

Abstract
Electron transfer from reduced nicotinamide adenine dinucleotide (NADH) to the hydroxylase component (MMOH) of soluble methane monooxygenase (sMMO) primes its non-heme diiron centers for reaction with dioxygen to generate high-valent iron intermediates that convert methane to methanol. This intermolecular electron-transfer step is facilitated by a reductase (MMOR), which contains [2Fe-2S] and flavin adenine dinucleotide (FAD) prosthetic groups. To investigate interprotein electron transfer, chemically reduced MMOR was mixed rapidly with oxidized MMOH in a stopped-flow apparatus, and optical changes associated with reductase oxidation were recorded. The reaction proceeds via four discrete kinetic phases corresponding to the transfer of four electrons into the two dinuclear iron sites of MMOH. Pre-equilibrating the hydroxylase with sMMO auxiliary proteins MMOB or MMOD severely diminishes electron-transfer throughput from MMOR, primarily by shifting the bulk of electron transfer to the slowest pathway. The biphasic reactions for electron transfer to MMOH from several MMOR ferredoxin analogues are also inhibited by MMOB and MMOD. These results, in conjunction with the previous finding that MMOB enhances electron-transfer rates from MMOR to MMOH when preformed MMOR-MMOH-MMOB complexes are allowed to react with NADH [Gassner, G. T.; Lippard, S. J. Biochemistry 1999, 38, 12768-12785], suggest that isomerization of the initial ternary complex is required for maximal electron-transfer rates. To account for the slow electron transfer observed for the ternary precomplex in this work, a model is proposed in which conformational changes imparted to the hydroxylase by MMOR are retained throughout the catalytic cycle. Several electron-transfer schemes are discussed with emphasis on those that invoke multiple interconverting MMOH populations.
Introduction
Long-range electron transfer is an essential process in many physiological pathways, including respiration, photosynthesis, and intermediary metabolism.1-3 Biological electron-transfer reactions often proceed via bimolecular interactions between donor and acceptor proteins containing redox-active prosthetic groups. To achieve efficient interprotein electron transfer, steps such as specific protein-protein binding, conformational rearrangements, and chemical transformations may be required.4-6 A common strategy in bacterial mono- and dioxygenase systems is separation of the reductase and oxygenase functionalities into individual protein components.7 In this manner, the rate of electron transfer to the oxygenase active site can be finely controlled by intricate intermolecular interactions. In addition, the reductant can be physically separated from, and thereby avoid being consumed by, active oxygen species generated in the oxygenase component.
Among the best-studied of these multicomponent oxygenase systems is soluble methane monooxygenase (sMMO), which catalyzes the selective oxidation of methane to methanol at ambient temperature and pressure (eq 1).8 By using methane
as their primary source of carbon and energy, methanotrophs significantly reduce the amount of this potent greenhouse gas that reaches the atmosphere.9 All methanotrophic bacteria can express a copper-containing membrane-bound MMO (pMMO),10 whereas the sMMO system is used by only a few species under copper-limited conditions.11 The relative ease of purifying and stabilizing the sMMO proteins has facilitated detailed reactivity, spectroscopic, structural, kinetic, and theoretical studies of these enzymes from Methylococcus capsulatus (Bath) and Methylosinus trichosporium OB3b.12,13
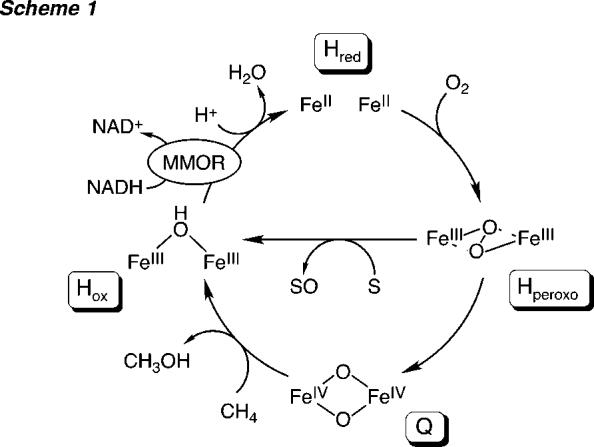
Both sMMO systems comprise three proteins that are required for optimal catalysis:14-16 a hydroxylase (MMOH, 251 kDa), a reductase (MMOR, 38.5 kDa), and a regulatory protein (MMOB, 15.9 kDa). The inclusion of a fourth protein (MMOD, 11.9 kDa) in the M. capsulatus (Bath) sMMO system was demonstrated recently.17 MMOH, a dimeric (αβγ)2 protein, houses in its R subunits carboxylate-bridged non-heme diiron sites where dioxygen activation and substrate hydroxylation occur. MMOR is an iron-sulfur flavoprotein that shuttles electrons from reduced nicotinamide adenine dinucleotide (NADH) to the hydroxylase to prime the enzyme system for reaction with dioxygen. A [2Fe-2S] cluster is located in the N-terminal reductase domain, which exhibits sequence homology with ferredoxins of plants, cyanobacteria, and archaebacteria.18 To begin the series of electron-transfer reactions necessary for steady-state sMMO turnover (Scheme 1), the flavin adenine dinucleotide (FAD) cofactor bound to the C-terminal portion of MMOR accepts two electrons from NADH. These electrons are then transmitted sequentially from the reduced flavin through the one-electron [2Fe-2S] center to the diiron(III) hydroxylase active sites.16,19-21 After reduction by MMOR, a complex formed between the diiron(II) state of MMOH, Hred, and MMOB reacts with dioxygen to generate a series of oxygenated high-valent diiron intermediates.22,23 The final transient species hydroxylates methane, producing methanol and water and returning the hydroxylase to the resting, diiron(III) state. To complete the cycle, the MMOH diiron centers are reduced by MMOR and methanol is discharged from the active site. Interactions between the three sMMO components modulate the rates, efficiency, and substrate regioselectivity of the reaction.16,23-26
Scheme 1.
MMOR is a member of a class of modular electron transferases, also called the ferredoxin:NADP+ oxidoreductase (FNR) family. These proteins are distinguished by a core flavin domain that transports electrons between a nicotinamide dinucleotide and a one-electron carrier domain, which may be either linked or dissociable (Figure 1A).27,28 Distinct flavin mononucleotide (FMN), NADH binding, and [2Fe-2S] domains are revealed in the crystal structure of Burkholderia cepacia phthalate dioxygenase reductase (PDR), demonstrating the structural and functional modularity of FNR proteins.28 To simplify and extend the characterization of MMOR, the ferredoxin (MMOR-Fd, referred to hereafter as MMOR-Fd(1-98)) and FAD/NADH (MMOR-FAD) domains were expressed as separate proteins.29 Except for interdomain electron transfer, the reductase domain proteins exhibit redox, spectroscopic, and kinetic properties nearly identical to those of full-length MMOR.29 In addition, NMR studies of MMOR-Fd(1-98)30 and MMOR-FAD31 show that the separated domains adopt compact structures in solution with folds similar to homologous FNR proteins.
Figure 1.
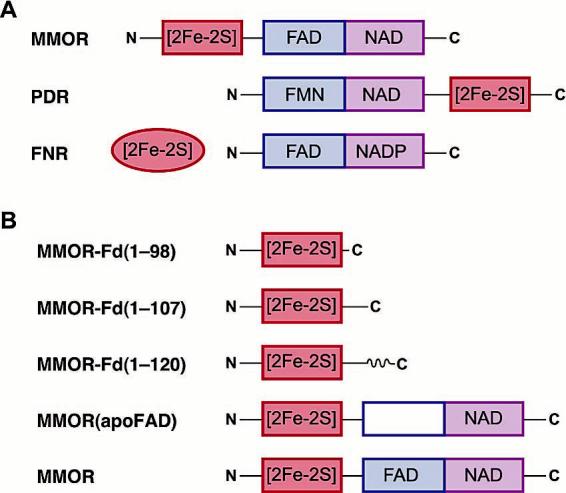
(A) Examples of alternative connectivities between the one-electron carrier and flavin/NAD(P) binding domains of flavoprotein electron transferases. (B) MMOR proteins examined in this study.
In previous studies of intermolecular electron transfer in the sMMO system, preformed MMOR-MMOH or MMOR-MMOH-MMOB complexes were allowed to react with NADH, and absorbance changes associated with multiple intra- and intermolecular kinetic steps were monitored.16 In the present work, reactions were conducted by rapidly mixing chemically reduced MMOR with oxidized MMOH, thereby affording direct observation of intermolecular electron-transfer events while avoiding the complicating kinetics of the reductive half-reaction of MMOR. Four MMOR-Fd analogues (Figure 1B) were also investigated to explore the features that modulate electron transfer to the hydroxylase. Mechanisms for the intermolecular electron-transfer reaction, and roles for MMOB and MMOD in regulating the rate and extent of electron transfer, are presented.
Experimental Section
Protein Purification. MMOH was isolated from M. capsulatus (Bath) cell paste by following a published protocol.16 Pure hydroxylase contained 3.8-4.0 mol of Fe/mol of protein dimer, as determined by ferrozine iron assays,32 and exhibited specific activities of 250-350 nmol min-1 mg-1 of MMOH for the conversion of propylene to propylene oxide at 45 °C. The proteins MMOR,21 MMOR-Fd(1-98),29 and MMOD17 were expressed in recombinant Escherichia coli systems and purified as previously reported. Recombinant MMOB was expressed in E. coli as described33 and purified by using a slightly modified protocol. FAD-depleted reductase, MMOR(apoFAD), was separated from MMOR by Sepharose 5′-AMP affinity chromatography.
Expression vectors for two longer ferredoxin domain proteins, MMOR-Fd(1-107) and MMOR-Fd(1-120), were constructed by using standard protocols (cloning details are provided as Supporting Information) and transformed into E. coli JM105 cells. Expression and purification were performed as described for recombinant MMOR-Fd(1-98),29 except that reducing the isopropyl thio-β-d-galactoside (IPTG) concentration (to 50 μM) and the expression temperature (to 25 °C) improved protein yields. The polypeptide masses (11 880 and 13 299 Da) were verified by mass spectrometry (MIT Biopolymers Laboratory).
Isothermal Titration Calorimetry. The binding affinity of the MMOH-MMOR-Fd(1-107) complex was measured with a VP-ITC isothermal titration calorimeter (MicroCal, Inc., Northampton, MA). All titrations were performed at 4.2 °C in 25 mM 3-(N-morpholino)-propanesulfonic acid (MOPS; pH 7.0) and 2 mM tri(2-carboxyethyl)-phosphine hydrochloride (TCEP; Sigma, St. Louis, MO). Titrant (453 μM MMOR-Fd(1-107)) was injected in 10-μL aliquots from a 250-μL stirred titration syringe into the 1.430-mL sample cell containing 12.1 μM MMOH. Stirring was maintained at 310 rpm throughout the titration. The experiment was repeated with buffer in the sample cell in order to measure the heat of dilution for MMOR-Fd(1-107). Data were integrated and fit with the MicroCal Origin v. 5.0 software package. This experiment was also performed for MMOR-Fd(1-98), MMOR-Fd(1-120), and MMOR. To examine MMOH-MMOR binding, the calorimetry studies were carried out in 25 mM MOPS (pH 7.0) and 1 mM dithiothreitol (DTT) because TCEP reacts with the FAD/NADH portion of MMOR.
Redox Potential Determinations for MMOR-Fd Analogues. The equilibrium midpoint potentials of the MMOR-Fd(1-107) and MMOR-Fd(1-120) proteins were determined by using reductive titrations including the redox-active indicator dye anthraquinone-2-sulfonate (E°′ = -226 mV). The MMOR-Fd domain (ca. 40 μM) and dye (ca. 35 μM), in a total volume of 2 mL of 25 mM potassium phosphate (pH 7.0) buffer, were placed in a sealed quartz cuvette under an anaerobic nitrogen atmosphere. After collection of a visible spectrum (300-900 nm) of the initial oxidized protein-dye mixture at 25 °C, aliquots (5 μL) of sodium dithionite (ca. 1 mM) were introduced via a gastight Hamilton syringe equipped with a repeating dispenser. Multiple spectra were recorded after each dithionite addition until no further optical changes were observed (usually 2-5 min after mixing). The final spectrum of the equilibrated system was saved for data analysis. This process was repeated until the ferredoxin and dye were reduced completely.
