

Abstract
Improved vaccines and adjuvants are being developed to reduce the threat posed by a terrorist attack involving aerosolized anthrax spores. Nevertheless, uncertainty persists concerning the relative benefits of inducing mucosal vs systemic immunity to host survival following inhalational exposure to anthrax spores. This work examines the effect of delivering the licensed human vaccine (Anthrax Vaccine Adsorbed, AVA) combined with a CpG oligodeoxynucleotide (ODN) adjuvant intraperitoneally or intranasally to A/J mice. Results indicate that protection from inhalational anthrax correlates with the induction of a strong systemic rather than mucosal immune response, and demonstrate that protection is significantly improved and accelerated by the addition of CpG ODN.
Keywords: Anthrax, vaccine, protection
1. INTRODUCTION
Bacillus anthracis is an aerobic gram-positive spore-forming bacterium found naturally in wild and domesticated animals [1]. B. anthracis spores are highly resistant to environmental degradation, and upon germination produce a tripartite toxin that reduces the ability of the host's immune system to eliminate the pathogen [1]. Human exposure to anthrax typically arises following contact with infected livestock, and generally results in a mild form of cutaneous disease [2]. In contrast, inhalational exposure to anthrax causes severe and rapidly progressive disease that can culminate in death [3;4]. Anthrax Vaccine Adsorbed (AVA) is the only anthrax vaccine licensed for human use in the US and was approved based on it's ability to reduce susceptibility to cutaneous anthrax exposure. The release of anthrax spores designed for aerosol delivery by bioterrorists in 2001, and the resultant morbidity, mortality, and panic, underscored the need to improve the speed and efficacy of vaccine-induced protection against inhalational exposure [5].
AVA is prepared by adsorbing the culture filtrate of an attenuated toxinogenic non-encapsulated strain of B. anthracis (V770-NP1-R) onto aluminum hydroxide [6]. Studies show that protective Ag (PA), the core of anthrax toxin, is the major immunogen of AVA. Antibody (Ab) against PA neutralize the toxin, inhibit spore germination, and improve the phagocytosis/killing of spores by macrophages [7-10]. The licensed AVA vaccine is administered as a series of 6 immunizations over 18 months followed by yearly boosters [11]. This schedule induces protective serum Ab titers somewhat slowly, and has been linked to adverse side effects [11-13].
Synthetic oligodeoxynucleotides (ODN) containing immunostimulatory “CpG motifs” have been shown to boost immunity to co-administered vaccines, including AVA [14-16]. CpG ODN induce the functional maturation of professional Ag presenting cells (APCs) and trigger the production of immunostimulatory cytokines and chemokines [17;18]. Although previous studies showed that adding CpG ODN to AVA boosted protection among animals challenged systemically with anthrax [16;19], their effect on mucosal immunity and protection against aerosolized anthrax spores was never evaluated. The current work examines whether AVA, alone or co-administered with CpG ODN, improves host resistance to inhalational anthrax, and examines the relative contribution of mucosal vs systemic immunity to host survival.
2. MATERIALS AND METHODS
2.1 Reagents
Phosphorothioate CpG ODN 1555 (GCTAGACGTTAGCGT) and control ODN 1612 (GCTAGAGCTTAGCGT) were synthesized at the CBER core facility [19]. Both were free of endotoxin and protein contamination. A single lot of clinical grade AVA was used in all experiments (BioPort Corporation, East Lansing, MI). Recombinant PA (rPA) was provided by USAMRIID (Fort Detrick, MD) and prepared as described [20]. B. anthracis strain 7702, which is toxinogenic (pXO1+) and non-encapsulated (pXO2), was used to prepare Sterne strain spores, as described [21].
2.2 Animals
Specific pathogen free male A/J mice were obtained from the NCI (Frederick, MD). They were housed in sterile micro-isolator cages in a barrier environment, and immunized at 8−12 wk of age. All animal experiments were conducted using ACUC approved protocols, and aerosol challenge studies were performed in a BL-3 facility.
2.3 Immunization and challenge studies
Male A/J mice were immunized intraperitoneally (i.p.) or intranasally (i.n.) with 2 or 10 ul of AVA ± 20 ug of CpG ODN in a final volume of 20 ul. AVA doses were selected on the basis of preliminary studies demonstrating that 2 ul of AVA induced a detectable but suboptimal IgG anti-PA response while 10 ul of AVA induced a response that protected 50% of mice from subsequent anthrax challenge [19]. The maximum dose of AVA used was limited by the volume of vaccine that could be safely administered i.n. to mice.
