

Abstract
Human immunodeficiency virus (HIV) entry into the CNS is an early event after infection, resulting in neurological dysfunction in a significant number of individuals. As people with acquired immunodeficiency syndrome (AIDS) live longer, the prevalence of cognitive impairment is increasing, despite antiretroviral therapy. The mechanisms that mediate CNS dysfunction are still not completely understood, and include inflammation, viral presence, and/or replication. In this report, we characterize a novel role of gap junctions in transmitting and thereby amplifying toxic signals originating from HIV-infected astrocytes that trigger cell death in uninfected astrocytes. HIV-infected astrocytes were resistant to apoptosis; however, uninfected astrocytes forming gap junctions with infected astrocytes were apoptotic. Gap junction blockers abolished apoptosis in uninfected astrocytes, supporting the role of these channels in amplifying cell death. Our findings describe a novel mechanism of toxicity within the brain, triggered by low numbers of HIV-infected astrocytes and amplified by gap junctions, contributing to the pathogenesis of NeuroAIDS.
Keywords: neuroinflammation, NeuroAIDS, apoptosis, chemokines, CCL2/MCP-1, Connexin
Introduction
Human immunodeficiency virus (HIV) enters the CNS early after infection and causes cognitive and motor impairment in 30–60% of infected individuals (Anthony et al., 2005). The cellular basis and mechanisms by which HIV-1 induces neuropathogy, or NeuroAIDS, are still not well understood (Brack-Werner, 1999; Gonzalez-Scarano and Martin-Garcia, 2005). Despite the initial drop in the incidence of encephalitis and/or dementia as a result of highly active antiretroviral therapy, the prevalence of cognitive impairment is again increasing as HIV-infected individuals are living longer, and therefore is becoming a significant public health issue (Nath and Sacktor, 2006; Ances and Ellis, 2007). Although microglia and perivascular macrophages are the predominant cell types productively infected by HIV-1 in the CNS in vivo (Wiley et al., 1986), HIV-infected astrocytes have also been detected in vivo and in vitro (Conant et al., 1994; Tornatore et al., 1994; Brack-Werner, 1999; Schweighardt and Atwood, 2001). The role of astrocytes in NeuroAIDS has not received extensive examination because astrocytes support low viral replication and only a few are infected.
Astrocytes actively participate in brain physiology, including maintenance of blood–brain barrier (BBB) integrity, neuronal support, and neuroimmune interactions (Becher et al., 2000). These functions are mediated in part by soluble factors, as well as by coordination of direct intercellular pathways through gap junction (GJ) channels (Saez et al., 2003). GJs are conglomerates of channels that allow the exchange of molecules between the cytoplasm of adjacent cells, including current-carrying ions, second messengers, various metabolites, and small peptides (Saez et al., 2003). Each channel is formed by the docking of two hemichannels, or connexons, located in apposing cell membranes, and each connexon is an assembly of six connexins (Cxs). GJs coordinate metabolic and electrical signals at long distances from the original stimulus site (Saez et al., 2003). In pathological conditions, inhibition of astrocyte GJs has been shown either to reduce (Garcia-Dorado et al., 1997; Rawanduzy et al., 1997; Cotrina et al., 1998) or to enhance (Blanc et al., 1998; Siushansian et al., 2001; Frantseva et al., 2002) damage. The mechanisms by which GJs mediate these effects could be by reducing the ability of the astrocytes to remove extracellular toxic soluble factors and/or by maintaining the propagation of proapoptotic or survival signals between dying and healthy cells.
There are no studies addressing the effects of HIV infection on GJ communication. In other studies, tumorigenic viruses or viral genes abolished expression and communication through GJs in different cell and animal systems (Atkinson et al., 1981; Danave et al., 1994; Jou et al., 1995; Faccini et al., 1996; Knabb et al., 2007). In contrast to those reports, here we describe a unique mechanism by which HIV “hijacks” gap junctional communication and uses it to spread toxic signals to uninfected astrocytes, rather than to abolish communication between cells as do other viruses.
Materials and Methods
Materials.
DMEM, fetal bovine serum, penicillin/streptomycin, and trypsin-EDTA were from Invitrogen (Carlsbad, CA). ATP, oxidized ATP (oATP), monoclonal antibody to GFAP, FITC- or Cy3-conjugated anti-rabbit IgG, and Cy3 or FITC anti-mouse IgG antibodies were from Sigma (St. Louis, MO). Purified mouse IgG2B and IgG1 myeloma proteins were from Cappel Pharmaceuticals (Cochranville, PA). Secondary antibodies conjugated to Cy5 were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA). Annexin 5 KIT, ethidium bromide-2, 4′,6′-diamidino-2-phenylindole (DAPI), and anti-mouse conjugated to Alexa-350 were from Invitrogen. In situ cell death detection kit [terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL)] was from Roche (Mannheim, Germany).
Astrocyte cultures.
Cortical human fetal tissue was obtained as part of an ongoing research protocol approved by the Albert Einstein College of Medicine. The preparation of cultures of astrocytes was performed as previously described (Eugenin et al., 2003, 2007).
HIV infection of primary cultures of astrocytes.
Confluent cultures of human astrocytes were infected by incubation with viral stocks (50–80 ng of p24/ml/1 × 106 cells), HIVADA, HIVJR-CSF, or HIV92UG021, using a previously described protocol (Ohagen et al., 1999). Briefly, astrocytes cultures were exposed to the virus for 24 h. Medium was removed and astrocytes were washed extensively to eliminate the unbound virus before addition of fresh medium. Immunofluorescence analyses for GFAP, CD68, and p24 indicate that the cells infected with HIV were astrocytes, and no contamination with microglia, CD68-positive cells, was detected.