Dilution corrections were applied to each spectrum, and difference spectra were generated by subtracting the starting oxidized spectrum from each spectrum in a titration data set. These titration difference spectra (350 ≤ λ ≤ 800 nm) were then fit with linear combinations of the previously determined component difference spectra, Fdred - Fdox and dyered - dyeox,29 in KaleidaGraph (v. 3.0 or 3.6, Synergy Software, Reading, PA). The concentration differences returned by fitting were readily converted to species concentrations by comparison to the total protein and dye concentrations. The solution potential and [2Fe-2S] midpoint potentials were calculated for each titration point with modified Nernst equations.21,29
Kinetic Measurements of Intermolecular Electron Transfer. Intermolecular electron transfer in the sMMO system was examined by stopped-flow optical spectroscopy. A Hi-Tech Scientific SF-61 DX2 double-mixing stopped-flow spectrophotometer configured for either single-wavelength photomultiplier or multiwavelength diode array collection was used for all experiments. To make the stopped-flow apparatus anaerobic, the syringes and flow cell were flushed first with a solution of ca. 5 mM sodium dithionite and then with anaerobic buffer. Proteins were exchanged into either 25 mM MOPS (pH 7.0) and 1 mM DTT (initial studies) or 25 mM MOPS (pH 7.0) with Biogel P6 desalting columns (Bio-Rad, Hercules, CA). A solution of oxidized MMOR, MMOR-Fd(1-98), MMOR-Fd(1-107), MMOR-Fd(1-120), or MMOR(apoFAD) in a sealed quartz cuvette was made anaerobic by 12 cycles of vacuum gas exchange with O2-free N2. Aliquots (5 μL) of 2-5 mM sodium dithionite were added with a gastight Hamilton titrating syringe. After each addition, optical spectra were recorded until no further changes were observed (typically less than 2 min). This process was repeated until the protein was reduced completely. Anaerobic MMOH solutions were prepared by 10-12 cycles of vacuum gas exchange with nitrogen. Concentrations of MMOH were 10 μM after mixing; reductase or ferredoxin concentrations were 20 μM, except where noted. The reaction of reduced MMOR or MMOR-Fd with oxidized MMOH was monitored at 458 nm (MMOR), 470 nm (MMOR-Fd), and 625 nm (MMOR) in single-wavelength photomultiplier mode. Data points (512 or 640) were collected on a logarithmic time scale (1-500 s, depending on the reaction). The experiment was repeated with the stopped-flow apparatus in multiwavelength diode array mode. For each shot (2-800 s), 160 spectra (380-800 nm) were recorded; during data sets that required long collection times, an anti-bleaching shutter apparatus was employed. The temperature was maintained at 4 °C with a constant temperature circulating water bath for all experiments, except where noted. The effects of MMOB and MMOD on intermolecular electron transfer were investigated by including these proteins in the same syringe as oxidized hydroxylase.
Single-wavelength absorbance data were fit to a sum of two (MMOR-Fd and MMOR(apoFAD)) or four (MMOR) exponentials with the program KinetAsyst (v. 2.2 or 3.04, Hi-Tech Limited, Salisbury, England). Rate constants and intermediate spectra were extracted from diode array data by using the global analysis routines resident in the Specfit software suite (v. 2.10U or 3.0.16, Spectrum Software Associates, Chapel Hill, NC). Compositions of intermediate spectra were calculated by fitting with linear combinations of [2Fe-2S] and FAD component spectra (Figure S1)29 in KaleidaGraph.
Results
MMOR Domain Separation. Although previous studies demonstrated that MMOR-Fd(1-98) retains biochemical characteristics essentially identical to those of the [2Fe-2S] domain in full-length MMOR,29 this protein exhibited surprisingly slow electron transfer to MMOH (vide infra). To generate better models for intermolecular electron-transfer reactions in the sMMO system, we cloned the longer MMOR-Fd(1-107) and MMOR-Fd(1-120) domains. Compared to the original MMOR-Fd(1-98) domain, MMOR-Fd(1-107) includes nine additional C-terminal amino acids (C99RISFGEVG) that comprise the linker between the [2Fe-2S] and FAD/NADH domains in MMOR (Figure 1B). MMOR-Fd(1-120) incorporates both the MMOR linker sequence and the first β strand of the reductase flavin domain (S108FEAEVVGLNWVS).31 FAD-depleted MMOR, MMOR(apoFAD), was also employed to study the initial events in the MMOR-MMOH electron-transfer reaction. All four ferredoxin proteins were competent for electron transfer to MMOH, indicating that the MMOR [2Fe-2S] domain can function as an independent modular unit.
Measurement of MMOH-MMOR-Fd Binding Affinity. The affinity of oxidized MMOR-Fd(1-107) for oxidized MMOH was determined by isothermal titration calorimetry. Figure 2A shows the power required to compensate for the exothermic binding reaction after each addition of titrant to the sample cell. The incremental heat change for each injection was calculated by integrating the heat pulses with respect to time. A plot of these data as a function of the MMOR-Fd(1-107)/MMOH molar ratio is presented in Figure 2B. Preliminary fitting of the enthalpy data with a non-interacting binding sites model in the Origin software suggested that only ca. 70% of the hydroxylase participated in binding MMOR-Fd(1-107). The same phenomenon was observed previously for full-length MMOR and MMOB interactions with MMOH.16 After correction of the MMOH concentration for the nonbinding fraction, a two-site sequential (interacting sites) model was used to obtain an excellent fit to the enthalpy data (solid line in Figure 2B). Thermodynamic parameters for the binding reaction are given in Tables 1 and S1. Similar results were obtained for MMOR-Fd(1-98) and MMOR-Fd(1-120) binding to MMOH (Figure S2; Tables 1 and S1). The very small heat pulses generated in the MMOH-MMOR ITC experiment (data not shown) afforded only a qualitative assessment that the Kd values are in the submicromolar range.
Figure 2.
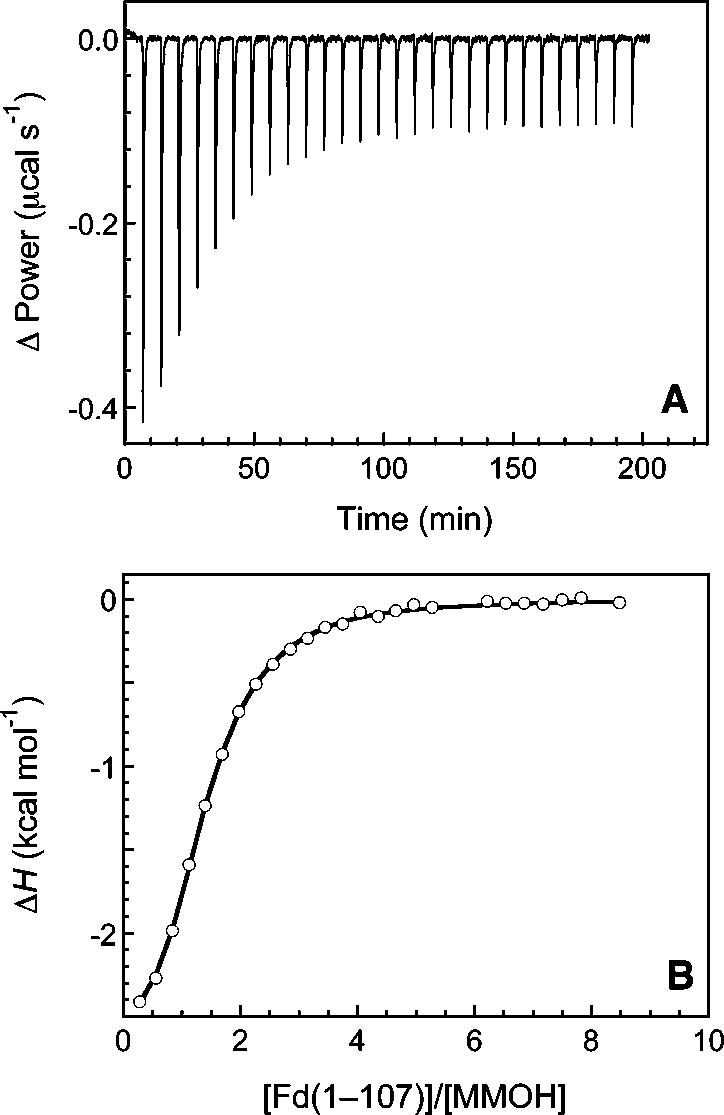
Determination of MMOH-MMOR-Fd(1-107) binding constants by isothermal titration calorimetry. (A) Data recorded for complex formation between MMOH (12.1 μM in the calorimeter cell) and MMOR-Fd(1-107) (453 μM in a 250-μL injection syringe) at 4.2 °C. Each heat pulse corresponds to a 10-μL injection into the 1.430-mL sample cell. (B) Integrated enthalpy data (circles) fit with an interacting sites binding model after correction of the MMOH concentration for the nonbinding fraction of hydroxylase (solid line).
Table 1.
Dissociation Constants for MMOH—MMOR-Fd and MMOH—MMOR Binding Interactionsa
| protein | Kd1 (μM) | Kd2 (μM) |
|---|---|---|
| MMOR-Fd(1-98)b | 0.6±0.2 | 3.2±0.2 |
| MMOR-Fd(1-107)b | 0.5±0.1 | 6.4±0.4 |
| MMOR-Fd(1-120)b | 3.8±1.1 | 9.7±0.6 |
| MMORc | 0.4 | 0.7 |
Isothermal titration data were fit with an interacting sites model after correcting for the nonbinding fraction of MMOH; see text for details. The number of binding sites, N, has an implicit value of 1 for each binding phase. For complete thermodynamic parameters determined for the binding interactions, see Table S1.
Measured at 4.2 °C in 25 mM MOPS (pH 7.0) and 2 mM TCEP.
From ref 16; measured at 3.3°C in 25 mM Tris (pH 7.0) and 1 mM DTT.
Redox Potential Determinations for Ferredoxin Analogues. Selected difference spectra from a reductive titration of MMOR-Fd(1-107) in the presence of the indicator anthraquinone-2-sulfonate are shown in Figure S3. Fitting the data with component difference spectra yielded equilibrium midpoint potentials of -200 ± 1 and -203 ± 3 mV for MMOR-Fd(1-107) and MMOR-Fd(1-120), respectively. These values are nearly identical to the [2Fe-2S] redox potentials determined previously for MMOR-Fd(1-98) (-205 ± 1 mV)29 and full-length MMOR (-209 ± 14 mV).21
Protein Concentration Dependence of Intermolecular Electron Transfer. Protein-protein binding and structural reorganization are prerequisites of efficient intermolecular electron-transfer reactions. The possibility that the kinetics of protein complex formation might affect electron-transfer reactions between sMMO components was investigated by reacting various concentrations of chemically reduced MMOR-Fd(1-107) with oxidized MMOH in the stopped-flow instrument at 4 °C (Figure S4A). The kinetic traces could not be fit well with single-exponential expressions; using two exponential decays yielded high-quality fits to the data (R > 0.999). A third exponential was required for ferredoxin:MMOH ratios greater than 2:1. As shown in Figure S4B, there is no significant effect of MMOR-Fd concentration on the observed rate constants of intermolecular electron transfer. Examination of the preexponential factors returned from data fitting showed that partitioning of electrons between the “fast” (32 s-1) and “slow” (8 s-1) reaction phases is altered by the concentration of MMOR-Fd (Figure S4C). Intermolecular electron-transfer rate constants for MMOR-Fd(1-98), MMOR-Fd(1-120), and full-length MMOR are also independent of protein concentration (data not shown).
Intermolecular Electron Transfer from Ferredoxin Analogues to MMOH. Because the [2Fe-2S] cofactor of MMOR is a one-electron carrier, the ferredoxin center must transfer two electrons sequentially to MMOH to reduce fully each diiron(III) hydroxylase active site. Several MMOR analogues lacking the FAD cofactor (Figure 1B) were implemented to observe the kinetics of the inaugural electron transfer from the ferredoxin domain to MMOH uncomplicated by the flavin moiety of the full-length reductase. All four ferredoxin proteins transferred electrons to the hydroxylase in a biphasic manner with varying rate constants (Table 2; see Discussion for reaction schemes). The temperature dependence of these electron-transfer reactions was investigated for all four ferredoxin analogues, and activation energies were calculated from Arrhenius plots (Figure 3, Table 3). The fractions of the total absorbance change associated with the rapid and slow pathways vary only slightly with temperature, as shown for MMOR-Fd(1-107) in Figure S5A. Analysis using electron-transfer theory expressions34 was performed by fitting temperature-dependence data to eqs 2 and 3 (Figure S5B, Table 4), where h is Planck’s constant, R is the
gas constant, T is temperature, k0 is the characteristic frequency of the nuclei, usually assigned a value of 1013 s-1, and r0 is the closest contact distance, generally set to 3.0 Å. HAB is the electronic coupling between redox centers, λ is the reorganizational energy, and r is the distance between the donor and acceptor sites. A value of 1.1 Å-1 was used for β,3,35 a global parameter related to the nature of the medium between electron-transfer partners. For the sMMO electron-transfer reactions, ΔG° was assigned as -27.1 kJ mol-1, which corresponds to a ΔE° value of +0.281 V (vide infra).
Table 2.
Intermolecular Electron-Transfer Rate Constants for MMOR-Fd and MMORa
| protein | aab | k1 (s−1) | k2 (s−1) | k3 (s−1) | k4 (s−1) |
|---|---|---|---|---|---|
| MMOR-Fd(1-98) | 98 | 1.0 ± 0.1 | 0.24 ± 0.04 | ||
| MMOR-Fd(1-107) | 107 | 32 ± 1 | 7.9 ± 0.4 | ||
| MMOR-Fd(1-120) | 120 | 32 ± 3 | 8.4 ± 0.8 | ||
| MMOR(apoFAD) | 348 | 17 ± 2 | 2.9 ± 0.3 | ||
| MMOR | 348 | 96 ± 10 | 15 ± 1 | 1.3 ± 0.2 | 0.07 ± 0.01 |
Measured at pH 7.0 and 4 °C in single-wavelength stopped-flow experiments.
Number of amino acid residues.
Figure 3.