Serum obtained by tail nicking was stored at −20° C until use. BAL was collected by instilling and then removing 0.7 ml of PBS into the lungs of anesthetized mice. Copra Ig was obtained by physically disrupting fecal pellets followed by suspension and votexing in a cocktail of protease inhibitors (including 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), E-64, bestatin, leupeptin, aprotinin, and EDTA (Sigma, St. Louis, MO)) as previously described [22]. Supernatants were collected by centrifugation at 16,000g for 10' and stored at −20° C until use.
Mice were challenged via the aerosol route with 20 LD50 (50% lethal doses) of STI spores suspended in dH2O (1 LD50 = 106 STI spores). The spore aerosol was generated using a six-jet Collison nebulizer (BGI Incorporated, Waltham, MA) and distributed to individual mice using a nose-only exposure system (CH Technologies, Westwood, NJ) as previously described [23]. Prior to challenge, mice were supplied with fresh air for 10' to allow respiratory rates to normalize. Survival was monitored for 21 days.
2.4 IgG and IgA anti-PA ELISA
IgG and IgA anti-PA Ab titers were monitored as described [19]. Briefly, 96-well microtiter plates (Immulon 1B, Thermo Labsystems, Franklin, MA) were coated with 1 ug/ml of rPA in PBS at 4° C overnight. The plates then were blocked with 5% non-fat dry milk in PBS containing 0.1% tween-20. Plates were washed, and overlaid with serially diluted BAL, serum or fecal extract for 2 h at room temperature. After thorough washing, bound Abs were detected by adding HRP-labeled goat anti-mouse IgG or IgA (Southern Biotechnology, Birmingham, AL) followed by ABTS substrate (Kirkegaard & Perry, Gaithersburg, MD). Ab titers were determined by comparison to a standard curve generated using pooled sera from hyper-immunized mice, and were expressed as the reciprocal of the end point dilution following background subtraction. All samples were analyzed in duplicate.
2.5 Statistics
Differences in the kinetic development of anti-PA immune responses were examined using two-way ANOVA. Differences in the IgG anti-PA response induced by various vaccine-adjuvant combinations were assessed by one-way ANOVA. Differences in survival were evaluated using log rank analysis of Kaplan-Meier curves. Correlation coefficients were determined by linear regression analysis.
3. RESULTS
3.1 Route of delivery influences the immunogenicity and protective activity of AVA
Initial studies compared the ability of AVA delivered by the systemic (i.p.) vs mucosal (i.n.) route to protect against aerosol challenge by anthrax spores. A/J mice were selected for evaluation because these animals i) mount strong Ab responses to AVA ± CpG ODN and ii) are highly susceptible to challenge by attenuated STI anthrax spores due to a defect in their complement cascade [16;24].
Mice were immunized with AVA ± CpG ODN. The magnitude of the resultant mucosal immune response was analyzed by monitoring anti-PA Abs present in pulmonary brochioalveolar lavage fluid (BAL) and/or in fecal pellets containing copra Ig secreted into the gut lumen. As seen in Table IA, Ag-specific copra IgA levels correlated closely with pulmonary IgA levels in vaccinated mice (R2 = .87, p <.001). Since mice do not survive the collection of BAL, experiments designed to assess the effect of vaccination on survival post challenge relied on copra anti-PA titers to monitor mucosal immunity. Systemic immunity was examined by monitoring serum IgG anti-PA titers. Previous studies showed that serum IgG anti-PA titers represent a sensitive and specific marker of protection against STI anthrax spores in A/J mice immunized with AVA ± CpG ODN [19].
Table I.
Influence of Dose, Route and Formulation on AVA-induced humoral immunity
| A. Mucosal response to intranasal vaccination. | |||
|---|---|---|---|
| Dose of AVA | CpG ODN | BAL | Fecal IgA |
| 2 | − | <10 | <10 |
| 2 | + | 24 | 34 |
| 10 | − | 57 | 180 |
| 10 | + | 2,220* | 2,150* |
| B. Systemic response to vaccination. | |||
|---|---|---|---|
| Dose of AVA (ug) | Route of Administration | CpG ODN | Serum IgG anti-PA titer |
| 2 | in | − | <10 |
| 2 | ip | − | 35 |
| 2 | in | + | 60 |
| 2 | ip | + | 3,000 |
| 10 | in | + | 5,500 |
| 10 | ip | + | 250,000 |
A/J mice were immunized with 2 or 10 ul of AVA ± 20 ug of CpG ODN. A) Geometric mean IgA anti-PA Ab titer of BAL and feces 3 wk post intranasal immunization of mice (N = 4/group). Note that both copra and BAL anti-PA titers were significantly higher in mice immunized with 10 ul of AVA + CpG ODN (p. <.01) than any other group. The correlation coefficient between BAL and copra IgA anti-PA titers was R2 = .87, p <.001. B) Serum IgG anti-PA titers in mice 14 days after either i.n. or i.p. immunization. 10 ul of AVA was more immunogenic than 2 ul (p <.01); i.p. vaccination was more immunogenic than i.n. (p < .02); and adding CpG ODN to AVA significantly increased the humoral immune response (p. <.01). N = 8 mice/group.