Immunofluorescence and apoptosis assay.
Astrocytes were grown on coverslips and fixed and permeabilized in 70% ethanol for 20 min at −20°C. Cells were incubated in TUNEL reaction mixture at 37°C for 1 h, washed three times in PBS, and incubated in blocking solution for 30 min at room temperature. Cells were incubated in diluted primary antibody (anti-p24, anti-Cx43, and anti-GFAP: 1:50, 1:2000, and 1:800, respectively) overnight at 4°C. Cells were washed several times with PBS at room temperature and incubated with the appropriate secondary antibodies conjugated to Alexa-350, FITC, Cy3, or Cy5 for 1 h at room temperature, followed by another wash in PBS for 1 h. Coverslips were then mounted using antifade reagent with DAPI, with the exception that when Alexa-350 was used, ethidium bromide-2 was used, to observe nuclei, and then the cells were examined by confocal microscopy. Antibody specificity was confirmed by replacing the primary antibody with a nonspecific myeloma protein of the same isotype or nonimmune serum. The percentage of apoptotic astrocytes was determined by double immunofluorescence staining for TUNEL and GFAP. The numbers of astrocytes, as well as the number of cells that were TUNEL positive in 10 fields per coverslip, were counted, and the data were expressed as percentage of apoptotic astrocytes (n = 15).
ELISA.
TNF-α, IL-1β, CCL2, and IL-6 were determined by ELISA according to the manufacturer's protocols (Biosource, Camarillo, CA). HIV-p24 concentrations were determined by ELISA using a commercial kit from Perkin-Elmer (Boston, MA).
Scrape loading assay.
The functionality of gap junction channels was evaluated by scrape loading using a previously published protocol that quantified the extent of diffusion of Lucifer yellow (LY) (el-Fouly et al., 1987). In brief, 0.05% Lucifer yellow (dye permeable to GJs) and rhodamine dextran (dye not permeable to GJs) dissolved in PBS were added to confluent cells, and a scrape using a blade was performed. The dye solution was left on the cells for 5 min and discarded, and the plate was rinsed with PBS. To examine unspecific uptake of LY, plates were prepared by exposing the cells under similar conditions to the dye mixture but without scraping. The damaged cells could pick up the dye mixture and were able to transfer Lucifer yellow into the neighboring cells through functional gap junctions. The marker dye, rhodamine dextran, remained entrapped, thus labeling the primary loaded cells at the edge of the scrape areas. The distance or LY diffusion from the damaged area was measured, and the percentage of coupling was calculated compared with uninfected conditions.
Measurement of extracellular glutamate.
The glutamate concentrations in astrocyte medium were determined spectrophotometrically using a commercially available kit according to the manufacturer's instructions (Sigma) with minimal modifications (Eugenin et al., 2003).
Statistical analysis.
In the case of GJ communication, mean differences were tested by nonparametric Kruskal–Wallis analysis. If a significant H value was obtained, means were compared with Bonferroni–Dunn multiple-comparison test. A value of p < 0.05 was considered significant. Student's two-tail or ANOVA (with Tukey's multiple comparisons) paired t test was used to compare the numbers of p24-infected cells, apoptosis levels, and glutamate and CCL2 concentrations. A value of p < 0.05 was considered significant.
Results
We determined that human astrocytes can be infected with both R5 and X4 strains of HIV. In agreement with other groups (Ohagen et al., 1999), low viral replication was detected 7, 14, 21, and 28 d after viral exposure to HIV92UG021 (X4), HIVADA (R5), or HIVJR-CSF (R5) (Fig. 1). Confocal microscopy analyses of HIV-infected astrocyte cultures confirmed HIV infection of a small number of GFAP-positive cells, as demonstrated by the presence of p24-positive cells in small clusters of 2–8 cells (Fig. 1A). These HIV-infected astrocytes represented only a small, but reproducible, percentage (4.7 ± 2.8%) of the total number of astrocytes. Viral replication of all viral isolates tested in these cultures was maximal after 14 d after infection, as determined by HIV-p24 ELISA, and remained stable until 28 d (n = 15) (Fig. 1B), the last time point assayed.
Figure 1.

X4 and R5-tropic viruses infect and have low replication in human primary astrocytes. A, Immunofluorescence analysis performed by confocal microscopy of cultures infected with HIV92UG021 (X4), HIVADA (R5), or HIVJR-CSF (R5). A representative set of pictures of astrocytes after 14 d of infection with HIVADA is shown stained for DAPI (nuclei labeling), GFAP (marker for astrocytes, red, Cy3), and p24 (an HIV-protein, green, FITC). The merge of these three colors (merge of the two cytoplasmic markers, p24 and GFAP, yellow plus nuclear staining) is also illustrated. Arrows denote the colocalization of p24 and GFAP staining demonstrating that HIV infects small numbers of astrocytes. Similar results were found using all three viral isolates. No staining of p24 was detected in uninfected cells (data not shown). Scale bar, 60 μm. B, Quantification of release of p24 into the medium of cultures of astrocytes, uninfected and HIV-infected, was performed by ELISA (n = 15) after 7, 14, 21, and 28 d after infection. No p24 was detected in uninfected cultures (data not shown). HIV92UG021, HIVADA, or HIVJR-CSF produced low levels of HIV replication (*p < 0.005; n = 15) compared with HIV-infected PBMCs with the same viral isolates (p24 = 1100 ± 150 pg/ml). HIV92UG021 (X4) replicates more efficiently in astrocytes compared with HIVADA or HIVJR-CSF (#p < 0.005; n = 15).