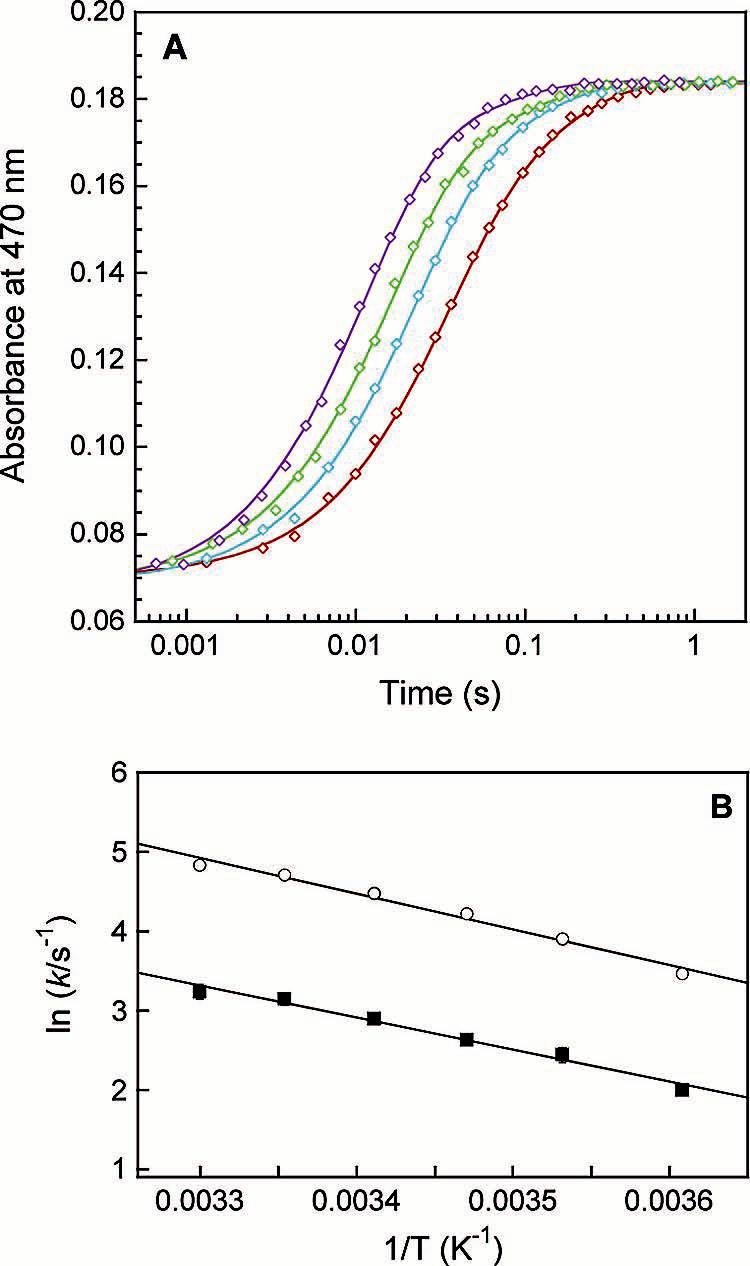
Effect of temperature on intermolecular electron transfer from reduced MMOR-Fd(1-107) to oxidized MMOH. (A) Parallel fits (solid lines) through kinetic data recorded at 470 nm for the reaction of 20 μM MMOR-Fd(1-107) with 10 μM MMOH at 4 (red), 10 (blue), 15 (green), and 25 °C (purple). (B) Arrhenius plot for determination of activation energies: k1 (circles) and k2 (squares).
Table 3.
Activation Energies for Intermolecular Electron Transfera
| protein | aab | Ea1 (kcal mol−1) | Ea2 (kcal mol−1) | Ea3 (kcal mol−1) |
|---|---|---|---|---|
| MMOR-Fd(1-98) | 98 | 11.6 ± 0.5 | 12.8 ± 0.5 | |
| MMOR-Fd(1-107) | 107 | 8.9 ± 0.6 | 8.0 ± 0.5 | |
| MMOR-Fd(1-120) | 120 | 8.6 ± 0.4 | 6.6 ± 0.2 | |
| MMOR(apoFAD) | 348 | 13.5 ± 0.7 | 11.8 ± 0.6 | |
| MMOR | 348 | 5.9 ± 0.2 | 6.4 ± 0.7 | 8.4 ± 0.5 |
Measured at pH 7.0 in single-wavelength stopped-flow experiments.
Number of amino acid residues.
Table 4.
Electron-Transfer Parameters from Temperature Dependence Studiesa
| reaction | rate constant (s−1)b | HAB (cm−1)c | λ (eV)c | r(Å)c |
|---|---|---|---|---|
| Fd(1-98)red + Hox | k1 (1.0) | 7.8 ± 3.4 | 2.8 ± 0.1 | 9.8 ± 0.8 |
| k2 (0.24) | 5.9 ± 1.4 | 2.8 ± 0.1 | 10.3 ± 0.4 | |
| Fd(1-107)red + Hox | k1 (32) | 0.8 ± 0.5 | 2.0 ± 0.1 | 13.7 ± 1.0 |
| k2 (7.9) | 0.2 ± 0.1 | 1.9 ± 0.1 | 16.1 ± 1.0 | |
| Fd(1-120)red + Hox | k1 (32) | 0.8 ± 0.2 | 2.0 ± 0.1 | 13.7 ± 0.5 |
| k2 (8.4) | 0.10 ± 0.02 | 1.7 ± 0.1 | 17.6 ± 0.4 | |
| MMOR3e− + Hox | k1 (96) | 0.22 ± 0.04 | 1.6 ± 0.1 | 16.1 ± 0.3 |
| k2 (15) | 0.08 ± 0.04 | 1.6 ± 0.1 | 17.9 ± 0.8 | |
| k3 (1.3) | 0.16 ± 0.08 | 2.0 ± 0.1 | 16.6 ± 0.9 | |
| k4 (0.07) | 19 000 ± 36 600 | 4.7 ± 0.4 | −4.1 ± 3.5 |
Measured at pH 7.0 in single-wavelength stopped-flow experiments.
Values in parentheses were measured at 4 °C.
Values of HAB, λ, and r were determined by fitting rate constant vs temperature plots with eq 2 or 3; ΔE° = +0.281 V (Fdox/red = −0.205 V, Hox/mv = +0.076 V), ΔG° = −27.1 kJ mol−1, k0 = 1013 s−1, β = 1.1 Å−1, and r0 = 3.0 Å. Because λ ⪢ ΔG°, altering ΔE° by ±0.1 V yields only small changes in HAB, λ, and r.
Because the extinction coefficients of the reduced and oxidized ferredoxin domains are known (Δε470 = 6040 M-1 cm-1),29 the extent of electron transfer to MMOH in single-wavelength stopped-flow studies can be determined. In the first electron-transfer phase from MMOR-Fd(1-107), the hydroxylase is reduced by 32% (with four-electron-reduced MMOH corresponding to 100%); after the second electron-transfer phase, 46% hydroxylase reduction is realized. Each ferredoxin protein can transfer only one electron to MMOH, so the theoretical maximum for hydroxylase reduction in this reaction is 50%. At the end of the electron-transfer reaction, total MMOH reduction was generally less than 50%, however. This result is in accord with other binding and kinetic data, demonstrating that a fraction (10-40%) of MMOH does not bind MMOB and MMOR.16 Therefore, if the nonbinding portion of MMOH is taken into account, full reduction of the active MMOH fraction was achieved. For both MMOR-Fd(1-107) and MMOR-Fd(1-120), ca. 70% of electron transfer proceeds through the rapid reaction phase, whereas this value is just 40% for MMOR-Fd(1-98) (Table 5).
Table 5.
Effects of MMOB and MMOD on Intermolecular Electron Transfer for MMOR Ferredoxin Analoguesa
| protein | k1 (s−1) | % k1b | k2 (s−1) | % k2 | k3 (s−1) | % k3 | ETc (s−1) | %d |
|---|---|---|---|---|---|---|---|---|
| MMOR-Fd(1-98) | 1.1 ± 0.1 | 38 | 0.24 ± 0.04 | 62 | 0.60 | 100 | ||
| + MMOB (2 equiv)e | 0.9 ± 0.1 | 9 | 0.038 ± 0.004 | 91 | 0.11 | 18 | ||
| MMOR-Fd(1-107) | 32 ± 2 | 70 | 7.9 ± 0.4 | 30 | 25 | 100 | ||
| + MMOB (2 equiv) | 10 ± 1 | 11 | 1.1 ± 0.1 | 14 | 0.035 ± 0.003 | 75 | 1.3 | 5 |
| + MMOD (0.2 equiv) | 28 ± 1 | 52 | 6.3 ± 0.6 | 32 | 1.7 ± 0.3 | 15 | 17 | 67 |
| MMOR-Fd(1-120) | 32 ± 3 | 68 | 8.4 ± 0.8 | 32 | 24 | 100 | ||
| + MMOB (2 equiv) | 13 ± 1 | 14 | 1.2 ± 0.1 | 16 | 0.037 ± 0.006 | 72 | 2.0 | 8 |
Measured at 4 °C and pH 7.0 in single-wavelength stopped-flow experiments.
Percentage of electrons that are transferred at rate constant k1.
Effective ET rate, represented by the weighted average of individual ET rate constants.
Percentage of the effective ET rate in the absence of auxiliary sMMO proteins.
Equilibrated mixture of MMOH and 2 mol equiv of MMOB.
Effects of MMOB and MMOD on Intermolecular Electron Transfer from Ferredoxins. Reactions that included MMOB in the same syringe as MMOH required a third exponential phase for fitting. Kinetic traces recording intermolecular electron transfer from MMOR-Fd(1-107) to MMOH in the presence of increasing concentrations of MMOB are presented in Figure 4A. Rate constants from parallel two- or three-exponential fits of these data are altered significantly as a function of MMOB concentration (Figure 5A, Table 5). The first equivalent of MMOB reduces the rate constant for rapid electron transfer by 50%; adding a second equivalent of MMOB decreases k1 by another 20%. The rate constant for the slow reaction phase decreases with increasing concentrations of MMOB such that an 8- to 10-fold reduction is observed at high [MMOB]. The extent of the fast and slow reaction phases, as measured by the fractional contributions of pre-exponential factors, is also highly dependent on MMOB concentration (Figure 5B). At high MMOB concentrations, electron transfer proceeds mostly (ca. 80%) through the third, slowest pathway. Thus, the effective ET rate decreases steadily with increasing MMOB concentration (diamonds, Figure 5A). Similar results were obtained for MMOR-Fd(1-98) and MMOR-Fd(1-120) (Table 5). Time-course profiles of MMOH reduction measured for two MMOR ferredoxin analogues and the effects of MMOB on those reactions are illustrated in Figure 6A.
Figure 4.
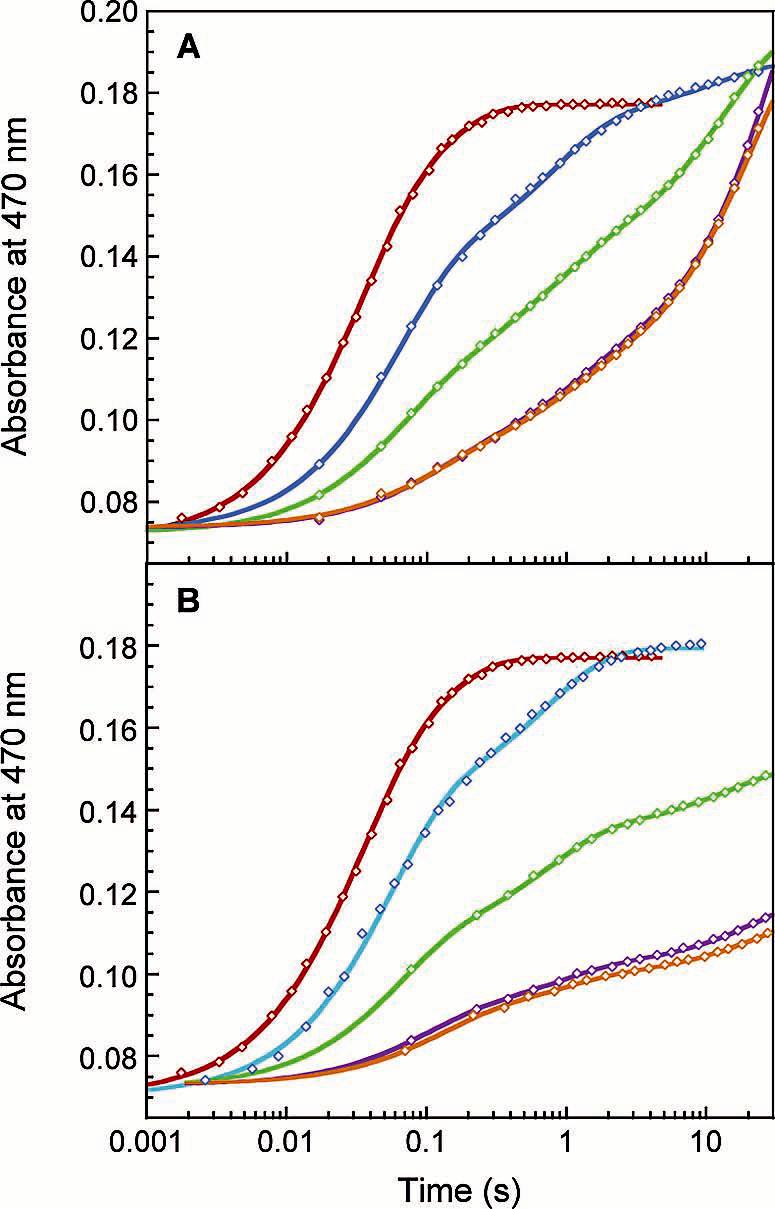
Effects of sMMO auxiliary proteins on electron transfer from reduced MMOR-Fd(1-107) to oxidized MMOH at 4 °C. Parallel fits (solid lines) through kinetic data recorded at 470 nm for the reaction of 20 μM MMOR-Fd(1-107) with 10 μM MMOH in the presence of (A) no MMOB (red), 5 μM MMOB (blue), 10 μM MMOB (green), 20 μM MMOB (purple), and 40 μM MMOB (orange); and (B) no MMOD (red), 5 μM MMOD (blue), 10 μM MMOD (green), 20 μM MMOD (purple), and 30 μM MMOD (orange).