Significantly higher anti-PA Ab titers were produced when mice were vaccinated i.n. with 10 rather than 2 ul of AVA (p < .01, Table I). Adding CpG ODN to intranasally administered AVA significantly boosted both BAL and copra IgA anti-PA titers (3−20 fold increase; p < .01, Table IA and Fig 1). Intranasal delivery of AVA + CpG ODN elicited a 3-fold higher and more consistent mucosal response than did systemic vaccination (Fig 1, p <.05). In contrast, i.p. immunization with AVA plus CpG ODN induced a significantly stronger serum IgG anti-PA response than did i.n. delivery of the same dose of vaccine (100 fold higher; p. <.01, Table IB and Fig 2). Adding CpG ODN to systemically administered AVA significantly boosted the serum Ab response by approximately 20-fold (p. <.001, Table IB and Fig. 2).
Figure 1. Copra IgA anti-PA Ab titers in AVA-vaccinated mice.
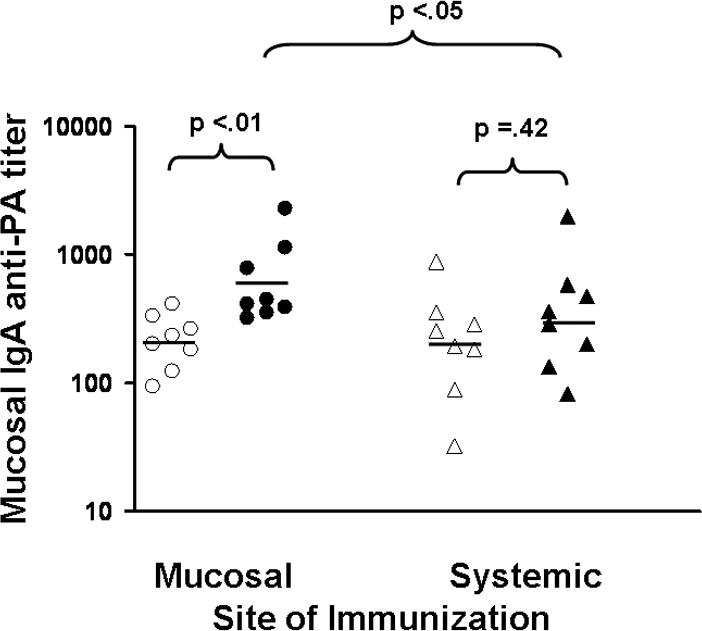
Male A/J mice (specific pathogen free) were immunized either i.n. (mucosal) or i.p. (systemic) with 10 ul of AVA (open symbols) or 10 ul of AVA + 20 ug of CpG ODN (solid symbols). Data show IgA anti-PA titers from 8 mice/group 6 weeks post immunization compiled by combining results from two independent experiments.
Figure 2. Serum IgG anti-PA Ab titers in AVA-vaccinated mice.
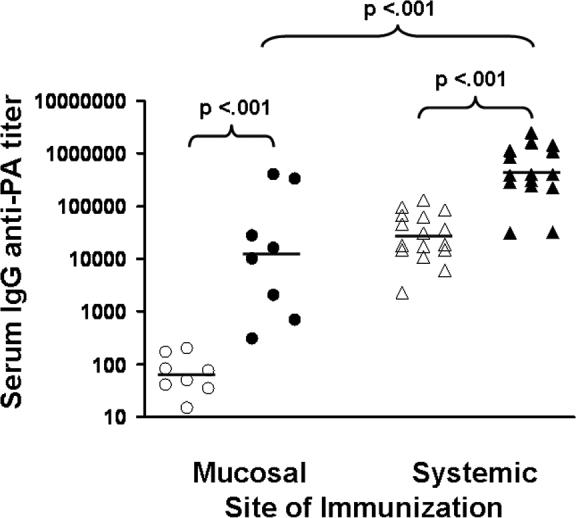
A/J mice were immunized either i.n. (mucosal) or i.p. (systemic) with 10 ul of AVA (open symbols) or 10 ul of AVA + 20 ug of CpG ODN (solid symbols). Data show IgG anti-PA titers from 8 − 16 independently studied mice/group 6 wk post immunization compiled by combining results from two independent experiments.
