143.7, 134.3, 133.8, 129.1, 119.1, 115.3, 113.9, 108.1, 70.8, 55.7, 37.8, 32.1. HRMS (M+H)+ calcd for C16H16O4, 272.1049; found, 272.1049. Mp. 128–129°C.
Plasmid Construction, Expression and Purification of Human Lipoxygenases
The histidine-tagged proteins, 12-hLO and 15-hLO-1 were expressed and purified as described previously.42,49 The SF9 expression vector for human prostate epithelial 15-lipoxygenase-2 (15-hLO-2) was constructed as follows. The previously published plasmid, pCRII-TOPO-15hLO-2,19 was cut with EcoRI, to liberate the 15-hLO-2 fragment. This 15-hLO-2 fragment was then ligated into EcoRI cut, pFastBac1 (GibcoBRL) to generate the complete plasmid, pFastBac-15-hLO-2, which was digested to determine the correct orientation. The pFastBac-15-hLO-2 was then transposed into a recombinant FastBac bacmid by DH10Bac cells (GibcoBRL) and then transfected into SF9 cells, as described in the product literature for pFastBac1 (GibcoBRL). The virus was subsequently amplified to ≈2×1010 plaque forming units (pfu), added to SF9 cells (≈2×106 cells/ml) at a concentration of ≈2×107 pfu/ml and allowed to shake for 72 hours. The cells were then harvested and frozen. 15-hLO-2 was purified by douncing the thawed cells and loading the cell extracts on to a 20 mL column of Macro-prep High Q, strong anion exchange column, from Bio-Rad. The protein was eluted with a 50–300 mM sodium chloride gradient. The collected fractions contained 95% purified protein and 5% glycerol and were frozen at −80 C. Subsequent thawing produced no decrease in enzymatic activity.
13-(S)-HPOD was prepared as before,50 while LA and AA were purchased from Aldrich Chemical, and BWB70C and BWA4C were purchased from Sigma Chemical. All other reagents were reagent grade or better and were used without further purification.
High-Throughput Inhibitor Screen
The high-throughput (HTP) screen was developed in 384 well plates where the hydroperoxide product was detected with iron/xylenol orange.40,51 Briefly, the lipid peroxide formed by the human lipoxygenase enzymes reacts with Fe2+ present in solution and converts it to Fe3+ which in turn forms an Fe3+-xylenol orange complex which absorbs at 560 nm. The HTP screen was performed as published earlier with the following changes.40 The buffer for 12-hLO was 25 mM HEPES (pH 8.0) with 0.1% titron and for 15-hLO-1 and 15-hLO-2 was 25 mM HEPES (pH 7.5) with 0.1% titron. Arachidonic acid was used as a substrate for 12-hLO and 15-hLO-2, while linoleic acid was used for 15-hLO-1. The first step is the addition of 45 μl of a 30 μM substrate buffer solution to each well using the Multidrop 384 by Thermo Labsystems. The inhibitor (1.2 μl at 2.5mg/ml in DMSO) was then added using a specially designed manual liquid pin transfer device from V & P Scientific. Addition of 5 μL of enzyme, which gives a rate of approximately 0.0020 abs/sec using the multidrop, started the reaction (approximately 40 nM for 12-hLO and 15-hLO-1 and 125 nM for 15-hLO-2, final concentration). The reaction was stopped well before completion (after approximately 5 minutes) by adding appropriate amount of reagents to give a final concentration of 100 μM of xylenol orange dye and 150 μM Fe2+ in 25 mM H2SO4, with a final volume of 100 μL in each well. The plates were stirred using a gyratory shaker in the dark for 30 minutes and read at 544 nm using Perkin-Elmer plate reader Victor2. The negative control for the assay was 0.2 μL of DMSO (with no inhibitor) added to reaction buffer, with no enzyme. The positive control for the assay was 0.2 μL of DMSO added to reaction buffer with enzyme, as mentioned above. All the assays were done in triplicate. The percent inhibition was calculated by subtracting the absorbance of the negative control from that of the reaction samples, and dividing by the absorbance of the positive control. The value was then subtracted from 1 and multiplied by 100 to get the percent inhibition. In order to confirm the accuracy of the HTP screen, all samples giving greater than 40 % inhibition by the HTP assay were screened manually to determine an accurate IC50 value (vida infra). From this additional screening, it was determined that compounds with less than 45 % inhibition from the HTP screen for all three hLO’s, had IC50 values greater than 100 μM.