To determine the consequences of HIV infection on astrocytes, apoptosis levels were assayed by TUNEL and Annexin 5 staining. An increase in astrocyte apoptosis was detected at 7–21 d after infection. Up to 15% of the total number of cells in the culture were TUNEL positive, even though only 4.7 ± 2.8% of the cells were HIV infected (Fig. 2A), suggesting that uninfected cells are being killed by a bystander process that is not a direct effect of HIV infection. Similar data were found with Annexin 5 staining (data not shown). At 28 d, the apoptosis levels declined, perhaps as a result of the proliferation of uninfected cells that replaced the dying astrocytes (Fig. 2A, supplemental Fig. 5, available at www.jneurosci.org as supplemental material). No alterations in the numbers of HIV-infected astrocytes were detected during the time course analyzed.
Figure 2.

HIV infection of astrocytes induces apoptosis in uninfected astrocytes by a gap junction-dependent mechanism. Data obtained with infection of astrocytes with HIVJR-CSF, a virus isolated from the CSF, are presented in the figure. The result for the other two viruses used are in the supplemental material (available at www.jneurosci.org). A, Quantification of apoptosis was performed by double labeling with TUNEL and GFAP staining. HIV infection of astrocyte cultures with HIVJR-CSF resulted in significantly increased apoptosis after 7, 14, and 21 d after infection (*p < 0.005; n = 15). B, Immunofluorescence staining and confocal analyses of the HIV-infected cultures 7 d after infection for ETBr-2 (nuclei labeling, red), TUNEL (apoptosis, green, FITC), and p24 staining (HIV, blue, Alexa350), shows that most p24-positive astrocytes were not apoptotic (red nuclei in the merge picture, 68 ± 8% of HIV-infected astrocytes survive HIV infection) compared with uninfected astrocytes in contact with HIV-infected cells that are apoptotic (yellow nuclear staining, 100% cells in contact with HIV-infected astrocytes were apoptotic). The monolayer of cells can be observed in the phase picture (Phase). HIV-infected astrocytes only corresponded to 4.7 ± 2.8% of the total number of astrocytes in the culture. Arrows indicate HIV-infected astrocytes that are not apoptotic, and arrowheads indicate uninfected apoptotic astrocytes. Scale bar, 40 μm. C, Whether HIVJR-CSF infection alters GJ communication was evaluated by a scrape loading technique. HIV infection reduced GJ communication ∼20–40% compared with uninfected astrocytes (100%), but GJs still remained functional (*p < 0.003; n = 11). D, To evaluate whether GJ communication participates in apoptosis of uninfected astrocytes in contact with HIV-infected astrocytes, gap junction blockers were used. Addition of AGA (50 μm) or octanol (500 μm) after 1, 2, and 3 d after infection reduced GJ communication by ∼70% compared with HIV-infected astrocytes cultures without the blockers. Similar results were found using octanol (data not shown). #p < 0.002, n = 17, AGA with respect to GJ communication shown in C. E, Blocking gap junction channels with AGA treatment resulted in significant reduction in astrocyte apoptosis mediated through HIV-infected cells (#p < 0.003; n = 17). These results demonstrate that GJ communications mediate transfer of proapoptotic signals between HIV-infected astrocytes and uninfected astrocytes that form GJs with infected cells. Gray diamonds represent the values of apoptosis shown by HIV-infected cultures without AGA (results shown in A), for comparison of apoptosis levels with AGA (E, #p < 0.0002; n = 17).
To identify which astrocytes, HIV infected or uninfected, were apoptotic, immunofluorescence and confocal microscopic analyses of HIV-infected cultures using ethidium bromide-2 (to label total nuclei), HIV-p24 (to identify viral particles), and TUNEL staining (to identify apoptotic astrocytes) (Fig. 2B) were performed. The results indicated that most HIV-infected, p24-positive astrocytes did not undergo apoptosis (only 19 ± 2.3% were TUNEL positive). However, neighboring, uninfected astrocytes, in contact with HIV-infected astrocytes, were 100% apoptotic (Fig. 2, supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Similar results were obtained for all three viral isolates used (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). These results indicate that HIV infection spares most HIV-infected astrocytes from apoptosis, but alters the survival of neighboring, uninfected cells through a bystander killing mechanism (Fig. 2B). As a result of this unusual finding of cell-contact-mediated bystander killing, we hypothesized that HIV-infected astrocytes generate intracellular toxic signals that diffuse through GJs and trigger apoptosis in neighboring, uninfected astrocytes.
Astrocytes are extensively coupled through gap junctions; however, under inflammatory conditions, gap junctional communication is usually lost (Rouach et al., 2002; Kielian and Esen, 2004). To examine whether HIV-infected cells use GJs to spread toxic signals to uninfected cells, we first evaluated the functionality of GJs in HIV-infected cultures of astrocytes. Gap junctional communication in cultures infected with HIV was ∼20–40% lower than for uninfected astrocytes; however, these GJs still remained functional as evaluated by scraping/loading technique (Fig. 2C, supplemental Fig. 5, available at www.jneurosci.org as supplemental material). This technique allows the quantification of functional GJ channels by measuring the diffusion distance of a fluorescent tracer (Lucifer yellow), permeable to GJs, from the scrape area to coupled cells in the culture dish as communicated through GJs, compared with dye diffusion in cultures with uncoupled cells using uncoupled cells, using chemical blocker, such as octanol or 18-α-glycyrrhetinic acid (AGA) and/or the impermeable dye, rhodamine dextran (see Materials and Methods).