Mice were vaccinated either i.n. or i.p. with AVA + CpG ODN, and then challenged 4 wk later with 20 LD50 of aerosolized Sterne strain anthrax spores (STI). This dose of STI spores was uniformly fatal to unimmunized controls. As seen in Fig 3, all mice vaccinated with 10 ul of AVA + CpG ODN by the i.p. route survived challenge, whereas half of those immunized with the same vaccine by the i.n. route (or with AVA alone i.p.) succumbed to infection (p. <.01). Significantly higher survival was also observed when the 2 ul dose of AVA + CpG ODN was delivered i.n. when compared to i.p. (p. =.02, Fig 3).
Figure 3. Survival varies by route of immunization.
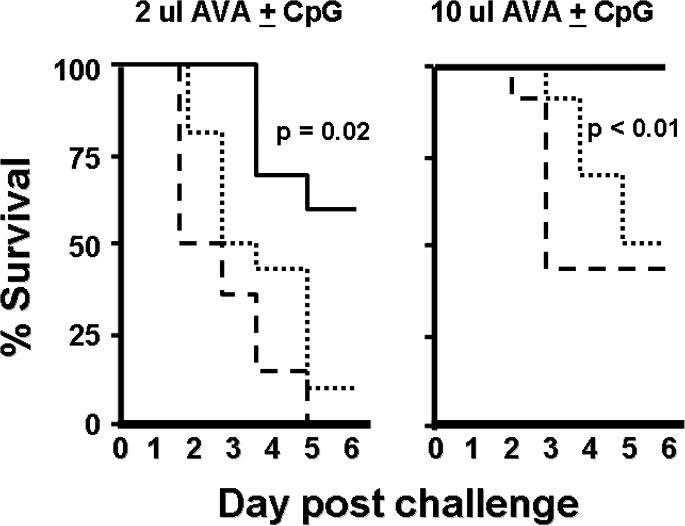
A/J mice were immunized with AVA ± 20 ug of CpG ODN and challenged 4 wk later with 20 LD50 of STI spores via the aerosol route. Mice immunized i.p. with AVA + CpG ODN (solid line) were significantly better protected than mice immunized i.p. with AVA alone (dashed line) or i.n. with AVA + CpG ODN (dotted line). Challenge was 100% fatal in unimmunized mice. Data show the percent survival of 10 mice/group compiled by combining results from two independent experiments.
3.2 Protection against aerosolized anthrax correlates with systemic but not mucosal anti-PA Ab titers
To pursue these observations, additional mice were vaccinated and their anti-PA Ab response evaluated immediately prior to STI spore challenge. Consistent with earlier findings, systemic immunity was enhanced by immunization with 10 vs 2 ul of AVA (p. <.01), vaccination via the i.p. rather than i.n. route (p. <.02), and inclusion of CpG ODN as the vaccine adjuvant (p. <.01, Table IB). Moreover, serum but not mucosal anti-PA titers predicted survival following aerosolized STI challenge (Fig 4, p <.001). Whereas nearly all mice with systemic IgG anti-PA titers >10,000 at the time of challenge survived, <10% of those with titers <2,500 survived (Fig 4). In contrast, there was no significant correlation between copra IgA anti-PA titers and survival (p = .49, Fig 4).
Figure 4. Serum IgG anti-PA titers predict survival following aerosol anthrax challenge.
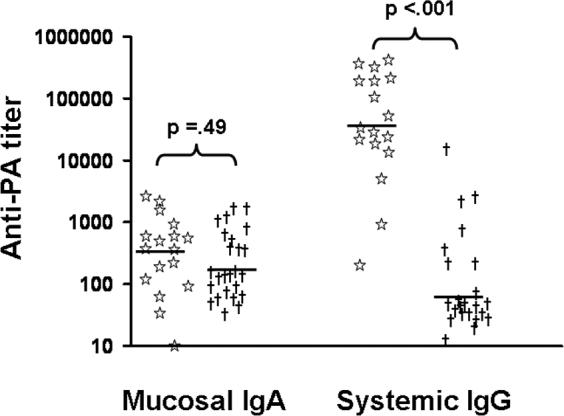
A/J mice were immunized i.n. or i.p. with 2 or 10 ul of AVA ± 20 ug of CpG ODN. Mucosal (copra) IgA and systemic (serum) IgG anti-PA titers were determined 2 weeks post immunization, and the mice challenged with 20 LD50 of STI spores via the aerosol route. Data show mucosal and systemic Ab titers in mice that survived (star) or succumbed (†) to challenge. Administering 10 ul of AVA + CpG ODN i.p. resulted in 92% survival, compared to 33% survival when administered i.n. Only 13% of mice immunized with 2 ul of AVA ± CpG ODN survived. Note that survival correlated significantly with serum IgG (p. <.001) but not with mucosal anti-PA titers.