Manual Absorption IC50 Inhibition Assay
IC50 inhibition constants for compounds with greater than 40% inhibition from the HTP screen were determined manually as described previously,52 with the following changes. The reaction buffer was 25 mM HEPES (pH 8.0) with 0.1% titron, for 12-hLO and was 25 mM HEPES (pH 7.5) with 0.1% titron for 15-hLO-1 and 15-hLO-2. Arachidonic acid was used as a substrate for 12-hLO (3 μM), and 15-hLO-2 (30 μM), while linoleic acid was used for 15-hLO-1 (3 μM). Enzyme rates were determined with approximately 40 nM for 12-hLO and 15-hLO-1 and 125 nM for 15-hLO-2, final concentration, by monitoring product formation at 234 nm (εM = 2.5 × 104) with an HP 8435 spectrophotometer at room temperature. Inhibitor (1 mg/mL in DMSO) was added to the substrate buffer and the reaction initiated by addition of enzyme after a stable baseline was observed at 234 nm. Control rates were measured using the same volume of DMSO as the volume of inhibitor. IC50 values were determined by measuring the enzymatic rate at a variety of inhibitor concentrations (depending on the inhibitor potency) and plotting their percent inhibition versus inhibitor concentration. The corresponding data were fit to a simple saturation curve using KaleidaGraph software (Synergy) on a Macintosh computer and the inhibitor concentration at 50% activity was calculated.
Pseudoperoxidase Activity Assay
The pseudoperoxidase activity was determined using a Perkin-Elmer Lambda 40 spectrophotometer by following the decrease in HPOD absorbance at 234 nm due to inhibitor-dependent consumption of HPOD.53 The reaction mixture contained 15-hLO-1 (185 μg), 25 mM Hepes pH 7.5, HPOD (5 μM), and inhibitor (10 μM). The reaction was initiated by adding enzyme at room temperature and the final volume of the reaction solution was 2 ml. BWB70C and BWA4C were used as positive controls for this experiment as they are known redox-type inhibitors for human 5-lipoxygenase.54,55
Molecular Modeling Studies
The 15-hLO-1 homology model was created using the Protein Local Optimization Program (PLOP, commercially distributed as Prime), which uses loop prediction,56 side chain prediction57, 58 and energy minimization to align the target and template sequences, as previously reported.41 Docking of inhibitors to this model has been verified previously by predicting experimental inhibitor potency based solely on docking parameters.41 The structures of compounds 25b, 25e, 29 and 30 were prepared for docking using the LigPrep (Schrödinger, Inc) ligand preparation software, which generates a minimized conformation of each ligand, and multiple protonation/tautomerization states when appropriate. Flexible ligand docking was performed using the Glide (Schrödinger, Inc.) program,59, 60 which uses a modified version of the Chemscore energy function to score the protein-ligand interactions.61
Acknowledgments
TRH gratefully acknowledges the gift of the original 15-hLO-2 gene from Dr. D. Tang. The 2-alkyl-4-H-benzopyran-4-ones described were synthesized by YVM during a stay in Prof. E. Breitmaier’s laboratory at the University of Bonn as the recipient of a DAAD fellowship. SSB thanks the MECESUP UCH 0116 program for the HRMS. The research was supported by NASA # 13171 (SSB), NIH (GM56062-6) (TRH) and the American Cancer Society (RPG-00-219-01-CDD) (TRH) grants.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yamamoto S. Mammalian lipoxygenases: molecular structures and functions. Biochim Biophys Acta. 1992;1128:117–131. doi: 10.1016/0005-2760(92)90297-9. [DOI] [PubMed] [Google Scholar]