Figure 5.
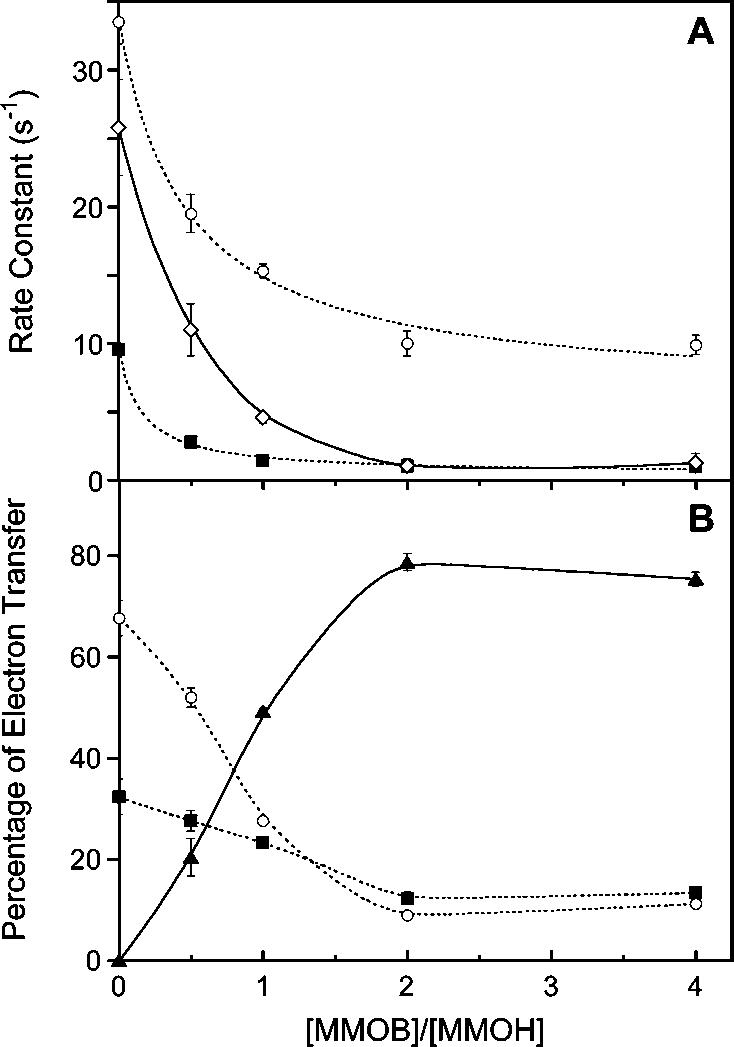
Effect of MMOB on electron transfer from reduced MMOR-Fd(1-107) to oxidized MMOH at 4 °C. (A) Variation of rate constants with MMOB concentration: k1 (circles), k2 (squares), and weighted average (diamonds). (B) Partitioning of electrons through the k1 (circles), k2 (squares), and k3 (triangles) reaction phases as a function of MMOB concentration. The lines shown here are for illustrative purposes only and do not represent fits to a model.
Figure 6.
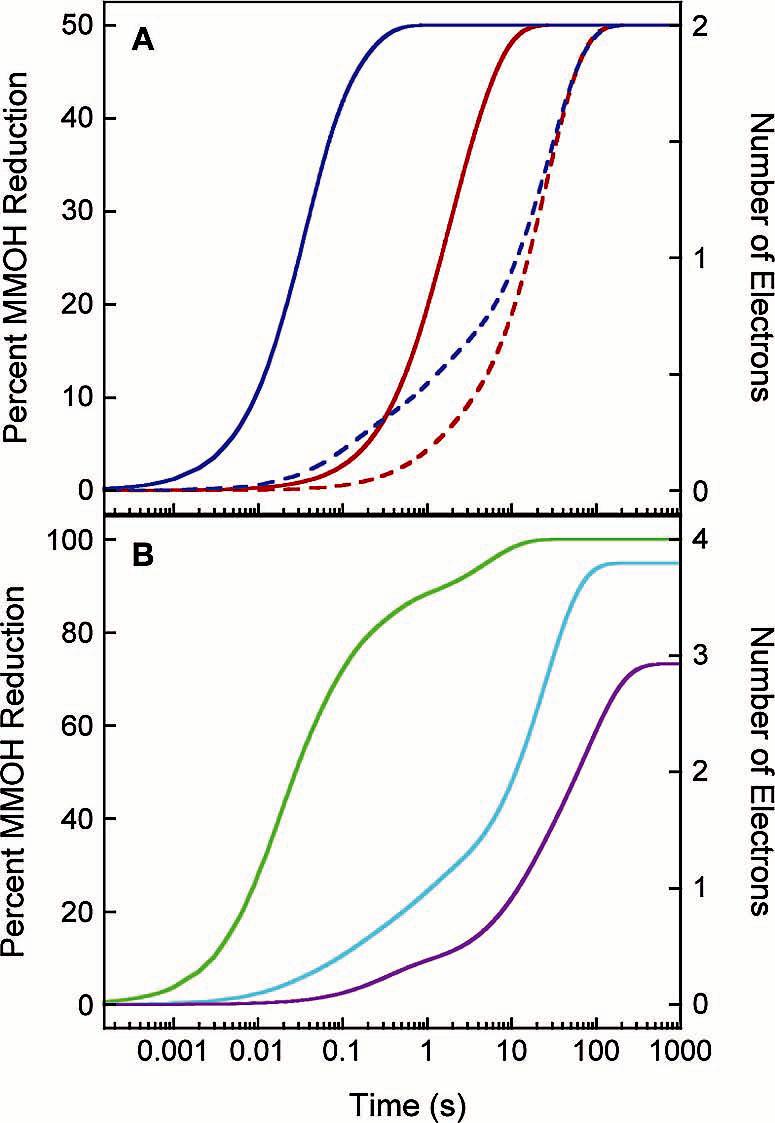
Time-course profiles for MMOH reduction by (A) MMOR ferredoxin analogues and (B) MMOR3e at 4 °C. (A) Electron-transfer reactions in the absence (solid lines) and presence (dashed lines) of 2 equiv of MMOB for MMOR-Fd(1-98) (red) and MMOR-Fd(1-107) (blue). (B) Progression of electron transfer from MMOR3e in reactions including MMOH alone (green), with 2 equiv of MMOB (cyan), or with 2 equiv of MMOD (purple).
MMOD is an even more potent inhibitor of electron transfer to the hydroxylase, as shown by visual inspection of the kinetic traces recorded for reactions including varying concentrations of MMOB (Figure 4A) and MMOD (Figure 4B). As observed for MMOB, increasing MMOD concentrations decrease the rate constants and shift the bulk of electron transfer to the slowest reaction phase. For the very slow reactions containing one or more equivalents of MMOD, fitting the kinetic data proved difficult, often requiring additional exponential phases and returning inconsistent values for the rate constants. At physiological MMOD:MMOH ratios (0.2:1),17 however, the effective electron-transfer rate is diminished by just 30% (Table 5).
Intermolecular Electron Transfer from MMOR3e- to MMOH. The reaction of 20 μM reduced MMOR (MMOR3e-) with 10 μM oxidized MMOH at 4 °C was studied by diode array stopped-flow spectroscopy. Intermediate spectra derived from globally fitting the data with a sequential five-component model are displayed in Figure 7A. Fits converged on rate constant values of k1 = 79 ± 10 s-1, k2 = 20 ± 1 s-1, k3 = 3.6 ± 0.5 s-1, and k4 = 0.19 ± 0.02 s-1. The oxidation state of each intermediate was determined by fitting the spectra with linear combinations of [2Fe-2S]ox, [2Fe-2S]red, FADox, FADsq, and FADhq component spectra (dashed lines in Figure 7A). In the four reaction phases, the hydroxylase diiron sites are reduced by 30%, 25%, 11%, and 11%, respectively, for a total of 77% reduction. After correction for the nonparticipating fraction of MMOH, the extent of hydroxylase reduction after each electron-transfer step was recalculated (Table 6). The first two reaction phases each account for the transfer of about 1.5 electrons to MMOH. The final electron is passed to MMOH in two slow steps. For comparison, the steady-state sMMO reaction with methane proceeds with a turnover number of ca. 0.3 s-1 at 4 °C.16 Therefore, except for k4 (which accounts for only a small fraction of the observed electron transfer), these pre-steadystate electron-transfer rates are compatible with steady-state kinetic measurements. Kinetic traces for the electron-transfer reaction from MMOR3e- to MMOH at varying temperatures (4-35 °C) were fit with sums of two (625 nm) or four (458 nm) exponentials (Figure 8A, Table 2). An Arrhenius plot was constructed (Figure 8B), and activation energies for the first three electron-transfer steps were determined (Table 3). Fitting the temperature-dependent data to eqs 2 and 3 generated values for the electron-transfer parameters HAB, λ, and r (Figure S6, Table 4).
Figure 7.
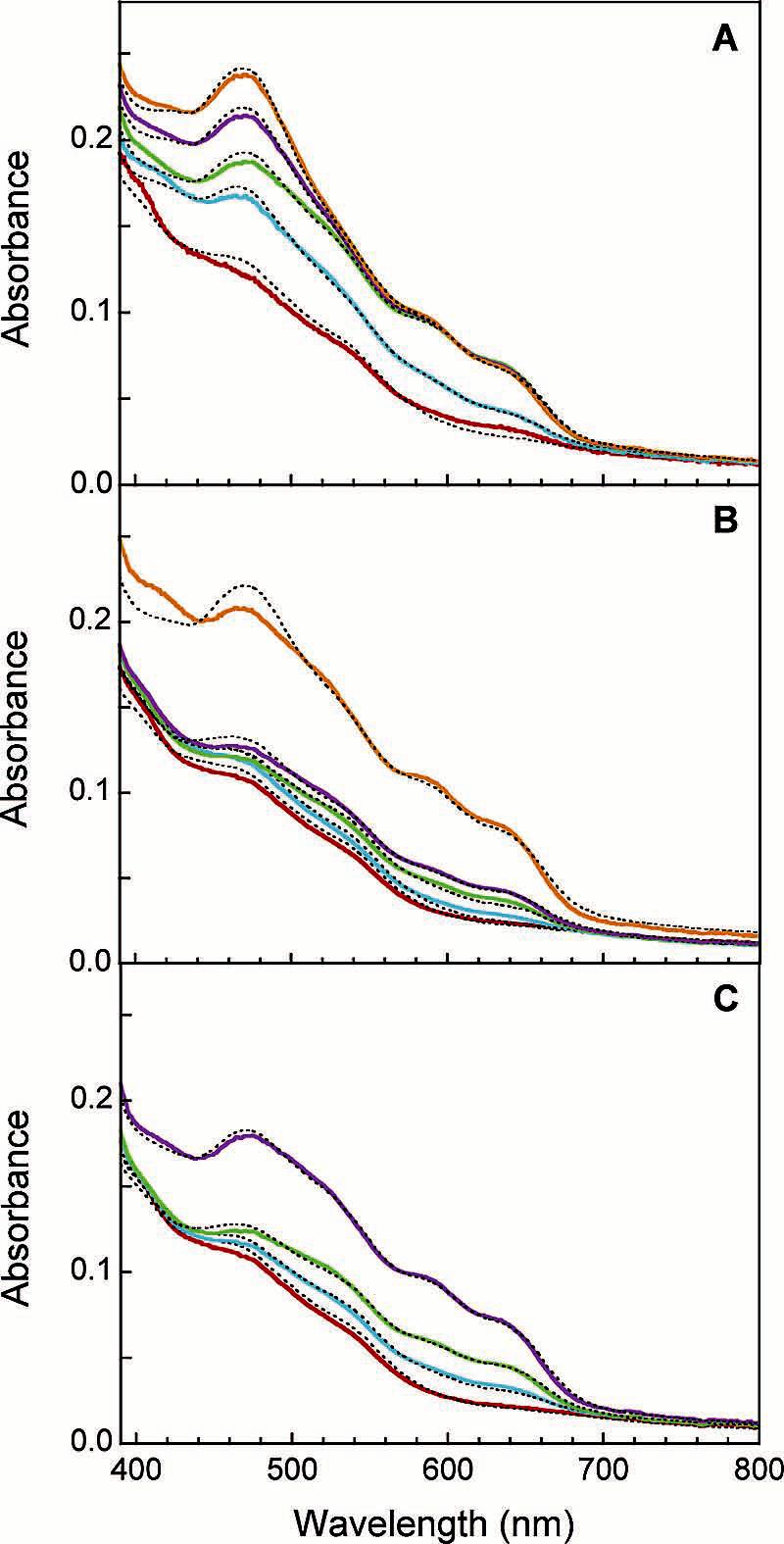
Spectra of intermediates resolved by global analysis of diode array kinetic data for the reaction of MMOR3e with (A) oxidized MMOH; (B) oxidized MMOH and 2 equiv of MMOB; and (C) oxidized MMOH and 2 equiv of MMOD. Intermediates A (red), B (blue), C (green), D (purple), and ≤ (orange) were fit (dashed lines) with linear combinations of MMOR component spectra (Fdox, Fdred, FADox, FADsq, and FADhq).