To examine whether GJs between HIV-infected and uninfected astrocytes are necessary to spread cell death signals, two well established gap junction blockers from two different families of blockers, AGA (50 μm) and octanol (500 μm) (Spray et al., 2002), were used. These blockers have a transient effect, and therefore multiple additions were required (three additions of blocker, at 1, 2 and 3 d after infection) to maintain a significant reduction in GJ coupling (Fig. 2D, supplemental Fig. 5, available at www.jneurosci.org as supplemental material). GJ communication in the presence of GJ blockers was reduced from 60% to 10–20% compared with HIV-infected astrocytes without GJ blockers (Fig. 2D, supplemental Fig. 5, available at www.jneurosci.org as supplemental material). No toxicity using the blockers alone and no changes in baseline apoptosis (2–4%) were detected during the time course examined (data not shown). Blocking functional GJs with AGA or octanol after HIV infection resulted in a dramatic decrease in astrocyte apoptosis induced by HIV-infected astrocytes (Fig. 2E, supplemental Fig. 5, available at www.jneurosci.org as supplemental material). This suggests that functional GJs are essential to spread apoptotic signals to uninfected cells (Fig. 2, compare A, E). At later time points (28 d after infection) apoptotic cells were replaced by proliferating uninfected cells, reducing the percentage of apoptotic cells almost to control levels (Fig. 2D, supplemental Fig. 5, available at www.jneurosci.org as supplemental material). In addition, experiments using mixed cultures of human neurons and astrocytes also demonstrated that HIV infection of astrocytes with the viruses 92UG021, JR-CSF, or ADA triggers apoptosis in 40 ± 6.2% neurons compared with 4.6 ± 2.1% of apoptosis in control conditions. The addition of gap junction blockers, octanol or AGA, to the mixed cultures reduced neuronal apoptosis to 6.8 ± 6.2%, suggesting that HIV infection of astrocytes also contributes to neuronal cell death. Thus, the mechanisms that initiate neuronal cell death are dependent on gap junctional communication, because by blocking gap junction channels, apoptosis of neurons as well as of uninfected astrocytes was also blocked.
The bystander killing effect induced by HIV-infected astrocytes was not observed in subconfluent astrocyte cultures, suggesting that membrane-to-membrane interactions are essential to spread death signals to uninfected astrocytes (data not shown). In addition, tissue culture media obtained from HIV-infected astrocyte cultures did not induce apoptosis in uninfected cultures, indicating that soluble factors released extracellularly by HIV-infected cells are not sufficient to trigger apoptosis in uninfected cells (data not shown).
It is unlikely that ATP receptors, especially P2X7 receptors, participate in the process of apoptosis triggered by HIV-infected astrocytes, for a number of reasons. First, confluent cultures are required to spread apoptosis, and ATP signaling does not require cell-to-cell contact (Guthrie et al., 1999). Second, when ATP receptors are activated, they mediate uptake of extracellular fluorescent dyes (Guthrie et al., 1999; Siushansian et al., 2001), and we did not observe nonspecific uptake of LY in our scrape loading assays. Third, we used two well established gap junction blockers, octanol and AGA, from two different families of blockers. Although AGA has never been reported to have any effect on ATP receptor, octanol has been demonstrated to block ATP receptor activation in astrocytes (Suadicani et al., 2006). Thus, to assure that ATP receptors and specifically P2X7 receptors do not participate in the apoptosis triggered by HIV-infected astrocytes, we performed experiments using HIV-infected cultures of astrocytes and treatments with ATP (3 mm, an agonist of ATP receptors) or oxidized ATP (0.3 and 3 mm, an ATP receptor blocker). ATP and oATP were added at different time points after infection (3, 7, 14, and 21 d) to assess changes in the time course and levels of apoptosis in astrocytes cultures. We did not detect any significant differences in the time course or level of apoptosis triggered by HIV-infected astrocytes in uninfected cells, suggesting no participation of ATP receptors (data not shown) in the transmission of toxic signals.
Immunofluorescence analyses of HIV-infected cultures for p24 and connexin43 (Cx43), the main connexin expressed by astrocytes, indicated that HIV-infected astrocytes, p24-positive cells, express higher levels of Cx43 than p24-negative cells (Fig. 3A). However, Western blot analyses of total cell lysates did not reflect this increase, and only minimal changes in the total amount of Cx43, and no changes in Cx43 phosphorylation, after HIV infection were detected (Fig. 3B). This may be because of the low total number of HIV-infected astrocytes in the culture that have increased Cx43 compared with the numbers of uninfected cells with control levels. These results indicate that HIV infection maintains/upregulates Cx43 in infected cells rather than abolishes it, as do other viruses, to maintain an active communication with uninfected, surrounding astrocytes.
Figure 3.