3.3 Effect of CpG ODN on the rapidity of AVA-induced protection
Previous studies suggested that the addition of CpG ODN to AVA both accelerated and boosted the induction of pathogen-specific immunity [16;19]. Those studies examined the susceptibility of mice to systemic anthrax challenge 10 − 21 days after AVA vaccination. To determine whether the addition of CpG ODN to AVA accelerated the induction of protective immunity against aerosolized spore challenge, A/J mice were immunized with AVA ± CpG ODN and their response to inhalational challenge evaluated 5, 10 and 15 days later.
Consistent with prior results, the serum IgG anti-PA response was magnified and accelerated by the addition of CpG ODN to AVA (Fig 5A). By day 10, serum anti-PA titers were 20-fold higher in the AVA + CpG ODN vaccinated group, and protection was significantly enhanced (15/16 vs 4/16, p< .01). Of note, although IgG Ab titers did not differ between groups on day 5 (p = .32), a significant difference in survival was still observed when mice were challenged at that early time point (p = .04, Wilcoxin log rank test). This finding suggests that host susceptibility to inhaled anthrax is influenced by the magnitude of the serum anti-PA response over the course of infection, rather than simply at the time of initial exposure.
Figure 5. Kinetics of immunity elicited by AVA immunization.
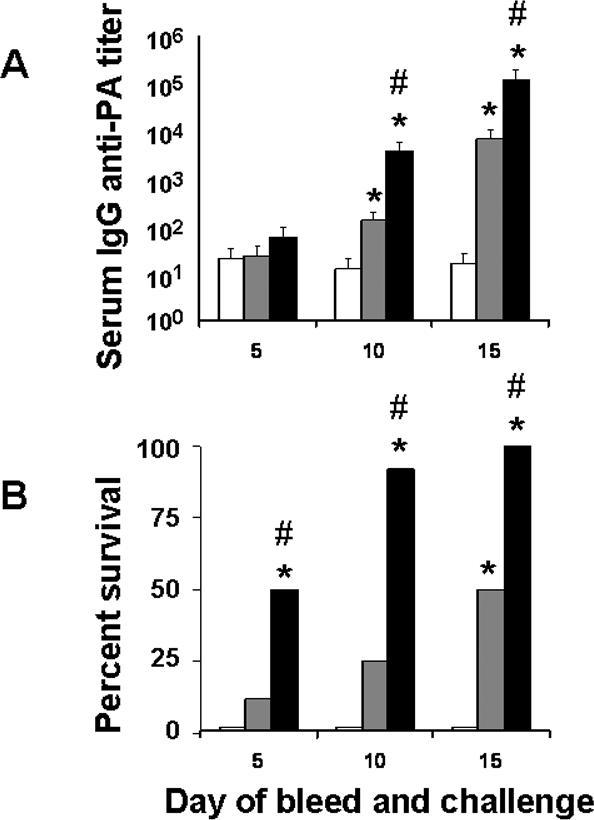
A/J mice (N = 24/group) were immunized i.p. with 20 ug of CpG ODN alone (□), 10 ul of AVA alone ( ) or 10 ul of AVA + 20 ug of CpG ODN (
) or 10 ul of AVA + 20 ug of CpG ODN ( ). Eight mice from each group were bled and then challenged with 20 LD50 of STI spores via the aerosol route on day 5, 10 or 15. Data show the serum IgG anti-PA titers (A) and survival (B) of animals in each group at each time point. *, p <.05 vs CpG ODN alone; #, p. <.05 vs AVA alone.
). Eight mice from each group were bled and then challenged with 20 LD50 of STI spores via the aerosol route on day 5, 10 or 15. Data show the serum IgG anti-PA titers (A) and survival (B) of animals in each group at each time point. *, p <.05 vs CpG ODN alone; #, p. <.05 vs AVA alone.
4. DISCUSSION
Anthrax spores designed for inhalational exposure were intentionally released in the US in 2001. The resultant morbidity and panic established the viability of anthrax as a bioterror weapon, and emphasized the need to improve the speed and efficiency with which protective immunity is induced against aerosolized anthrax [5]. To supplement the development of novel vaccines and adjuvants (such as CpG ODN), it is of value to clarify the role of mucosal and systemic immunity in providing protection against inhalational anthrax.