Table 6.
Distribution of Electrons in Intermolecular Electron-Transfer Intermediatesa
| % total Fe2S2 |
% total FADb |
% MMOH reductionb |
||||||
|---|---|---|---|---|---|---|---|---|
| species | Kf (S−1) | Fdox | Fdred | FADox | FADsq | FADhq | stepc | totalc |
| 2R3e− + Hox | ||||||||
| A | 6 | 94 | 0 | 2 | 98 | 4(0) | ||
| B | 79 | 40 | 60 | 3 | 21 | 75 | 30(39) | 34(39) |
| C | 20 | 41 | 59 | 10 | 57 | 34 | 25(32) | 59(71) |
| D | 4 | 55 | 45 | 15 | 55 | 30 | 12(15) | 70(86) |
| E | 0.2 | 65 | 35 | 22 | 53 | 25 | 11(14) | 81(100) |
| 2R3e− + Hox/2Bd | ||||||||
| A | 0 | 100 | 0 | 0 | 100 | 0(0) | ||
| B | 50 | 4 | 96 | 0 | 1 | 99 | 3(4) | 3(4) |
| C | 9 | 4 | 96 | 0 | 14 | 86 | 6(8) | 9(12) |
| D | 1.5 | 2 | 98 | 6 | 24 | 70 | 10(13) | 19(25) |
| E | 0.04 | 52 | 48 | 16 | 63 | 21 | 54(70) | 73(95) |
| 2R3e− + Hox/2De | ||||||||
| A | 1 | 99 | 0 | 0 | 100 | 1(0) | ||
| B | 3 | 2 | 98 | 0 | 11 | 89 | 6(8) | 7(8) |
| C | 0.08 | 2 | 98 | 3 | 31 | 66 | 13(17) | 20(25) |
| D | 0.01 | 38 | 62 | 7 | 62 | 31 | 37(48) | 57(73) |
Measured at pH 7.0 and 4 °C in diode array stopped-flow experiments. Intermediates are for sequential kinetic schemes.
In some cases, percentages do not total 100 due to rounding.
Numbers in parentheses represent percent MMOH reduction after correction for the nonbinding hydroxylase fraction.
MMOB pre-equilibrated with MMOH.
MMOD pre-equilibrated with MMOH.
Figure 8.
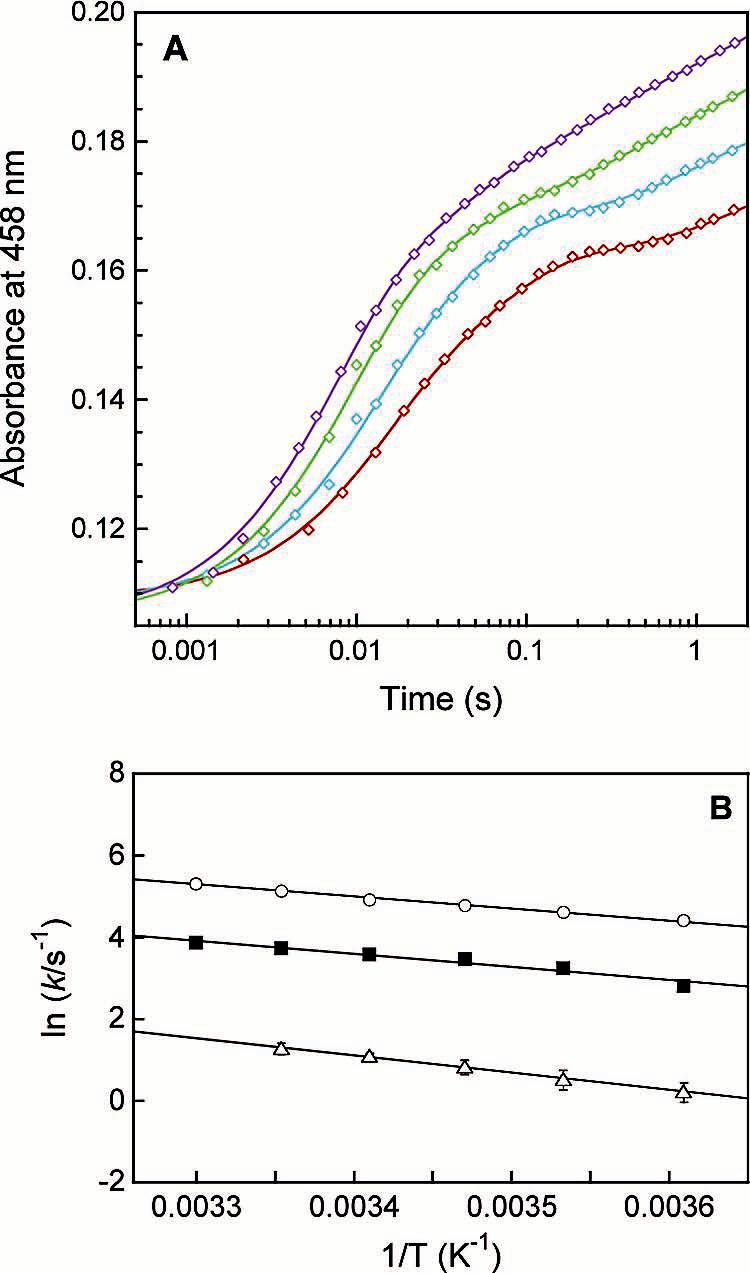
Effect of temperature on intermolecular electron transfer from reduced MMOR3e to oxidized MMOH. (A) Four-exponential parallel fits (solid lines) through kinetic data recorded at 458 nm for the reaction of 20 μM MMOR3e with 10 μM MMOH at 4 (red), 10 (blue), 20 (green), and 30 °C (purple). (B) Arrhenius plot for determination of activation energies: k1 (circles), k2 (squares), and k3 (triangles).
Effects of MMOB and MMOD on Electron Transfer from MMOR3e- to MMOH. When 2 equiv of MMOB or MMOD was added to MMOH before mixing with MMOR3e- in the stopped-flow instrument, intermolecular electron transfer was inhibited. Intermediate spectra obtained from global analysis of diode array data for the reaction of 20 μM MMOR3e- with 10 μM MMOH/20 μM MMOB at 4 °C are shown in Figure 7B. Rate constants for the four electron-transfer steps are about 50% lower (k1 = 52 ± 7 s-1, k2 = 10.5 ± 1.5 s-1, k3 = 1.5 ± 0.5 s-1, k4 = 0.03 ± 0.01 s-1) than those determined in the absence of MMOB. The inhibitory effects of MMOB on intermolecular electron transfer arise primarily from a large shift in the fraction of electrons traveling through the four different reaction phases. The initial three electron-transfer steps combine to transmit a single electron to MMOH; the remaining three electrons are transferred at the slowest rate (Table 6). MMOD is an even more potent inhibitor of intermolecular electron transfer in the sMMO system. Diode array data recorded for the reaction of 20 μM MMOR3e- with 10 μM MMOH/20 μM MMOD were fit with a sequential four-component model to give rate constants of k1 = 3 ± 1 s-1, k2 = 0.06 ± 0.02 s-1, and k3 = 0.009 ± 0.003 s-1 (Figure 7C). These rate constants are substantially diminished compared to those determined in the absence of MMOD. Like MMOB, MMOD induces a change in electron partitioning between the various reaction phases (Table 6). During the first two steps, a total of one electron is transferred to the two hydroxylase diiron sites. Two more electrons are transmitted to MMOH in the slow third step, resulting in 75% reduced hydroxylase. Transfer of a fourth electron was not observed but may occur eventually beyond the examined time range (400 s). The full extent of electron-transfer inhibition by MMOB and MMOD can be appreciated by plotting percent MMOH reduction vs time (Figure 6B).
The MMOB concentration dependence for electron transfer from MMOR3e- to oxidized MMOH was investigated with single-wavelength stopped-flow experiments. Kinetic traces (470 nm) for this reaction were collected at 4 °C with MMOB included in the same syringe as MMOH (Figure 9A). Rate constants extracted from parallel four-exponential fitting are only mildly perturbed by increasing concentrations of MMOB (Figure 9B). As noted for MMOR-Fd(1-107), the extent of the fast and slow reaction phases exhibits considerable dependence on MMOB concentration, such that electron transfer proceeds nearly completely via slow pathways with saturating MMOB (Figure 9C). Including MMOB in the syringe with MMOR3e- produced a similar but smaller interconversion effect. At high MMOB concentrations, 30% of electron transfer occurs through the rapid pathway compared to <5% when MMOB is premixed with MMOH.
Figure 9.
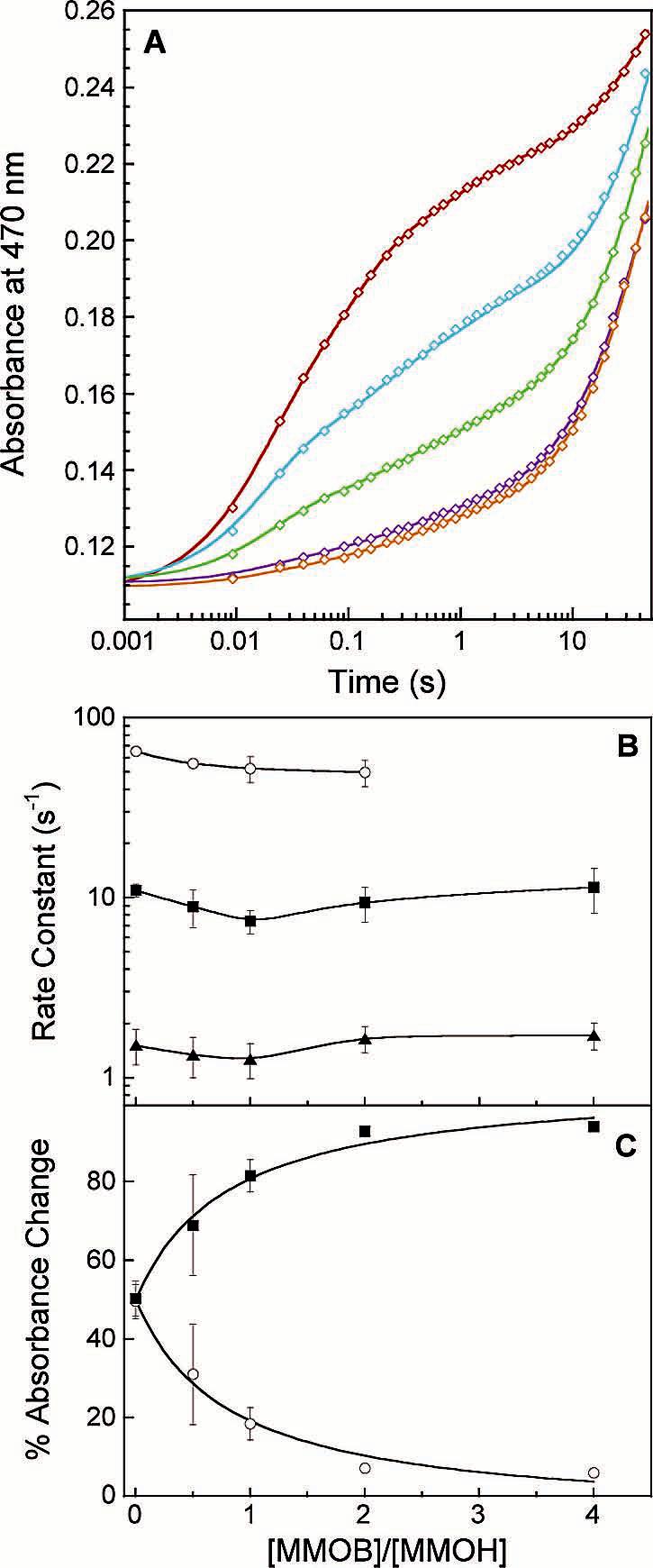
Effect of MMOB on electron transfer from MMOR3e to oxidized MMOH at 4 °C. (A) Parallel fits (solid lines) through kinetic data recorded at 470 nm for the reaction of 20 μM MMOR with 10 μM MMOH in the presence of no MMOB (red), 5 μM MMOB (blue), 10 μM MMOB (green), 20 μM MMOB (purple), and 40 μM MMOB (orange). (B) Variation of rate constants with MMOB concentration: k1 (circles), k2 (squares), and k3(triangles). (C) Partitioning of electrons through the rapid (k1 and k2, circles) and slow (k3 and k4, squares) reaction phases as a function of MMOB concentration.
Discussion
Separating the ferredoxin portion from the multidomain MMOR protein facilitates characterization of electron transfer to MMOH in the absence of spectral and redox contributions from the flavin and NADH cofactors. The four MMOR-Fd analogues (Figure 1B) studied here provide considerable insight into the complex intermolecular electron-transfer reactions in the sMMO system. Our results with these ferredoxin proteins establish the multiphasic nature of electron transfer to the hydroxylase and reveal multiple donor-acceptor populations. The effects of the sMMO auxiliary proteins MMOB and MMOD on electron transfer from full-length MMOR, also examined in this study, are modeled well by the ferredoxin analogues.