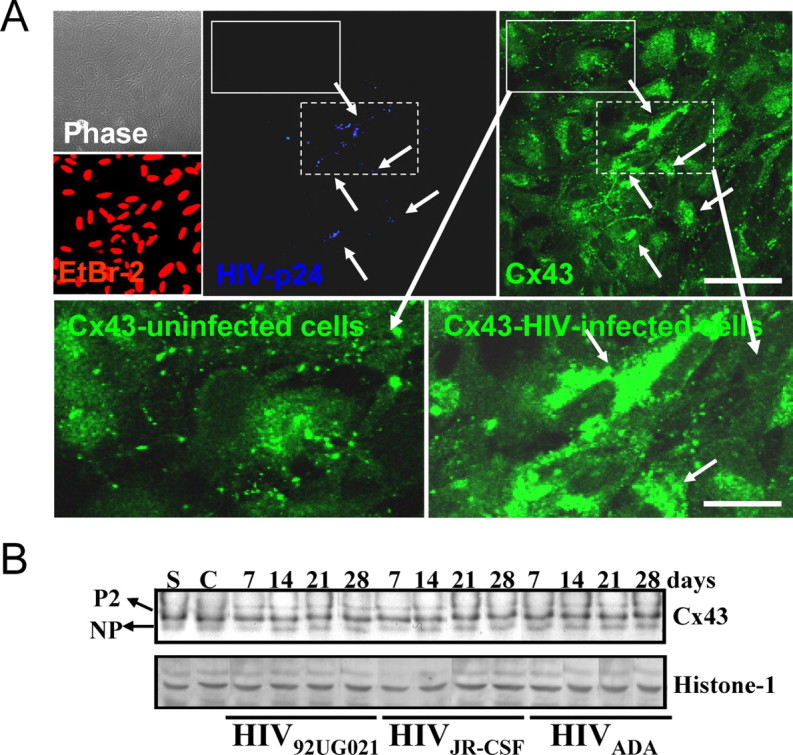
HIV alters the levels and distribution of Cx43 in HIV-infected astrocytes cultures but does not alter their total protein amount or phosphorylated isoforms. A, Immunofluorescence of uninfected and HIV-infected astrocytes cultures and confocal microscopy to identify nuclei (EtBr-2, red), p24 (blue, Alexa350), and Cx43 (green, FITC) demonstrate that HIV-positive astrocytes (p24-positive cells) have higher membrane and intracellular Cx43 staining than uninfected cells. Arrows denote HIV-infected astrocytes and the corresponding high Cx43 staining. Uninfected cells, negative for p24 staining, located around the HIV-infected astrocyte clusters, express typical levels of Cx43. The lower panels of immunofluorescence are enlarged areas of uninfected and HIV-infected astrocytes (see corresponding dotted boxes in p24 and Cx43 staining pictures) and the corresponding Cx43 staining. Scale bar, 160 μm. B, Western blot analysis for Cx43 and histone-1 was performed on lysates obtained from cultures of uninfected (C, control cells) and astrocytes infected with HIV92UG021, HIVJR-CSF, or HIVADA for 7, 14, 21, and 28 d. Histone-1 was used as loading control. No significant differences in amount and phosphorylation (NP, not phosphorylated; P2, isoforms phosphorylated-2) of Cx43 were observed with HIV infection, perhaps because of the low number of HIV-infected cells, 4.7 ± 2.8%, with respect to the numbers of uninfected cells. “S” corresponds to a standard lysate of human astrocytes used as positive control (n = 15).
To evaluate the participation of GJs in the neuroimmune consequences of HIV infection of astrocytes, even when only 4.7 ± 2.8% of the cells are infected, five factors that play significant roles in the pathogenesis of NeuroAIDS (Gonzalez-Scarano and Martin-Garcia, 2005), IL-1β, IL-6, TNF-α, CCL2, and glutamate, were examined. Elevated levels of CCL2 (Fig. 4A, supplemental Fig. 6, available at www.jneurosci.org as supplemental material) and glutamate (Fig. 4B, supplemental Fig. 6, available at www.jneurosci.org as supplemental material) in the culture medium of HIV-infected cells compared with media from uninfected astrocytes were found. IL-1β, IL-6, and TNF-α were not detected in the medium of infected or uninfected cultures (data not shown). The enhanced extracellular accumulation of CCL2 and glutamate induced by HIV was dependent on functional GJs, because the addition of the GJ blocker, AGA (Fig. 4A,B, supplemental Fig. 6, available at www.jneurosci.org as supplemental material) or octanol (data not shown), reduced the extracellular levels of CCL2 and increased the extracellular levels of glutamate. These results suggest that GJ communication in HIV-infected astrocyte cultures promotes inflammation by facilitating the release of high levels of the chemokine CCL2 (Fig. 4B, supplemental Fig. 6, available at www.jneurosci.org as supplemental material). CCL2 has been shown to recruit monocytes, infected and uninfected, into the CNS and to participate in disruption of the blood–brain barrier (Eugenin et al., 2006; Persidsky et al., 2006). The GJ blockers did not completely reduce CCL2 secretion in HIV-infected astrocyte cultures, perhaps because of the reversibility of the GJ blockers used, providing only a partial blocking effect. These data also suggest that extracellular glutamate is not a key mediator of apoptosis in our culture system because HIV infection increases glutamate, but the addition of GJ blockers that are protective against HIV-induced apoptosis (Fig. 2, supplemental Fig. 6, available at www.jneurosci.org as supplemental material) further increased the accumulation of glutamate (Fig. 4A, supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Thus, extracellular glutamate is not directly causing cell death in our cultures of astrocytes. However, the extracellular glutamate generated by astrocytes in vivo in the infected CNS could contribute to neuronal cell death. Control uninfected astrocyte cultures with only GJ blockers did not exhibit altered CCL2 or glutamate release (data not shown).
Figure 4.