- 2.Brash AR. Lipoxygenases: Occurrence, Functions, Catalysis and Acquisition of Substrate. J Biol Chem. 1999;274:23679–23682. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]
- 3.Solomon EI, Zhou J, Neese F, Pavel EG. New Insights from Spectroscopy into the structure/function relationships of lipoxygenases. Chem Biol. 1997;4:795–808. doi: 10.1016/s1074-5521(97)90113-7. [DOI] [PubMed] [Google Scholar]
- 4.Prigge ST, Boyington JC, Faig M, Doctor KS, Gaffney BJ, Amzel LM. Structure and mechanism of lipoxygenases. Biochimie. 1997;79:629–636. doi: 10.1016/s0300-9084(97)83495-5. [DOI] [PubMed] [Google Scholar]
- 5.Kuhn H. Structural basis for the positional specificity of lipoxygenases. Prostaglandins Other Lipid Mediat. 2000;62:255–270. doi: 10.1016/s0090-6980(00)00084-8. [DOI] [PubMed] [Google Scholar]
- 6.Yoshimoto T, Takahashi Y. Arachidonate 12-lipoxygenases. Prostaglandins Other Lipid Mediat. 2002:68–69. 245–262. doi: 10.1016/s0090-6980(02)00034-5. [DOI] [PubMed] [Google Scholar]
- 7.Dailey LA, Imming P. 12-Lipoxygenase: classification, possible therapeutic benefits from inhibition, and inhibitors. Curr Med Chem. 1999;6:389–398. [PubMed] [Google Scholar]
- 8.Honn KV, Tang DG, Gao X, Butovich IA, Liu B, Timar J, Hagmann W. 12-lipoxygenases and 12(S)-HETE: role in cancer metastasis. Cancer Metastasis Rev. 1994;13:365–396. doi: 10.1007/BF00666105. [DOI] [PubMed] [Google Scholar]
- 9.Arenberger P, Raap A, Armah B, Kemény L, Ruzicka T. The lipoxygenase inhibitor 2-phenylmethyl-1-naphthol (DuP 654) is a 12(S)-hydroxyeicosatetraenoic acid receptor antagonist in the human epidermal cell line SCL-II. Skin Pharmacol. 1993;6:148–151. doi: 10.1159/000211099. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Wang H, Li J, Jimenez DA, Levitan ES, Aizenman E, Rosenberg PA. Peroxynitrite-induced neuronal apoptosis is mediated by intracellular zinc release and 12-lipoxygenase activation. J Neurosci. 2004;24:10616–10627. doi: 10.1523/JNEUROSCI.2469-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuhn H, Walther M, Kuban RJ. Mammalian arachidonate 15-lipoxygenases structure, function, and biological implications. Prostaglandins Other Lipid Mediat. 2002:68–69. 263–290. doi: 10.1016/s0090-6980(02)00035-7. [DOI] [PubMed] [Google Scholar]
- 12.Cathcart MK, Folcik VA. Lipoxygenases and atherosclerosis: protection versus pathogenesis. Free Radic Biol Med. 2000;28:1726–1734. doi: 10.1016/s0891-5849(00)00230-6. [DOI] [PubMed] [Google Scholar]
- 13.Neuzil J, Upston JM, Witting PK, Scott KF, Stocker R. Secretory phospholipase A2 and lipoprotein lipase enhance 15-lipoxygenase-induced enzymic and nonenzymic lipid peroxidation in low-density lipoproteins. Biochemistry. 1998;37:9203–9210. doi: 10.1021/bi9730745. [DOI] [PubMed] [Google Scholar]
- 14.Schewe T. 15-lipoxygenase-1: a prooxidant enzyme. Biol Chem. 2002;383:365–374. doi: 10.1515/BC.2002.041. [DOI] [PubMed] [Google Scholar]
- 15.Ikawa H, Kamitani H, Calvo BF, Foley JF, Eling TE. Expression of 15-lipoxygenase-1 in human colorectal cancer. Cancer Res. 1999;59:360–366. [PubMed] [Google Scholar]