Towards that end, the effect of systemic vs mucosal delivery of AVA ± CpG ODN was evaluated. A strong systemic IgG anti-PA Ab response was elicited following intraperitoneal immunization, consistent with previous reports (Fig 2 and [16;19]). Intranasal vaccination was used to elicit mucosal immunity, based on previous reports showing that this technique effectively stimulates pulmonary Ab production (Table 1 and [25;26]). The alternative, to administer AVA intratracheally, was considered unnecessarily invasive by the CBER Animal Care and Use Committee. The utility of i.n. vaccination was established during initial studies showing that all mice immunized with 10 ul of AVA + CpG ODN mounted strong mucosal responses, with high levels of IgA anti-PA Abs detected in both their lungs and GI tract (Table 1 and Fig 1). Consistent with previous reports, BAL and copra Ab titers correlated significantly following i.n. vaccination (Table 1 and [25;26]). Since copra Ab levels could be monitored without harm to the mice (whereas the collection of BAL is fatal), the former was relied upon to assess mucosal immunity in mice subsequently utilized for pathogen challenge studies.
The induction of humoral immunity was evaluated by comparing the relative change in copra, BAL and serum Ab levels induced by vaccination. Each source of Abs was evaluated independently, since it is difficult to compare Ab containing fluid collected from different sites. As noted in the methods section, BAL is harvested by infusing the lungs with PBS and copra Ig by diluting fecal pellets in a cocktail of protease inhibitors. Abs collected from mucosal sites are thus more dilute that those present in the serum. In addition, Abs secreted into the GI tract persist for only a short period (dictated by their transit time through the gut) whereas Abs secreted into the serum can accumulate over a period of weeks.
The current work establishes that systemic Ab titers correlate with resistance to aerosolized anthrax spores, while mucosal Ab titers do not. Even among mice protected from challenge by i.n. immunization, resistance correlated with systemic rather than mucosal Ab levels. When the same dose of AVA ± CpG was delivered i.n. or i.p., protection was elicited more effectively by systemic vs mucosal immunization (Figs 3, 4).
Current findings support several additional conclusions. First, intraperitoneal immunization induced a higher systemic but lower mucosal Ab response than did intranasal immunization (Table I and Figs 1,2). Second, the addition of CpG ODN significantly increased the magnitude of the anti-PA response elicited by both routes of immunization (Tables I, 2 and Figs 1, 2 and 5). Finally, the addition of CpG ODN to AVA significantly reduced host susceptibility to infection by aerosolized anthrax within 5 days of administration (Fig 5). Results indicate that this effect was not mediated through the induction of an innate immune response by CpG ODN, since none of the mice treated with CpG ODN alone survived challenge. Rather, we hypothesize that survival was influenced by the cumulative serum anti-PA response over the course of infection rather than solely at the time of pathogen exposure. Thus, vaccines capable of accelerating the induction of protective Abs when the host is subsequently infected may reduce morbidity and mortality long after the initial Ab response has waned. Studies designed to test this hypothesis, and determine whether co-administering CpG-adjuvanted AVA plus antibiotics can protect mice immediately after anthrax exposure, are underway.
CpG ODN interact with immune cells that express TLR 9, triggering an innate immune response characterized by the production of pro-inflammatory and Th1 cytokines, chemokines, and the functional maturation of APCs [17;18]. Previous studies showed that combining CpG ODN with systemically administered AVA accelerated and increased the production of serum IgG anti-PA Abs, thereby improving protection against systemic anthrax challenge [16;19]. Results from a phase I clinical study showed that adding CpG ODN to AVA boosted the serum IgG anti-PA responses of normal human volunteers by 6−8 fold and accelerated the induction of immunity by approximately 3 weeks (p. <.001 for both parameters) [27]. In that trial, no significant difference was reported in the frequency of severe adverse events in recipients of AVA plus CpG ODN vs AVA alone, although mild-moderate adverse events (including pain, warmth, and swelling at the injection site, and systemic effects such as fever and fatigue) were slightly more frequent in the former group [27].