Comparison of MMOH-MMOR-Fd and MMOH-MMOR Binding Interactions. To ensure that the MMOR-Fd proteins retain an affinity for MMOH similar to that of full-length MMOR, the MMOH-MMOR-Fd binding interactions at 4.2 °C were examined by isothermal titration calorimetry. By fitting the calorimetric data with an interacting sites binding model, dissociation constants of 0.5 and 6.4 μM were determined for the first and second MMOR-Fd(1-107) binding interactions, respectively, with the dimeric MMOH protein. Although one of the binding constants is the same, the second Kd value is about 9-fold greater, indicating weaker binding, than that measured by the same method for the MMOH-MMOR interaction at 3.3 °C (0.7 μM).16 MMOR-Fd(1-120) binds MMOH yet more weakly, with 10- to 14-fold increases in both Kd values compared to the MMOH-MMOR values. Furthermore, the MMOR-Fd proteins bind to MMOH in exothermic reactions, whereas the MMOH-MMOR binding reaction is endothermic (Table S1). Possible reasons for these differences in thermodynamic binding parameters include different coverage of the MMOH surface owing to the larger full-length MMOR protein, altered structural features in MMOR-Fd compared to the [2Fe-2S] domain and adjacent peptide regions of MMOR, and more extensive conformational changes induced by MMOH-MMOR binding.
Our previous NMR titration studies of MMOR-Fd(1-98) with MMOH revealed a face of the ferredoxin protein, including Asp26 and Glu73, involved in binding to the hydroxylase.30 Mutating either of these residues to alanine in full-length MMOR resulted in decreased affinity for MMOH, demonstrating that MMOR-Fd(1-98) and MMOR contact the hydroxylase in a similar fashion.36 Both MMOR-Fd(1-98) and MMOR from M. capsulatus (Bath) cross-link to the MMOH alpha subunit, providing further evidence for shared binding interactions.37 We therefore conclude that MMOR-Fd(1-98) and MMOR minimally bind to the same sites on MMOH. Additional protein contacts involving the MMOH-MMOR interface and/or changes in structure as a consequence of complex formation are probably responsible for the altered thermodynamic properties noted in the calorimetry experiments.
Formation of Electron-Transfer Complexes. Protein-protein binding typically precedes intermolecular electron transfer in biological systems. Thus, for stopped-flow experiments in which the donor and acceptor pair is combined by mixing, rate constants associated with protein-protein binding and orientation may contribute to the observed rate constant for intermolecular electron transfer. This feature was avoided in previous studies of electron transfer from MMOR to MMOH by allowing the preformed MMOH-MMOR complex to react with NADH.16 When fully reduced MMOR-Fd or MMOR is rapidly mixed with MMOH, the measured rate constants for the intermolecular electron-transfer reactions are independent of protein concentration (Figure S4B). This result indicates that the ferredoxin analogues and MMOR associate rapidly with MMOH to form a tight complex prior to the electron-transfer reactions. Consequently, fitting these electron-transfer data with first-order kinetic equations is warranted.
The rate constants determined for the first two electron-transfer steps from MMOR3e- to MMOH at 4 °C (k1 = 96 ± 10 s-1 and k2 = 15 ± 1 s-1) match closely those observed for the initial electron transfers from MMOR to MMOH in the NADH-initiated reaction (k1 = 106 ± 13 s-1, k2 = 8 or 23 s-1, k3 = 7or12s-1, and k4 = 5s-1).16 The subsequent two electron-transfer phases measured in this work (k3 = 1.3 ± 0.2 s-1 and k4 = 0.07 ± 0.01 s-1) are substantially slower, however. We propose that the initial rapid association of MMOH and MMOR is followed by a slower reorganization of the protein complex that limits the latter observed rate constants for intermolecular electron transfer. On the basis of kinetic and thermodynamic studies of MMOH complex formation with MMOB and MMOR, a similar two-step binding process was postulated.16 Isomerization of the initial MMOH-MMOR precomplex at 25 °C was estimated to occur with maximum forward and reverse rate constants of kf = 0.38 s-1 and kr = 0.057 s-1, respectively.16 Rate constants for electron transfer from MMOR3e- to MMOH at 25 °C determined in this study are k1 = 170 ± 2 s-1, k2 = 42 ± 4 s-1, k3 = 3.6 ± 0.5 s-1, and k4 = 0.35 ± 0.10 s-1. The comparable values of k4 and the approximate MMOH-MMOR precomplex forward isomerization rate constant suggest that the last intermolecular electron-transfer step, corresponding to the transmission of ca. 0.5 electron to MMOH (Table 6), may be controlled primarily by slow complex reorganization. This assignment is supported by ET theory analysis (vide infra), which demonstrates that k4 describes an adiabatic process. The initial three reaction phases may not require as stringent cofactor positioning in the MMOH-MMOR precomplex.
Modeling Intermolecular Electron Transfer in the sMMO System. Multiple kinetic steps are detected for electron transfer from chemically reduced MMOR or MMOR-Fd to oxidized MMOH. The data reported here were fit by using both parallel (single-wavelength data) and sequential (diode array data) mechanisms for the intermolecular electron-transfer reactions. Rate constants returned from both fitting methods were quite similar. In addition, global fitting of diode array data with sequential models yielded internally consistent sets of composite intermediate spectra, which are well described by linear combinations of the previously determined oxidized and reduced iron-sulfur and flavin component spectra.29 Multiwavelength data sets for the four-step reaction of MMOR3e- with MMOHox could also be fit well with various alternative mechanisms (vide infra), but these models generally failed to converge upon unique solutions.
For the simple case of MMOR ferredoxin analogues transferring electrons to MMOH, a discrete hydroxylase intermediate that is 35% reduced and a final state that is 50% reduced are observed. These results are consistent with an asynchronous mechanism (half-sites reactivity) in which a mixed-valent diiron center is generated at one hydroxylase protomer in the first reaction phase and at the other protomer in the second reaction phase (Scheme S1A, upper pathway). A concerted mechanism (Scheme S1A, lower pathway), defined by simultaneous electron transfer to both MMOH protomers, is also possible and cannot be ruled out with certainty on the basis of the present data. A third option invokes two hydroxylase populations that exhibit intrinsically different intermolecular electron-transfer rates (Scheme S1B). Because MMOR-Fd concentration variations can alter the partitioning of electrons through the rapid and slow reaction pathways, the electron-transfer steps are interchangeable, probably by means of conformational changes in the hydroxylase component. Multiple reductase binding sites and/or orientations on MMOH may also give rise to separate populations of protein-protein complexes with different electron-transfer characteristics, as reported for several other biological electron-transfer systems.6,38 In this scenario, excess MMOR-Fd would saturate the binding site(s) that support “fast” electron transfer, thus shifting electron-transfer throughput to the faster reaction phase (Figure S4C).
The sequence of electron-transfer steps and the species compositions determined for the MMOH-MMOR reaction at 4 °C are presented graphically in Schemes 2 and S2. The first two reaction phases each correspond to the transmission of 1.5 electrons to MMOH. The final electron is transferred in two distinct steps. The oxidation levels of the MMOR cofactors were assessed throughout the electron-transfer reaction by fitting the optical spectra of intermediates with combinations of the well-established reductase [2Fe-2S] and FAD component spectra.16,29 The oxidized, mixed-valent, and reduced forms of MMOH are virtually silent in the visible region of the spectrum examined in these stopped-flow experiments. Although the total number of electrons transferred to MMOH can be calculated with precision from the MMOR composition, the distribution of electrons within the hydroxylase diiron sites cannot be ascertained. As described for the MMOR-Fd analogues, both asynchronous (Scheme S2) and parallel (Scheme 2) electron-transfer mechanisms are plausible. Furthermore, because there are four discrete steps, various types of half-sites reactivity can be imagined, including differential or alternating electron transfer to the two hydroxylase protomers (Scheme S2). Further spectroscopic studies of the MMOH diiron center will be necessary to distinguish between these mechanisms. Since X-ray structural investigations have revealed substantial amino acid side-chain repositioning during reduction of Hox to Hmv to Hred,39 it is likely that these conformational changes contribute to the kinetic features observed in the present work.
Scheme 2.

Interconversion of Rapid and Slow Electron-Transfer Pathways. The binding of MMOB to the hydroxylase induces structural changes that are coupled energetically to the redox equilibria of the MMOH diiron centers.40-42 For the sMMO system from M. trichosporium OB3b, the equilibrium midpoint potential of MMOH (Em = +48 mV, relative to the normal hydrogen electrode) is decreased by 132 mV (Em = -84 mV) upon binding MMOB. Adding MMOR to either MMOH or an MMOH-MMOB complex raises the hydroxylase midpoint potential to ca. +100 mV.25 Moreover, MMOR inverts the relative values of the two MMOH redox potentials such that the transfer of two electrons is thermodynamically favored.41 Both MMOB and MMOD alter the structure of the MMOH diiron site, as indicated by several spectroscopic techniques. EPR, MCD, and EXAFS studies provide evidence for MMOB-induced changes at the hydroxylase active site.24,26,43,44 Optical spectroscopy reveals that a small fraction (ca. 15%) of hydroxo-bridged diiron sites is converted to (μ-oxo)diiron(III) centers when MMOD binds to MMOH.17 Formation of the oxo-bridged species is also supported by EXAFS spectroscopy.45 In contrast, MMOR does not alter the spectroscopic properties of the hydroxylase diiron center.13 MMOB and MMOR profoundly affect steady-state sMMO catalysis and efficiency, single-turnover reaction rates, and product distribution by means of intricate interactions that are still not fully understood.16,23-26 By competing with MMOB for binding to the hydroxylase, MMOD inhibits sMMO activity in steady-state assays.17
A role for MMOB in modulating the rate of intermolecular electron transfer in steady-state sMMO reactions has been suggested.15,19,46 Previous stopped-flow studies of electron transfer from MMOR to MMOH involved rapidly mixing preassembled MMOR-MMOH or MMOR-MMOH-MMOB complexes with NADH and monitoring absorbance changes associated with MMOR reduction by NADH and reoxidation as electrons were transferred to MMOH.16 Four kinetic steps corresponding to the transfer of two electrons into a single hydroxylase diiron site were observed. Increasing concentrations of MMOB in the reactions increased the latter three electron-transfer rate constants to a point of saturation. In contrast to these findings, when chemically reduced MMOR is allowed to react with premixed MMOH and MMOB, electron transfer to the hydroxylase is substantially slower than in the absence of MMOB (Figure 6B). This inhibitory effect is produced primarily by shifting the bulk of electron transfer from the initial, faster reaction phases to the fourth, slowest phase. We propose that MMOB may play a key role in the sMMO catalytic cycle by preventing undesired electron and/or proton transfer to the intermediates that form at the hydroxylase active sites and are required for substrate oxidation.47
MMOB and MMOR from M. capsulatus (Bath) do not bind to each other and have separate binding sites on MMOH.16 Therefore, the effect of MMOB on intermolecular electron transfer must be transmitted through structural changes of the hydroxylase component. The fully equilibrated MMOR-MMOH-MMOB complex exhibits relatively fast intermolecular electron-transfer rates (k1 = 106 s-1, k2 = 24.5 s-1, k3 = 15.8 s-1, and k4 = 6.4 s-1) when reduced by NADH at 4 °C.16 The significantly lower rate constants (k1 = 52 s-1, k2 = 10.5 s-1, k3 = 1.5 s-1, and k4 = 0.03 s-1) measured in the present study for electron transfer from MMOR3e- to a preformed MMOH-MMOB complex suggest that isomerization of the initial ternary complex is required for maximal intermolecular electron-transfer rates. By extrapolation from binding data collected at 25 °C (vide supra),16 the rate constant for this isomerization reaction may correspond to k4. Thus, in the presence of MMOB, 75% of electron transfer from MMOR3e- to MMOH may be gated by a slow conformational change, compared to just 14% without MMOB (Table 6). This effect can be explained by a simple model that includes two interconvertible populations of MMOH, Hox and Hox′, which exhibit rapid and slow electron-transfer rates, respectively (Scheme 2). Addition of MMOB to this heterogeneous MMOH mixture would shift the equilibrium between the two species toward Hox′, such that more electrons are transferred by the slow mechanism. The presence of MMOR would counteract the effect of MMOB by moving the equilibrium toward Hox. This intermolecular electron-transfer model mimics closely the changes in the hydroxylase redox potentials produced by MMOB and/or MMOR.25,40
There are several spectroscopic indications of heterogeneity in both oxidized and reduced hydroxylase samples. In freeze-quench Mössbauer studies of the reaction of Hred/2B with dioxygen, two distinct populations of Hred were observed.23 Only the minority species (30-40% of the iron that participated in the reaction) generated high-valent intermediates at rates fast enough to support steady-state turnover numbers. In addition, a third fraction (ca. 12% of the total iron) did not react with dioxygen at all. The occurrence of two populations of Hox from M. capsulatus (Bath) was previously detected by EPR spectral analysis of cryoreduced samples.48 Moreover, this study indicated that the effects of MMOB did not involve significant alterations in the primary coordination sphere of the diiron center. EXAFS spectroscopy of oxidized hydroxylase from M. trichosporium OB3b revealed two populations of diiron sites with Fe-Fe distances of 3.01 (60%) and 3.36 Å (40%).49 Only ca. 15% of MMOHox is converted to the μ-oxo species by MMOD, even though two equivalents of MMOD bind to each hydroxylase molecule.17 The multiple populations of hydroxylase proposed for intermolecular electron-transfer reactions (Schemes 2 and S4) are fully consistent with these prior observations. An extended discussion about how these interconverting MMOH populations might affect intermolecular electron transfer in the sMMO system, including Scheme S3 (midpoint potentials for sMMO complexes) and Scheme S4 (electron-transfer reaction scheme involving three hydroxylase populations), is provided as Supporting Information.