- 16.Kelavkar UP, Glasgow W, Olson SJ, Foster BA, Shappell SB. Overexpression of 12/15-lipoxygenase, an ortholog of human 15-lipoxygenase-1, in the prostate tumors of TRAMP mice. Neoplasia. 2004;6:821–830. doi: 10.1593/neo.04286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brash AR, Boeglin WE, Chang MS. Discovery of a second 15S-lipoxygenase in humans. Proc Natl Acad Sci U S A. 1997;94:6148–6152. doi: 10.1073/pnas.94.12.6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shappell SB, Boeglin WE, Olson SJ, Kasper S, Brash AR. 15-lipoxygenase-2 (15-LOX-2) is expressed in benign prostatic epithelium and reduced in prostate adenocarcinoma. Am J Pathol. 1999;155:235–245. doi: 10.1016/S0002-9440(10)65117-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang S, Bhatia B, Maldonado CJ, Yang P, Newman RA, Liu J, Chandra D, Traag J, Klein RD, Fischer SM, Chopra D, Shen J, Zhau HE, Chung LW, Tang DG. Evidence that arachidonate 15-lipoxygenase 2 is a negative cell cycle regulator in normal prostate epithelial cells. J Biol Chem. 2002;277:16189–16201. doi: 10.1074/jbc.M111936200. [DOI] [PubMed] [Google Scholar]
- 20.Vavrecková C, Gawlik I, Müller K. Benzophenanthridine alkaloids of Chelidonium majus; I. Inhibition of 5- and 12-lipoxygenase by a non-redox mechanism. Planta Med. 1996;62:397–401. doi: 10.1055/s-2006-957924. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki H, Ueda T, Juránek I, Yamamoto S, Katoh T, Node M, Suzuki T. Hinokitiol, a selective inhibitor of the platelet-type isozyme of arachidonate 12-lipoxygenase. Biochem Biophys Res Commun. 2000;275:885–889. doi: 10.1006/bbrc.2000.3390. [DOI] [PubMed] [Google Scholar]
- 22.Alanko J, Kurahashi Y, Yoshimoto T, Yamamoto S, Baba K. Panaxynol, a polyacetylene compound isolated from oriental medicines, inhibits mammalian lipoxygenases. Biochem Pharmacol. 1994;48:1979–1981. doi: 10.1016/0006-2952(94)90598-3. [DOI] [PubMed] [Google Scholar]
- 23.Cho H, Ueda M, Tamaoka M, Hamaguchi M, Aisaka K, Kiso Y, Inoue T, Ogino R, Tatsuoka T, Ishihara T, et al. Novel caffeic acid derivatives: extremely potent inhibitors of 12-lipoxygenase. J Med Chem. 1991;34:1503–1505. doi: 10.1021/jm00108a039. [DOI] [PubMed] [Google Scholar]
- 24.Müller K, Altmann R, Prinz H. 2-arylalkyl-substituted anthracenones as inhibitors of 12-lipoxygenase enzymes. 1. Structure-activity relationships of the terminal aryl ring. Eur J Med Chem. 2001;36:569–575. doi: 10.1016/s0223-5234(01)01248-x. [DOI] [PubMed] [Google Scholar]
- 25.Segraves EN, Shah RR, Segraves NL, Johnson TA, Whitman S, Sui JK, Kenyon VA, Cichewicz RH, Crews P, Holman TR. Probing the activity differences of simple and complex brominated aryl compounds against 15-soybean, 15-human, and 12-human lipoxygenase. J Med Chem. 2004;47:4060–4065. doi: 10.1021/jm049872s. [DOI] [PubMed] [Google Scholar]
- 26.Cichewicz RH, Kenyon VA, Whitman S, Morales NM, Arguello JF, Holman TR, Crews P. Redox inactivation of human 15-lipoxygenase by marine-derived meroditerpenes and synthetic chromanes: archetypes for a unique class of selective and recyclable inhibitors. J Am Chem Soc. 2004;126:14910–14920. doi: 10.1021/ja046082z. [DOI] [PubMed] [Google Scholar]
- 27.Amagata T, Whitman S, Johnson T, Stessmann CC, Carroll J, Loo C, Clardy J, Lobkovsky E, Crews P, Holman TR. Sponge Derived Terpenoids with Selectivity towards Human 15-Lipoxygenase versus Human 12-Lipoxygenase. J Nat Prod. 2003;66:230–235. doi: 10.1021/np020462l. [DOI] [PubMed] [Google Scholar]