HIV infection of astrocytes alters glutamate metabolism and CCL2 secretion by a gap junction-dependent mechanism. To examine the functional consequences of HIV infection in astrocytes and the role of gap junctions in control extracellular levels of glutamate and CCL2, quantification of extracellular levels of glutamate and CCL2 was performed by ELISA. A, Extracellular levels of glutamate from uninfected astrocytes were stable during the time course examined (■), and HIV infection of cultures of astrocytes with JR-CSF (▴) virus (HIVADA and HIV92UG021 results are presented in the supplemental material, available at www.jneurosci.org) resulted in a time-dependent accumulation of extracellular glutamate in the media compared with control conditions (*p < 0.0002; n = 9). The gap junction blocker, AGA (×), reduced the increase in extracellular glutamate triggered by HIV infection (#p < 0.0005 compared with viral infection alone; n = 9). B, CCL2 secretion was highly increased by HIV infection, and addition of AGA to HIV-infected cultures resulted in ∼50% reduction in the amount of CCL2 secretion induced by HIV infection alone (*p < 0.0002 compared with control conditions, and # p < 0.0002 compared with HIV infection alone; n = 9). These results indicate that HIV infection of just a few astrocytes results in dysregulation of glutamate metabolism and enhanced secretion of CCL2 into the extracellular medium by a gap junction-dependent mechanism. C, Proposed model illustrating that HIV-infected astrocytes trigger apoptosis in uninfected astrocytes. Proapoptotic signals are generated in HIV-infected astrocytes that diffuse through gap junction channels to uninfected cells, resulting in apoptosis of uninfected astrocytes in close contact with HIV-infected cells.
Discussion
We demonstrate that HIV-infected astrocytes use gap junction channels to spread toxic signals to uninfected astrocytes. Our overall hypothesis is that in vivo, HIV-infected astrocytes impair CNS function by inducing both apoptosis in surrounding cells and inflammation, and that the surviving cells, mainly HIV-infected astrocytes, subsequently serve as a reservoir of HIV for infection of other cells types that are more susceptible to HIV infection and support high viral replication, such as microglia and monocytes/macrophages. Despite the low numbers of HIV-infected astrocytes, important alterations in astrocyte physiology occur, in addition to apoptosis, such as enhanced secretion of the chemokine CCL2 and of glutamate.
Astrocytes are a critical cell type in many CNS functions, including neuronal excitability, control of extracellular synaptic metabolites, syncytial signaling, synaptic plasticity, immune activation, inflammation, and maintenance of the BBB (Becher et al., 2000; Rouach et al., 2002; Kielian and Esen, 2004). In vivo, using postmortem tissue, HIV-infected astrocytes were detected in the CNS (Conant et al., 1994; Tornatore et al., 1994; An et al., 1999). This infection and its contribution to the neuropathogenesis of AIDS have received only limited examination. We suggest that even low levels of infection in astrocytes can have major consequences within the CNS.
The bystander killing between HIV-infected and uninfected astrocytes was dependent on GJs, but not on P2X7 ATP receptors, and was associated with the enhanced/maintained expression of Cx43 in HIV-infected astrocytes as well with their functional coupling. In general, most inflammatory stimuli abolish gap junctional communication; however, after HIV infection, gap junction communication in our culture system was still functional, suggesting that in some way HIV infection is maintaining/enhancing Cx43 expression. Cx43 is the main connexin in astrocytes, and these channels are permeable to small molecules, such as IP3, small peptides, cyclic nucleotides, siRNA, cytochrome C, calcium, ATP, glucose, and glutamate (Harris, 2007). These small molecules are potential mediators of uninfected astrocyte dysfunction, but the nature of the signal(s) that trigger the bystander killing initiated by HIV-infected astrocytes is unknown and is under investigation. In addition, we propose that HIV proteins generated as a result of low viral replication maintain viability of HIV-infected astrocytes by uncharacterized mechanisms.
A similar mechanism of bystander killing through GJs has been developed as a therapeutic approach in gene therapy to kill cancer cells. This has been used to improve killing of glioma tumors, using the herpes simplex virus gene thymidine kinase in combination with ganciclovir treatment and cotransfection of Cx43 to amplify the killing through GJ-coupled tumor cells without thymidine kinase (Krutovskikh et al., 2002).
HIV infection of astrocytes, even at such low levels, generated high secretion of key factors involved in NeuroAIDS, such as CCL2 and glutamate. CCL2 plays a significant role in HIV-CNS dysfunction (Buckner et al., 2006) and our current results indicate that HIV-infected astrocyte cultures release high levels of this chemokine. Enhanced expression of CCL2 may amplify inflammation, trigger recruitment of leukocytes into the CNS, and disrupt the blood–brain barrier (Eugenin et al., 2006). Alternatively or additionally, increased levels of virally induced CCL2 may also function as a mediator of neuronal-glial protection against apoptosis induced by viral proteins, such as tat, released by HIV-infected cells (Eugenin et al., 2003). We previously demonstrated that HIV-tat induced apoptosis of cultured astrocytes/neurons is inhibited early in the process by CCL2 (Eugenin et al., 2003, 2007).
Glutamate is the main neurotransmitter within the brain, and excess of this molecule has been associated with NeuroAIDS. Our results indicate that, although HIV infection of astrocytes enhanced extracellular accumulation of glutamate, gap junction blockers enhanced extracellular glutamate levels even more, indicating that glutamate is not a key mediator of apoptosis in our culture system, because we showed that blocking gap junctions also reduced apoptosis.
Our current studies provide evidence of a mechanism of glial toxicity within the CNS mediated by HIV-infected astrocytes and amplified by GJs. The low viral replication and numbers of HIV-infected astrocytes are compensated for by an active GJ communication that allows toxic signals generated in HIV-infected astrocytes to diffuse and kill neighboring uninfected astrocytes (see proposed model, Fig. 4C). Despite low infection and viral replication, cytokines described in NeuroAIDS that are released by inflammatory cells, such as TNF-α, IL-1β, and/or IFN-γ, can induce reactivation of viral replication and transfer the virus to cells that support high viral replication (Brack-Werner, 1999; Schweighardt and Atwood, 2001). This mechanism of cross-infection is termed dissemination in trans (Geijtenbeek et al., 2000; Burleigh et al., 2006). We propose that HIV-infected astrocytes may participate in this trans process.