The multiple electron-transfer pathways observed in the current study may reflect the positioning of the hydroxylase surface residue Asn214, a universally conserved amino acid in bacterial multicomponent monooxygenases.33,50 This amino acid adopts redox-dependent side-chain orientations in MMOH crystal structures, projecting outward toward the solvent in MMOHox and inward toward the active site in MMOHred.39 MMOB has long been proposed to bind to the central hydroxylase canyon region, where Asn214 is situated, and may transmitits effects on the diiron site via this residue.33,39,43,51,52 In the homologous toluene/o-xylene monooxygenase (ToMO) system from Pseudomonas stutzeri OX1, the coupling protein significantly enhances electron-transfer throughput from the Rieske protein to the hydroxylase component. Mutating the analogous Asn202 residue in the ToMO hydroxylase to either alanine or tyrosine considerably reduces binding affinity for the coupling protein, thereby inhibiting intermolecular electron transfer.53 Although the sMMO and ToMO coupling proteins exert different effects on electron-transfer reactions in their respective systems, these results may indicate a universal role in regulating electron transfer for the coupling proteins in bacterial multicomponent monooxygenases.
Incubating MMOH with 2 equiv of MMOD inhibits electron transfer from MMOR3e- even more severely than MMOB (Figure 6B). Several possibilities exist for inhibition of intermolecular electron transfer by MMOD: (i) MMOD binding to MMOH obscures the MMOR binding site; (ii) MMOD binding to MMOH induces a conformational change in MMOH that makes MMOR binding and/or electron transfer unfavorable; (iii) MMOD alters the MMOH redox potentials; (iv) MMOD binds to MMOR; and (v) MMOD alters the MMOR redox potentials. The inclusion of MMOD in the sMMO enzyme system was established only recently;17 for this reason, its component interactions have not been studied in as much detail as those of MMOB and MMOR. MMOD competes with MMOB for binding sites on the hydroxylase and cross-links to both the MMOH alpha and beta subunits.17 There is no evidence for binding to MMOR or MMOB, a finding that argues against proposals iv and v. The effect of MMOD on the hydroxylase redox potentials is currently unknown. After reaction with MMOR3e- in the presence of MMOD, the hydroxylase is reduced by only 75%. Therefore, a large decrease in the midpoint potential of the MMOH-MMOD complex that cannot be overcome by MMOR binding is possible.
Hysteresis in Intermolecular Electron-Transfer Reactions. In the absence of MMOB or MMOD, MMOR3e- transmits two electrons to MMOH at 4 °C in about 28 ms, which corresponds to a maximum turnover number of 36 s-1 for the complete reduction of one of the two hydroxylase diiron centers. Premixing 2 equiv of MMOB with MMOH increases the time for two-electron transfer to 11.2 s (maximum turnover number of 0.089 s-1). In steady-state sMMO reactions with methane, a turnover number of ca. 0.3 s-1 is achieved with similar ratios of the MMO components at 4 °C.16 If the effects of reductase binding to the MMOH-MMOB complex rapidly dissipated upon dissociation of MMOR, steady-state catalysis would be limited by electron transfer to a rate at least 3-fold lower than actually observed. Therefore, either MMOR does not dissociate from the ternary MMOR-MMOH-MMOB complex during steady-state turnover or relaxation of the MMOH-MMOR complex to the free hydroxylase conformational state is slow with respect to the binding and dissociation of MMOR. Because steady-state sMMO activity can be maximized in the presence of only 0.1-0.2 equiv of MMOR per hydroxylase,15,16,24 a single reductase molecule must service multiple MMOH molecules during each turnover of the enzyme. This process requires rapid binding to and dissociation from MMOH-MMOB complexes. We conclude that hysteresis of the conformational changes imparted to MMOH by MMOR is necessary for sustained rapid intermolecular electron transfer in the sMMO system. By this mechanism, the ungated hydroxylase structure would be retained throughout the catalytic cycle so that slow MMOH-MMOR isomerization reactions do not inhibit turnover. Furthermore, the equilibrated MMOHox:2MMOB complex cannot be a catalytically relevant participant in steady-state sMMO reactions because electron transfer to this species is too slow to support the observed reaction rates. This complex may represent a subset of the hydroxylase population but cannot be the majority species. The present work was performed at 4 °C; it is possible that at higher temperatures a step other than MMOH-MMOR isomerization becomes rate-limiting. A comprehensive study of component interactions over a wide temperature range (4-45 °C) suggested that the regulatory mechanism of hydroxylase activity by MMOB and MMOR is conserved with respect to temperature, however.16 An alternative and seemingly unlikely explanation is that a large portion of MMOH does not participate in steady-state turnover.
Hysteresis has been invoked for several other aspects of the sMMO system. For example, just 0.6 equiv of MMOB effects the full conversion of the reduced MMOH EPR spectrum to that of the MMOHred-MMOB complex.26 Substrate hydroxylation regioselectivity is also controlled by substoichiometric quantities of MMOB.26 The complete effects of MMOR on the hydroxylase redox potentials, single-turnover product yields, and MMOHred reactivity with O2 are achieved with a 0.2:1 MMOR: MMOH ratio.25 On the basis of these hysteretic changes induced by MMOR, a mechanism in which MMOR converts MMOH to a different conformation (MMOH″) with distinct physical properties was proposed.25 At physiological ratios of sMMO components (1:2:0.2 for MMOH:MMOB:MMOR),15,25 the hydroxylase would exist primarily in the MMOH″ form with increased and inverted redox potentials, thereby favoring two-electron transfer. In addition to these features of MMOH″, we propose that this hydroxylase conformation supports rapid intermolecular electron transfer in the initial MMOH-MMOR or MMOR-MMOH-MMOB precomplex.
MMOR Ferredoxins as Models for Intermolecular Electron Transfer. Several reductase ferredoxin analogues ranging in length from a 98-residue MMOR-Fd truncate to FAD-depleted full-length MMOR were examined as models for intermolecular electron transfer in the sMMO system (Figure 1B). Electron transfer to MMOH from all four ferredoxin constructs proceeds via biphasic reactions similar to the multiple phases observed for wild-type MMOR. Rate constants for these electron-transfer steps vary significantly from protein to protein (Figure 6A, Table 2). The particularly low electron-transfer rates measured for MMOR-Fd(1-98) (k1 = 1.0 s-1 and k2 = 0.24 s-1) were somewhat surprising, considering the spectroscopic (optical, EPR, and EXAFS) and redox potential studies demonstrating that the properties of the [2Fe-2S] center of MMOR-Fd(1-98) are identical to those of the corresponding cofactor in MMOR.29,54 The NMR structure of this short MMOR-Fd domain reveals a fold typical of plant-type ferredoxins.30 Addingjust nine amino acids to the C-terminus of this core ferredoxin domain enhances electron-transfer rates by a factor of 30, as observed for MMOR-Fd(1-107). These findings implicate the MMOR domain linker sequence, C99RISFGEVG, in intermolecular electron transfer either by direct involvement or by orienting the protein cofactors for efficient electron transfer. Further lengthening the ferredoxin analogue does not provide additional increases in electron-transfer rates, as seen for MMOR-Fd(1-120), and even slightly inhibits electron transfer in the extreme case of MMOR(apoFAD). We postulate that this domain linker region may be more structurally ordered in MMOR, thereby affording 2- to 3-fold faster intermolecular electron transfer than for MMOR-Fd(1-107) or MMOR-Fd(1-120).
Application of ET Theory to Intermolecular Electron Transfer in sMMO. ET theory may be applied to interprotein electron-transfer reactions, provided that the observed rate constants are not limited by preceding adiabatic processes such as proton transfer. Because it is not practical to vary ΔG° systematically in most biological systems, ET parameters can be determined instead by examining the reaction at varying temperatures.55 There are several important caveats to this type of investigation, which are included as Supporting Information. Nevertheless, analysis of interprotein electron-transfer reactions by ET theory can be a useful diagnostic tool to distinguish between true, gated, and coupled electron transfer.5,55
A value for ΔE° (+281 mV) was selected to represent the initial electron transfer from the [2Fe-2S] cluster (E°′ = -205 mV)29 to oxidized MMOH (E1°′ = +76 mV).40 Other possible ΔE° values for the sMMO system range from +226 to +319 mV, corresponding to a difference in ΔG° values of only 9.0 kJ mol-1, a potential error which is not significant in this analysis. Except for the slowest MMOR-MMOH reaction phase, the HAB, λ, and r values determined for electron transfer from three MMOR-Fd analogues and MMOR to MMOH are all reasonable for true electron-transfer reactions (Table 4). These results provide support for treating the observed rate constants as excellent estimates of kET, the microscopic rate constants associated with electron transmission. In the case of the slow step of MMOR-MMOH electron transfer (represented by k4), however, the extremely large HAB value (19 000 cm-1) and negative distance determined in the analysis reveal that this electron-transfer phase is gated by an adiabatic process. The somewhat larger HAB and λ values obtained for MMOR-Fd(1-98), in conjunction with the low magnitudes of the observed rate constants, may indicate that these electron-transfer steps are coupled kinetically with other preceding reactions. The ET parameters for the remaining five reactions are fairly similar to each other.
Of particular interest is r, the distance between the reductase [2Fe-2S] and hydroxylase diiron centers in the MMOH-MMOR complex. The average r value of 16.0 ± 1.7 Å determined for MMOR, MMOR-Fd(1-107), and MMOR-Fd(1-120) falls within the 20-Å requirement for efficient electron transfer in biological systems.2,56 In addition, the shortest path from the MMOH diiron site to the hydroxylase surface is ca. 12 Å.51 If the ET theory-derived r values are accurate, MMOR must bind in the central hydroxylase canyon region, as suggested previously.37,51 The rather large λ values determined for intermolecular electron transfer (1.6-2.0 eV) suggest that considerable local conformational changes accompany hydroxylase reduction. Oxidation of the reductase [2Fe-2S] is not expected to require significant reorganization. Upon formation of reduced and mixed-valent MMOH, carboxylate shifts and severe distortion of the diiron site occur, respectively, as observed in crystal structures of the hydroxylase in different oxidation states.39,57 These types of sizable structural perturbations at the MMOH active site could account for the large λ values. Alternatively, the observed electron-transfer rate constants could represent kinetically coupled ET reactions, in which case λ would contain contributions from both the electron-transfer event and the preceding reaction step.58
Concluding Remarks. Electron transfer from chemically reduced MMOR to oxidized MMOH occurs in four discrete kinetic phases corresponding to the transfer of four electrons into the two dinuclear iron centers of the hydroxylase. Both MMOB and MMOD severely diminish intermolecular electron-transfer rates by shifting the majority of electron transfer to the slowest reaction phase, suggesting a function for one or both of these proteins in regulating electron transfer in the sMMO system. A model in which MMOR-induced conformational changes to MMOH are retained throughout the steady-state catalytic cycle is proposed to account for slow intermolecular electron transfer observed in the ternary MMOR-MMOH-MMOB precomplex. To distinguish between the various possible asynchronous, concerted, and parallel electron-transfer mechanisms, freeze-quench EPR, Mössbauer, and XAS spectra are required to characterize the MMOH diiron sites throughout the reactions. Using flash photolysis to initiate electron transfer in preformed MMOH-MMOR complexes may be valuable for determining intrinsic intermolecular electron-transfer rates in the absence of slow binding isomerization reactions.
Acknowledgments
Acknowledgment. This work was supported by National Institutes of Health Grant GM32134 (S.J.L.). J.L.B. was a Howard Hughes Medical Institute predoctoral fellow, and G.T.G. was an NIH postdoctoral fellow. We acknowledge Dr. Maarten Merkx and Lisa L. Chatwood for providing the MMOD and some of the MMOR used in this study and the Multiuser Facility for the Study of Complex Macromolecular Systems (NSF-0070319) for access to an isothermal titration calorimeter.