- 28.Whitman S, Gezginci M, Timmermann BN, Holman TR. Structure-activity relationship studies of nordihydroguaiaretic acid inhibitors toward soybean, 12-human, and 15-human lipoxygenase. J Med Chem. 2002;45:2659–2661. doi: 10.1021/jm0201262. [DOI] [PubMed] [Google Scholar]
- 29.Malterud KE, Farbrot TL, Huse AE, Sund RB. Antioxidant and radical scavenging effects of anthraquinones and anthrones. Pharmacology. 1993;47(Suppl 1):77–85. doi: 10.1159/000139846. [DOI] [PubMed] [Google Scholar]
- 30.Schewe T, Sadik C, Klotz LO, Yoshimoto T, Kuhn H, Sies H. Polyphenols of cocoa: inhibition of mammalian 15-lipoxygenase. Biol Chem. 2001;382:1687–1696. doi: 10.1515/BC.2001.204. [DOI] [PubMed] [Google Scholar]
- 31.Deschamps JD, Kenyon VA, Holman TR. Baicalein is a potent in vitro inhibitor against both reticulocyte 15-human and platelet 12-human lipoxygenases. Bioorg Med Chem. 2006;14:4295–4301. doi: 10.1016/j.bmc.2006.01.057. [DOI] [PubMed] [Google Scholar]
- 32.Hsieh RJ, German JB, Kinsella JE. Relative inhibitory potencies of flavonoids on 12-lipoxygenase of fish gill. Lipids. 1988;23:322–326. doi: 10.1007/BF02537342. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto H, Sakakibara J, Nagatsu A, Sekiya K. Inhibitors of arachidonate lipoxygenase from defatted perilla seed. J Agric Food Chem. 1998;46:862–865. [Google Scholar]
- 34.Gutierrez-Lugo MT, Deschamps JD, Holman TR, Suarez E, Timmermann BN. Lipoxygenase inhibition by anadanthoflavone, a new flavonoid from the aerial parts of Anadenanthera colubrina. Planta Med. 2004;70:263–265. doi: 10.1055/s-2004-818920. [DOI] [PubMed] [Google Scholar]
- 35.Sekiya K, Okuda H, Arichi S. Selective inhibition of platelet lipoxygenase by esculetin. Biochim Biophys Acta. 1982;713:68–72. [PubMed] [Google Scholar]
- 36.Sadik CD, Sies H, Schewe T. Inhibition of 15-lipoxygenases by flavonoids: structure-activity relations and mode of action. Biochem Pharmacol. 2003;65:773–781. doi: 10.1016/s0006-2952(02)01621-0. [DOI] [PubMed] [Google Scholar]
- 37.Nussbaumer P, Lehr P, Billich A. 2-Substituted 4-(thio)chromenone 6-O-sulfamates: potent inhibitors of human steroid sulfatase. J Med Chem. 2002;45:4310–4320. doi: 10.1021/jm020878w. [DOI] [PubMed] [Google Scholar]
- 38.Fougerousse A, Gonzalez E, Brouillard R. A convenient method for synthesizing 2-aryl-3-hydroxy-4-oxo-4H-1-benzopyrans or flavonols. J Org Chem. 2000;65:583–586. doi: 10.1021/jo990735q. [DOI] [PubMed] [Google Scholar]
- 39.Sepúlveda-Boza S, Walizei SHG, Rezende MC, Vásquez Y, Mascayano C, Mejías L. The preparation of new isoflavones. Synth Commun. 20012001;31:1933–1940. [Google Scholar]
- 40.Waslidge NB, Hayes DJ. A colorimetric method for the determination of lipoxygenase activity suitable for use in a high throughput assay format. Anal Biochem. 1995;231:354–358. doi: 10.1006/abio.1995.0063. [DOI] [PubMed] [Google Scholar]
- 41.Kenyon V, Chorny I, Carvajal WJ, Holman TR, Jacobson MP. Novel human lipoxygenase inhibitors discovered using virtual screening with homology models. J Med Chem. 2006;49:1356–1363. doi: 10.1021/jm050639j. [DOI] [PubMed] [Google Scholar]