Current findings establish that serum rather than mucosal Ab levels predict resistance to aerosolized anthrax. They also indicate that serum Ab titers may provide a surrogate marker for protection against aerosolized anthrax spores. The combination of AVA plus CpG ODN significantly increased anti-PA Ab titers and accelerated the induction of protective immunity (conferring a significant increase in survival within 5 days of vaccination). Following the 2001 anthrax attacks, less than half of potentially exposed individuals completed the recommended course of antibiotics [28]. Thus, vaccinating individuals at risk of exposure to anthrax with CpG-adjuvanted AVA, perhaps in combination with short-term antibiotic therapy, might prevent pathogen dissemination and optimize the speed and reliability with which these individuals develop a protective immune response. In mice, this protection developed even before serum Ab titers began to rise, indicating that the magnitude of the Ab response over the course of the infection rather than solely at the time of challenge determines host survival.
5. ACKNOWLEDGMENTS
The assertions herein are the private ones of the authors and are not to be construed as official or as reflecting the views of DTRA or the NCI at large. Support for this work was provided in part by the Joint Science and Technology Office for Chemical and Biological Defense of the Defense Threat Reduction Agency.
Abbreviations
- AVA
Anthrax Vaccine Adsorbed
- Ab
antibody
- ODN
oligodeoxynucleotide
- BAL
bronchioalveolar lavage fluid
- STI
Sterne strain anthrax spores
- Ag
antigen
- PA
protective antigen
- APC
antigen presenting cell
- LD50
50% lethal dose
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. REFERENCES
- 1.Hanna P. Anthrax pathogenesis and host response. Curr Top Microbiol Immunol. 1998;225:13–35. doi: 10.1007/978-3-642-80451-9_2. [DOI] [PubMed] [Google Scholar]
- 2.Quinn CP, Turnbull PC, Anthrax . In: Topley and Wilson's Microbiology and Microbial Infections. Ballow M, Sussman M M, editors. Collier, London: 1998. pp. 799–818.1. [Google Scholar]
- 3.Fritz DL, Jaax NK, Lawrence WB, Davis KJ, Pitt ML, Ezzell JW, Friedlander AM. Pathology of experimental inhalation anthrax in the rhesus monkey. Lab Invest. 1995;73:691–702. [PubMed] [Google Scholar]
- 4.Zaucha GM, Pitt LM, Estep J, Ivins BE, Friedlander AM. The pathology of experimental anthrax in rabbits exposed by inhalation and subcutaneous inoculation. Arch Pathol Lab Med. 1998;122:982–92. [PubMed] [Google Scholar]
- 5.Lane HC, Montagne JL, Fauci AS. Bioterrorism: a clear and present danger. Nat Med. 2001;7:1271–3. doi: 10.1038/nm1201-1271. [DOI] [PubMed] [Google Scholar]
- 6.Ivins BE, Welkos SL. Recent advances in the development of an improved, human anthrax vaccine. Eur J Epidemiol. 1998;4:12–9. doi: 10.1007/BF00152686. [DOI] [PubMed] [Google Scholar]
- 7.Ivins BE, Welkos SL, Little SF, Crumrine MH, Nelson GO. Immunization against anthrax with Bacilus anthracis protective antigen combined with adjuvants. Infect Immun. 1992;60:662–8. doi: 10.1128/iai.60.2.662-668.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Little SF, Ivins BE. Molecular pathogenesis of Bacillus anthracis infection. Microbes Infect. 1999;1:131–9. doi: 10.1016/s1286-4579(99)80004-5. [DOI] [PubMed] [Google Scholar]
- 9.Welkos S, Little SF, Friedlander AM, Fritz D, Fellows P. The role of antibodies to Bacillus anthracis and anthrax toxin components in inhibiting the early stages of infection by anthrax spores. Microbiology. 2001;147:1677–85. doi: 10.1099/00221287-147-6-1677. [DOI] [PubMed] [Google Scholar]
- 10.Welkos SL, Friedlander AM. Comparative safety and efficacy against Bacillus anthracis of protective antigen and live vaccines in mice. Microb Pathol. 1998;5:127–39. doi: 10.1016/0882-4010(88)90015-0. [DOI] [PubMed] [Google Scholar]
- 11.Pittman PR, Gibbs PH, Cannon TL, Friedlander AM. Anthrax vaccine: short-term safety experience in humans. Vaccine. 2001;12:972–8. doi: 10.1016/s0264-410x(01)00387-5. [DOI] [PubMed] [Google Scholar]
- 12.Ready T. US soldiers refuse to fall in line with anthrax vaccination scheme. Nat Med. 2004;10:112. doi: 10.1038/nm0204-112b. [DOI] [PubMed] [Google Scholar]