Footnotes
Supporting Information Available: Description of cloning procedures, extended discussion, Table S1, Schemes S1-S4, and Figures S1-S6, as described in the text (PDF). This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Moser CC, Page CC, Farid R, Dutton PL. J. Bioenerg. Biomembr. 1995;27:263–274. doi: 10.1007/BF02110096. [DOI] [PubMed] [Google Scholar]; (b) Mauk AG. In: Essays in Biochemistry. Ballou DP, editor. Vol. 34. Portland Press, Ltd.; London: 1999. pp. 101–124. [DOI] [PubMed] [Google Scholar]
- 2.Gray HB, Winkler JR. Q. ReV. Biophys. 2003;36:341–372. doi: 10.1017/s0033583503003913. [DOI] [PubMed] [Google Scholar]
- 3.Gray HB, Winkler JR. Proc. Natl. Acad. Sci. U.S.A. 2005;102:3534–3539. doi: 10.1073/pnas.0408029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Hoffman BM, Ratner MA. J. Am. Chem. Soc. 1987;109:6237–6243. [Google Scholar]; (b) Nocek JM, Zhou JS, De Forest S, Priyadarshy S, Beratan DN, Onuchic JN, Hoffman BM. Chem. ReV. 1996;96:2459–2489. doi: 10.1021/cr9500444. [DOI] [PubMed] [Google Scholar]; (c) Page CC, Moser CC, Dutton PL. Curr. Opin. Chem. Biol. 2003;7:551–556. doi: 10.1016/j.cbpa.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Davidson VL. Acc. Chem. Res. 2000;33:87–93. doi: 10.1021/ar9900616. [DOI] [PubMed] [Google Scholar]
- 6.(a) Pletneva EV, Fulton DB, Kohzuma T, Kostić NM. J. Am. Chem. Soc. 2000;122:1034–1046. [Google Scholar]; (b) Leys D, Basran J, Talfournier F, Sutcliffe MJ, Scrutton NS. Nat. Struct. Biol. 2003;10:219–225. doi: 10.1038/nsb894. [DOI] [PubMed] [Google Scholar]
- 7.Batie CJ, Ballou DP, Correll CC. In: Chemistry and Biochemistry of FlaVoenzymes. Müller F, editor. III. CRC Press; Boca Raton, FL: 1992. pp. 543–556. [Google Scholar]
- 8.(a) Higgins IJ, Best DJ, Hammond RC, Scott D. Microbiol. ReV. 1981;45:556–590. doi: 10.1128/mr.45.4.556-590.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hanson RS, Hanson TE. Microbiol. ReV. 1996;60:439–471. doi: 10.1128/mr.60.2.439-471.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higgins IJ, Best DJ, Hammond RC. Nature. 1980;286:561–564. doi: 10.1038/286561a0. [DOI] [PubMed] [Google Scholar]
- 10.(a) Murrell JC. Biodegradation. 1994;5:145–159. doi: 10.1007/BF00696456. [DOI] [PubMed] [Google Scholar]; (b) Nguyen H-HT, Elliott SJ, Yip JH-K, Chan SI. J. Biol. Chem. 1998;273:7957–7966. doi: 10.1074/jbc.273.14.7957. [DOI] [PubMed] [Google Scholar]; (c) Lieberman RL, Shrestha DB, Doan PE, Hoffman BM, Stemmler TL, Rosenzweig AC. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3820–3825. doi: 10.1073/pnas.0536703100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lieberman RL, Rosenzweig AC. Nature. 2005;434:177–182. doi: 10.1038/nature03311. [DOI] [PubMed] [Google Scholar]
- 11.Prior SD, Dalton H. J. Gen. Microbiol. 1985;131:155–163. [Google Scholar]
- 12.(a) Feig AL, Lippard SJ. Chem. ReV. 1994;94:759–805. [Google Scholar]; (b) Wallar BJ, Lipscomb JD. Chem. ReV. 1996;96:2625–2657. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]; (c) Baik M-H, Newcomb M, Friesner RA, Lippard SJ. Chem. ReV. 2003;103:2385–2419. doi: 10.1021/cr950244f. [DOI] [PubMed] [Google Scholar]
- 13.Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Müller J, Lippard SJ.Angew. Chem., Int. Ed 2001402782–2807.and references therein [DOI] [PubMed] [Google Scholar]
- 14.Colby J, Dalton H. Biochem. J. 1978;171:461–468. doi: 10.1042/bj1710461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fox BG, Froland WA, Dege JE, Lipscomb JD. J. Biol. Chem. 1989;264:10023–10033. [PubMed] [Google Scholar]
- 16.Gassner GT, Lippard SJ. Biochemistry. 1999;38:12768–12785. doi: 10.1021/bi990841m. [DOI] [PubMed] [Google Scholar]
- 17.Merkx M, Lippard SJ. J. Biol. Chem. 2002;277:5858–5865. doi: 10.1074/jbc.M107712200. [DOI] [PubMed] [Google Scholar]
- 18.Stainthorpe AC, Lees V, Salmond GPC, Dalton H, Murrell JC. Gene. 1990;91:27–34. doi: 10.1016/0378-1119(90)90158-n. [DOI] [PubMed] [Google Scholar]
- 19.Lund J, Woodland MP, Dalton H. Eur. J. Biochem. 1985;147:297–305. doi: 10.1111/j.1432-1033.1985.tb08750.x. [DOI] [PubMed] [Google Scholar]
- 20.Green J, Dalton H. Biochem. J. 1989;259:167–172. doi: 10.1042/bj2590167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kopp DA, Gassner GT, Blazyk JL, Lippard SJ. Biochemistry. 2001;40:14932–14941. doi: 10.1021/bi015556t. [DOI] [PubMed] [Google Scholar]
- 22.(a) Lee S-K, Nesheim JC, Lipscomb JD. J. Biol. Chem. 1993;268:21569–21577. [PubMed] [Google Scholar]; (b) Shu L, Nesheim JC, Kauffmann K, Münck E, Lipscomb JD, Que L., Jr. Science. 1997;275:515–518. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]; (c) Valentine AM, Stahl SS, Lippard SJ. J. Am. Chem. Soc. 1999;121:3876–3887. [Google Scholar]
- 23.Liu KE, Valentine AM, Wang D, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. J. Am. Chem. Soc. 1995;117:10174–10185. [Google Scholar]
- 24.Fox BG, Liu Y, Dege JE, Lipscomb JD. J. Biol. Chem. 1991;266:540–550. [PubMed] [Google Scholar]
- 25.Liu Y, Nesheim JC, Paulsen KE, Stankovich MT, Lipscomb JD. Biochemistry. 1997;36:5223–5233. doi: 10.1021/bi962743w. [DOI] [PubMed] [Google Scholar]
- 26.Froland WA, Andersson KK, Lee S-K, Liu Y, Lipscomb JD. J. Biol. Chem. 1992;267:17588–17597. [PubMed] [Google Scholar]
- 27.(a) Andrews SC, Shipley D, Keen JN, Findlay JBC, Harrison PM, Guest JR. FEBS Lett. 1992;302:247–252. doi: 10.1016/0014-5793(92)80452-m. [DOI] [PubMed] [Google Scholar]; (b) Karplus PA, Daniels MJ, Herriott JR. Science. 1991;251:60–66. [PubMed] [Google Scholar]
- 28.Correll CC, Batie CJ, Ballou DP, Ludwig ML. Science. 1992;258:1604–1610. doi: 10.1126/science.1280857. [DOI] [PubMed] [Google Scholar]
- 29.Blazyk JL, Lippard SJ. Biochemistry. 2002;41:15780–15794. doi: 10.1021/bi026757f. [DOI] [PubMed] [Google Scholar]
- 30.Müller J, Lugovskoy AA, Wagner G, Lippard SJ. Biochemistry. 2002;41:42–51. doi: 10.1021/bi015668k. [DOI] [PubMed] [Google Scholar]
- 31.Chatwood LL, Müller J, Gross JD, Wagner G, Lippard SJ. Biochemistry. 2004;43:11983–11991. doi: 10.1021/bi049066n. [DOI] [PubMed] [Google Scholar]
- 32.(a) Massey V. J. Biol. Chem. 1957;229:763–770. [PubMed] [Google Scholar]; (b) Stookey LL. Anal. Chem. 1970;42:779–781. [Google Scholar]
- 33.Coufal DE, Blazyk JL, Whittington DA, Wu WW, Rosenzweig AC, Lippard SJ. Eur. J. Biochem. 2000;267:2174–2185. doi: 10.1046/j.1432-1327.2000.01210.x. [DOI] [PubMed] [Google Scholar]
- 34.Marcus RA, Sutin N. Biochim. Biophys. Acta. 1985;811:265–322. [Google Scholar]
- 35.(a) Moser CC, Keske JM, Warncke K, Farid RS, Dutton PL. Nature. 1992;355:796–802. doi: 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]; (b) Gray HB, Winkler JR. Annu. ReV. Biochem. 1996;65:537–561. doi: 10.1146/annurev.bi.65.070196.002541. [DOI] [PubMed] [Google Scholar]
- 36.Blazyk JL.Electron Transfer and Protein Engineering Studies of the Soluble Methane Monooxygenase from Methylococcus capsulatus (Bath) September2003Massachusetts Institute of Technology; Cambridge, MA: Ph.D. ThesisMMOR mutants D26A and E73A exhibit 2-fold and 3-fold decreases, respectively, in MMOH-MMOR binding affinity and/or frequency in steady-state sMMO assays using propylene as a substrate [Google Scholar]
- 37.Kopp DA, Berg EA, Costello CE, Lippard SJ. J. Biol. Chem. 2003;278:20939–20945. doi: 10.1074/jbc.M301581200. [DOI] [PubMed] [Google Scholar]
- 38.(a) Stemp EDA, Hoffman BM. Biochemistry. 1993;32:10848–10865. doi: 10.1021/bi00091a041. [DOI] [PubMed] [Google Scholar]; (b) Liang Z-X, Nocek JM, Huang K, Hayes RT, Kurnikov IV, Beratan DN, Hoffman BM. J. Am. Chem. Soc. 2002;124:6849–6859. doi: 10.1021/ja0127032. [DOI] [PubMed] [Google Scholar]; (c) Miyashita O, Okamura MY, Onuchic JN. Proc. Natl. Acad. Sci. U.S.A. 2005;102:3558–3563. doi: 10.1073/pnas.0409600102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whittington DA, Lippard SJ. J. Am. Chem. Soc. 2001;123:827–838. doi: 10.1021/ja003240n. [DOI] [PubMed] [Google Scholar]
- 40.Paulsen KE, Liu Y, Fox BG, Lipscomb JD, Münck E, Stankovich MT. Biochemistry. 1994;33:713–722. doi: 10.1021/bi00169a013. [DOI] [PubMed] [Google Scholar]
- 41.Liu KE, Lippard SJ. J. Biol. Chem. 1991;266:12836–12839. [PubMed] [Google Scholar]
- 42.Liu KE, Lippard SJ. In: AdVances in Inorganic Chemistry. Sykes AG, editor. Vol. 42. Academic Press; San Diego: 1995. pp. 263–289. [Google Scholar]
- 43.Pulver SC, Froland WA, Lipscomb JD, Solomon EI. J. Am. Chem. Soc. 1997;119:387–395. [Google Scholar]
- 44.DeWitt JG, Rosenzweig AC, Salifoglou A, Hedman B, Lippard SJ, Hodgson KO. Inorg. Chem. 1995;34:2505–2515. [Google Scholar]
- 45.Rudd DJ, Sazinsky MH, Merkx M, Lippard SJ, Hedman B, Hodgson KO. Inorg. Chem. 2004;43:4579–4589. doi: 10.1021/ic049716b. [DOI] [PubMed] [Google Scholar]
- 46.Green J, Dalton H. J. Biol. Chem. 1985;260:15795–15801. [PubMed] [Google Scholar]
- 47.Beauvais LG, Lippard SJ. J. Am. Chem. Soc. 2005;127:7370–7378. doi: 10.1021/ja050865i. [DOI] [PubMed] [Google Scholar]
- 48.Davydov R, Valentine AM, Komar-Panicucci S, Hoffman BM, Lippard SJ. Biochemistry. 1999;38:4188–4197. doi: 10.1021/bi982391o. [DOI] [PubMed] [Google Scholar]
- 49.Shu L, Liu Y, Lipscomb JD, Que L., Jr. J. Biol. Inorg. Chem. 1996;1:297–304. [Google Scholar]
- 50.Leahy JG, Batchelor PJ, Morcomb SM. FEMS Microbiol. ReV. 2003;27:449–479. doi: 10.1016/S0168-6445(03)00023-8. [DOI] [PubMed] [Google Scholar]
- 51.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 52.(a) MacArthur R, Sazinsky MH, Kühne H, Whittington DA, Lippard SJ, Brudvig GW. J. Am. Chem. Soc. 2002;124:13392–13393. doi: 10.1021/ja0279904. [DOI] [PubMed] [Google Scholar]; (b) Sazinsky MH, Merkx M, Cadieux E, Tang S, Lippard SJ. Biochemistry. 2004;43:16263–16276. doi: 10.1021/bi048140z. [DOI] [PubMed] [Google Scholar]
- 53.Cadieux E, McCormick MS, Sazinsky MH, Lippard SJ.unpublished results
- 54.Rudd DJ, Merkx M, Lippard SJ, Hedman B, Hodgson KO.unpublished results
- 55.Davidson VL. Biochemistry. 1996;35:14035–14039. doi: 10.1021/bi961577p. [DOI] [PubMed] [Google Scholar]
- 56.Page CC, Moser CC, Chen X, Dutton PL. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 57.(a) Rosenzweig AC, Nordlund P, Takahara PM, Frederick CA, Lippard SJ. Chem. Biol. 1995;2:409–418. [PubMed] [Google Scholar]; (b) Rosenzweig AC, Frederick CA, Lippard SJ. In: Microbial Growth on C1 Compounds. Lidstrom ME, Tabita FR, editors. Kluwer Academic Publishers; Dordrecht, The Netherlands: 1996. pp. 141–149. [Google Scholar]
- 58.Blazyk JL, Lippard SJ.Initial work in our laboratory indicates that the rate constants for ET from MMOR-Fd(1-107) to MMOH decrease at pH values above 7.0, suggesting that proton transfer accompanies, and may be coupled with, this ET reactionunpublished work [Google Scholar]