This is a novel mechanism of bystander toxicity coordinated by gap junctions allowing few infected astrocytes to cause considerable damage within the CNS. These cells also generate high concentrations of CCL2 that recruit additional microglia and circulating monocyte/macrophages, thereby facilitating and/or reactivating their infection within the CNS. Thus, GJs play an important role in the pathogenesis of NeuroAIDS and may serve as potential therapeutic target to reduce the devastating consequences of NeuroAIDS.
Footnotes
This work was supported by National Institute of Mental Health Grants MH075679 and MH070297 (J.W.B.), National Institutes of Health (NIH) Grant NS11920 (J.W.B.), and National Institute of Mental Health KO1 Grant MH076679 (E.A.E.). We thank the Fetal Tissue Repository, the Analytical Imaging Facility, and the NIH Centers for AIDS Research Grant AI-051519 (especially the Virology and the Immunology/Pathology Cores) at the Albert Einstein College of Medicine.
References
- An SF, Groves M, Gray F, Scaravilli F. Early entry and widespread cellular involvement of HIV-1 DNA in brains of HIV-1 positive asymptomatic individuals. J Neuropathol Exp Neurol. 1999;58:1156–1162. doi: 10.1097/00005072-199911000-00005. [DOI] [PubMed] [Google Scholar]
- Ances BM, Ellis RJ. Dementia and neurocognitive disorders due to HIV-1 infection. Semin Neurol. 2007;27:86–92. doi: 10.1055/s-2006-956759. [DOI] [PubMed] [Google Scholar]
- Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE. Influence of HAART on HIV-related CNS disease and neuroinflammation. J Neuropathol Exp Neurol. 2005;64:529–536. doi: 10.1093/jnen/64.6.529. [DOI] [PubMed] [Google Scholar]
- Atkinson MM, Menko AS, Johnson RG, Sheppard JR, Sheridan JD. Rapid and reversible reduction of junctional permeability in cells infected with a temperature-sensitive mutant of avian sarcoma virus. J Cell Biol. 1981;91:573–578. doi: 10.1083/jcb.91.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher B, Prat A, Antel JP. Brain-immune connection: immuno-regulatory properties of CNS-resident cells. Glia. 2000;29:293–304. [PubMed] [Google Scholar]
- Blanc EM, Bruce-Keller AJ, Mattson MP. Astrocytic gap junctional communication decreases neuronal vulnerability to oxidative stress-induced disruption of Ca2+ homeostasis and cell death. J Neurochem. 1998;70:958–970. doi: 10.1046/j.1471-4159.1998.70030958.x. [DOI] [PubMed] [Google Scholar]
- Brack-Werner R. Astrocytes: HIV cellular reservoirs and important participants in neuropathogenesis. Aids. 1999;13:1–22. doi: 10.1097/00002030-199901140-00003. [DOI] [PubMed] [Google Scholar]
- Buckner CM, Luers AJ, Calderon TM, Eugenin EA, Berman JW. Neuroimmunity and the blood-brain barrier: molecular regulation of leukocyte transmigration and viral entry into the nervous system with a focus on NeuroAIDS. J Neuroimmune Pharmacology. 2006;1:160–181. doi: 10.1007/s11481-006-9017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burleigh L, Lozach PY, Schiffer C, Staropoli I, Pezo V, Porrot F, Canque B, Virelizier JL, Arenzana-Seisdedos F, Amara A. Infection of dendritic cells (DCs), not DC-SIGN-mediated internalization of human immunodeficiency virus, is required for long-term transfer of virus to T cells. J Virol. 2006;80:2949–2957. doi: 10.1128/JVI.80.6.2949-2957.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant K, Tornatore C, Atwood W, Meyers K, Traub R, Major EO. In vivo and in vitro infection of the astrocyte by HIV-1. Adv Neuroimmunol. 1994;4:287–289. doi: 10.1016/s0960-5428(06)80269-x. [DOI] [PubMed] [Google Scholar]
- Cotrina ML, Kang J, Lin JH, Bueno E, Hansen TW, He L, Liu Y, Nedergaard M. Astrocytic gap junctions remain open during ischemic conditions. J Neurosci. 1998;18:2520–2537. doi: 10.1523/JNEUROSCI.18-07-02520.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danave IR, Tiffany-Castiglioni E, Zenger E, Barhoumi R, Burghardt RC, Collisson EW. Feline immunodeficiency virus decreases cell-cell communication and mitochondrial membrane potential. J Virol. 1994;68:6745–6750. doi: 10.1128/jvi.68.10.6745-6750.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Fouly MH, Trosko JE, Chang CC. Scrape-loading and dye transfer. A rapid and simple technique to study gap junctional intercellular communication. Exp Cell Res. 1987;168:422–430. doi: 10.1016/0014-4827(87)90014-0. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, D'Aversa TG, Lopez L, Calderon TM, Berman JW. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J Neurochem. 2003;85:1299–1311. doi: 10.1046/j.1471-4159.2003.01775.x. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood–brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci. 2006;26:1098–1106. doi: 10.1523/JNEUROSCI.3863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, King JE, Nath A, Calderon TM, Zukin RS, Bennett MV, Berman JW. HIV-tat induces formation of an LRP-PSD-95-NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci USA. 2007;104:3438–3443. doi: 10.1073/pnas.0611699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faccini AM, Cairney M, Ashrafi GH, Finbow ME, Campo MS, Pitts JD. The bovine papillomavirus type 4 E8 protein binds to ductin and causes loss of gap junctional intercellular communication in primary fibroblasts. J Virol. 1996;70:9041–9045. doi: 10.1128/jvi.70.12.9041-9045.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantseva MV, Kokarovtseva L, Naus CG, Carlen PL, MacFabe D, Perez Velazquez JL. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J Neurosci. 2002;22:644–653. doi: 10.1523/JNEUROSCI.22-03-00644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Dorado D, Inserte J, Ruiz-Meana M, Gonzalez MA, Solares J, Julia M, Barrabes JA, Soler-Soler J. Gap junction uncoupler heptanol prevents cell-to-cell progression of hypercontracture and limits necrosis during myocardial reperfusion. Circulation. 1997;96:3579–3586. doi: 10.1161/01.cir.96.10.3579. [DOI] [PubMed] [Google Scholar]
- Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GC, Middel J, Cornelissen IL, Nottet HS, KewalRamani VN, Littman DR, Figdor CG, van Kooyk Y. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100:587–597. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Guthrie PB, Knappenberger J, Segal M, Bennett MV, Charles AC, Kater SB. ATP released from astrocytes mediates glial calcium waves. J Neurosci. 1999;19:520–528. doi: 10.1523/JNEUROSCI.19-02-00520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AL. Connexin channel permeability to cytoplasmic molecules. Prog Biophys Mol Biol. 2007;94:120–143. doi: 10.1016/j.pbiomolbio.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jou YS, Layhe B, Matesic DF, Chang CC, de Feijter AW, Lockwood L, Welsch CW, Klaunig JE, Trosko JE. Inhibition of gap junctional intercellular communication and malignant transformation of rat liver epithelial cells by neu oncogene. Carcinogenesis. 1995;16:311–317. doi: 10.1093/carcin/16.2.311. [DOI] [PubMed] [Google Scholar]
- Kielian T, Esen N. Effects of neuroinflammation on glia-glia gap junctional intercellular communication: a perspective. Neurochem Int. 2004;45:429–436. doi: 10.1016/j.neuint.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Knabb MT, Danielsen CA, McShane-Kay K, Mbuy GK, Woodruff RI. Herpes simplex virus-type 2 infectivity and agents that block gap junctional intercellular communication. Virus Res. 2007;124:212–219. doi: 10.1016/j.virusres.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutovskikh VA, Piccoli C, Yamasaki H. Gap junction intercellular communication propagates cell death in cancerous cells. Oncogene. 2002;21:1989–1999. doi: 10.1038/sj.onc.1205187. [DOI] [PubMed] [Google Scholar]
- Nath A, Sacktor N. Influence of highly active antiretroviral therapy on persistence of HIV in the central nervous system. Curr Opin Neurol. 2006;19:358–361. doi: 10.1097/01.wco.0000236614.51592.ca. [DOI] [PubMed] [Google Scholar]
- Ohagen A, Ghosh S, He J, Huang K, Chen Y, Yuan M, Osathanondh R, Gartner S, Shi B, Shaw G, Gabuzda D. Apoptosis induced by infection of primary brain cultures with diverse human immunodeficiency virus type 1 isolates: evidence for a role of the envelope. J Virol. 1999;73:897–906. doi: 10.1128/jvi.73.2.897-906.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Heilman D, Haorah J, Zelivyanskaya M, Persidsky R, Weber GA, Shimokawa H, Kaibuchi K, Ikezu T. Rho-mediated regulation of tight junctions during monocyte migration across the blood-brain barrier in HIV-1 encephalitis (HIVE) Blood. 2006;107:4770–4780. doi: 10.1182/blood-2005-11-4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawanduzy A, Hansen A, Hansen TW, Nedergaard M. Effective reduction of infarct volume by gap junction blockade in a rodent model of stroke. J Neurosurg. 1997;87:916–920. doi: 10.3171/jns.1997.87.6.0916. [DOI] [PubMed] [Google Scholar]
- Rouach N, Avignone E, Meme W, Koulakoff A, Venance L, Blomstrand F, Giaume C. Gap junctions and connexin expression in the normal and pathological central nervous system. Biol Cell. 2002;94:457–475. doi: 10.1016/s0248-4900(02)00016-3. [DOI] [PubMed] [Google Scholar]
- Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83:1359–1400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- Schweighardt B, Atwood WJ. HIV type 1 infection of human astrocytes is restricted by inefficient viral entry. AIDS Res Hum Retroviruses. 2001;17:1133–1142. doi: 10.1089/088922201316912745. [DOI] [PubMed] [Google Scholar]
- Siushansian R, Bechberger JF, Cechetto DF, Hachinski VC, Naus CC. Connexin43 null mutation increases infarct size after stroke. J Comp Neurol. 2001;440:387–394. doi: 10.1002/cne.1392. [DOI] [PubMed] [Google Scholar]
- Spray DC, Rozental R, Srinivas M. Prospects for rational development of pharmacological gap junction channel blockers. Curr Drug Targets. 2002;3:455–464. doi: 10.2174/1389450023347353. [DOI] [PubMed] [Google Scholar]
- Suadicani SO, Brosnan CF, Scemes E. P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J Neurosci. 2006;26:1378–1385. doi: 10.1523/JNEUROSCI.3902-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornatore C, Chandra R, Berger JR, Major EO. HIV-1 infection of subcortical astrocytes in the pediatric central nervous system. Neurology. 1994;44:481–487. doi: 10.1212/wnl.44.3_part_1.481. [DOI] [PubMed] [Google Scholar]
- Wiley CA, Schrier RD, Nelson JA, Lampert PW, Oldstone MB. Cellular localization of human immunodeficiency virus infection within the brains of acquired immune deficiency syndrome patients. Proc Natl Acad Sci USA. 1986;83:7089–7093. doi: 10.1073/pnas.83.18.7089. [DOI] [PMC free article] [PubMed] [Google Scholar]