- 42.Amagata T, Whitman S, Johnson TA, Stessman CC, Loo CP, Lobkovsky E, Clardy J, Crews P, Holman TR. Exploring sponge-derived terpenoids for their potency and selectivity against 12-human, 15-human, and 15-soybean lipoxygenases. J Nat Prod. 2003;66:230–235. doi: 10.1021/np020462l. [DOI] [PubMed] [Google Scholar]
- 43.Kilty I, Logan A, Vickers PJ. Differential characteristics of human 15-lipoxygenase isozymes and a novel splice variant of 15S-lipoxygenase. Eur J Biochem. 1999;266:83–93. doi: 10.1046/j.1432-1327.1999.00818.x. [DOI] [PubMed] [Google Scholar]
- 44.Nelson MJ. Catecholate Complexes of Ferric Soybean Lipoxygenase-1. Biochemistry. 1988;27:4273–4278. [Google Scholar]
- 45.Nelson MJ, Batt DG, Thompson JS, Wright SW. Reduction of the active-site iron by potent inhibitors of lipoxygenases. J Biol Chem. 1991;266:8225–8229. [PubMed] [Google Scholar]
- 46.Kemal C, Louis-Flamberg P, Krupinski-Olsen R, Shorter AL. Reductive inactivation of soybean lipoxygenase 1 by catechols: a possible mechanism for regulation of lipoxygenase activity. Biochemistry. 1987;26:7064–7072. doi: 10.1021/bi00396a031. [DOI] [PubMed] [Google Scholar]
- 47.Voss C, Sepulveda-Boza S, Zilliken FW. New isoflavonoids as inhibitors of porcine 5-lipoxygenase. Biochem Pharmacol. 1992;44:157–162. doi: 10.1016/0006-2952(92)90049-o. [DOI] [PubMed] [Google Scholar]
- 48.Kostanecki S, Tambor J. Chem Ber. 1901;34:1691. [Google Scholar]
- 49.Holman TR, Zhou J, Solomon E. Spectroscopic and functional characterization of a ligand coordination mutant of soybean lipoxygenase: First coordination sphere analogue of human 15-lipoxygenase. J Am Chem Soc. 1998:120. [Google Scholar]
- 50.Lewis ER, Johansen E, Holman TR. Large competitive kinetic isotope effects in human 15-LO catalysis measured by a novel HPLC method. J Am Chem Soc. 1999;121:1395–1396. [Google Scholar]
- 51.Gay C, Collins J, Gebicki JM. Hydroperoxide assay with the ferric-xylenol orange complex. Anal Biochem. 1999;273:149–155. doi: 10.1006/abio.1999.4208. [DOI] [PubMed] [Google Scholar]
- 52.Carroll J, Jonsson EN, Ebel R, Hartman MS, Holman TR, Crews P. Probing sponge-derived terpenoids for human 15-lipoxygenase inhibitors. J Org Chem. 2001;66:6847–6851. doi: 10.1021/jo015784t. [DOI] [PubMed] [Google Scholar]
- 53.Riendeau D, Falgueyret JP, Guay J, Ueda N, Yamamoto S. Pseudoperoxidase activity of 5-lipoxygenase stimulated by potent benzofuranol and N-hydroxyurea inhibitors of the lipoxygenase reaction. Biochem J. 1991;274(Pt 1):287–292. doi: 10.1042/bj2740287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Falgueyret JP, Hutchinson JH, Riendeau D. Criteria for the identification of non-redox inhibitors of 5-lipoxygenase. Biochem Pharmacol. 1993;45:978–981. doi: 10.1016/0006-2952(93)90185-y. [DOI] [PubMed] [Google Scholar]
- 55.Hussey HJ, Tisdale MJ. Inhibition of tumour growth by lipoxygenase inhibitors. Br J Cancer. 1996;74:683–687. doi: 10.1038/bjc.1996.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 57.Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol. 2002;320:597–608. doi: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 58.Jacobson MP, Kaminski GA, Friesner RA, Rapp CS. Force Field Validation Using Protein Side Chain Prediction. J Phys Chem B. 2002;106:11673–11680. [Google Scholar]
- 59.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 60.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 61.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]