- 13.Geier DA, Geier MR. Anthrax vaccination and joint related adverse reactions in light of biological warfare scenarios. Clin Exp Rheumatol. 2002;20:217–20. [PubMed] [Google Scholar]
- 14.Jones TR, Obaldia N, Gramzinski RA, Charoenvit Y, Kolodny N, Kitov S, et al. Synthetic oligodeoxynucleotides containing CpG motifs enhance immunogenic vaccine in Aotus monkeys. Vaccine. 1999;17:3065–71. doi: 10.1016/s0264-410x(99)00145-0. [DOI] [PubMed] [Google Scholar]
- 15.Davis HL, Suparto IL, Weeratna RR, Junintarto DD, Chamzah SS, Ma'ruf AA, et al. CpG DNA overcomes hyporesponsiveness to hepatitis B vaccine in orangutans. Vaccine. 2000;18:1920–4. doi: 10.1016/s0264-410x(99)00443-0. [DOI] [PubMed] [Google Scholar]
- 16.Klinman DM, Xie H, Little SF, Currie D, Ivins BE. CpG oligonucleotides improve the protective immune response induced by the anthrax vaccination of rhesus macaques. Vaccine. 2004;22:2881–6. doi: 10.1016/j.vaccine.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 17.Klinman DM, Yi A, Beaucage SL, Conover J, Krieg AM. CpG motifs expressed by bacterial DNA rapidly induce lymphocytes to secrete IL-6, IL-12 and IFNg. Proc Natl Acad Sci USA. 1996;93:2879–83. doi: 10.1073/pnas.93.7.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krieg AM, Yi A, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–8. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 19.Xie H, Gursel I, Ivins BE, Singh M, O'Hagan DT, Ulmer JB, et al. CpG oligodeoxynucleotides adsorbed onto polylactide-co-glycolide microparticles improve the immunogenicity and protective activity of the licensed anthrax vaccine. Infect Immun. 2005;73:828–33. doi: 10.1128/IAI.73.2.828-833.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ivins BE, Pitt ML, Fellows PF, Farchaus JS, Benner GE, Waag DM, et al. Comparative efficacy of experimental anthrax vaccine candidates against inhalation anthrax in rhesus macaques. Vaccine. 1998;16:1141–8. doi: 10.1016/s0264-410x(98)80112-6. [DOI] [PubMed] [Google Scholar]
- 21.Dixon TC, Meselson M, Guillemin J, Hanna PC, Anthrax N Engl J Med. 1999;341:815–26. doi: 10.1056/NEJM199909093411107. [DOI] [PubMed] [Google Scholar]
- 22.Grewal HM, Hemming KT, Vetvik H, Ahr MC, Gjessing HK, Sommerfelt H, et al. Measurement of specific IgA in faecal extracts and intestinal lavage fluid for monitoring of mucosal immune responses. J Immunol Meth. 2000;239:53–62. doi: 10.1016/s0022-1759(00)00171-x. [DOI] [PubMed] [Google Scholar]
- 23.Pickering AK, Osorio M, Lee GM, Grippe VK, Bray M, Merkel TJ. Cytokine response to infection with Bacillus anthracis spores. Infect Immun. 2004;72:6382–9. doi: 10.1128/IAI.72.11.6382-6389.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harvill ET, Lee G, Grippe VK, Merkel TJ. Complement depletion renders C57BL/6 mice sensitive to the Bacillus anthracis Sterne strain. Infect Immun. 2005;73:4420–2. doi: 10.1128/IAI.73.7.4420-4422.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heckert RA, Saif LJ, Mengel JP, Myers GW GW. Isotype-specific antibody responses to bovine coronavirus structural proteins in serum, feces, and mucosal secretions from experimentally challenge-exposed colostrum-deprived calves. Am J Vet Res. 1991;52:692–9. [PubMed] [Google Scholar]
- 26.Garcia-Diaz A, Lopez-Andujar P, Rodriguez DJ, Montava R, Torres BC, Ribes JM, et al. Nasal immunization of mice with a rotavirus DNA vaccine that induces protective intestinal IgA antibodies. Vaccine. 2004;23:489–98. doi: 10.1016/j.vaccine.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 27.Rynkiewicz D, Rathkopf M, Ransom J, Sim I, Giri L, Quinn J, et al. Marked enhancement of antibody response to anthrax vaccine adsorbed with CpG 7909 in healthy volunteers, ICAAC abstract LB-25. 2005. [DOI] [PubMed]
- 28.Jefferds MD, Laserson K, Fry AM, Roy S, Hayslett J, Grummer-Strawn L, et al. Adherence to antimicrobial inhalational anthrax prophylaxis among postal workers, Washington, D.C. in 2001. Emerg Infect Dis. 2002;8:1138–44. doi: 10.3201/eid0810.020331. [DOI] [PMC free article] [PubMed] [Google Scholar]