

Abstract
Transglutaminase type 2 (TG2) has recently been implicated in crosslinking of mutant huntingtin protein into aggregates. Here we show that TG2 also crosslinks spinocerebellar ataxia-1 (SCA1) gene product ataxin-1. HeLa cell lysates expressing GFP tagged ataxin-1 with 2, 30 or 82 glutamines showed covalent crosslinking of ataxin-1 when incubated with exogenously added TG2. This crosslinking was inhibited by TG2 inhibitor cystamine. SCA1 transgenic mice which overexpress the mutant ataxin-1 in cerebellar Purkinje cells showed elevated nuclear TG2 in the absence of ataxin-1 nuclear aggregates. The addition of purified TG2 to the nuclear extracts or addition of SCA1 nuclear TG2 to GFP-Q82 HeLa cell lysates resulted in the formation of insoluble aggregates. These data indicate that ataxin-1 is a substrate of TG2. Further, in SCA1 TG2 may translocate to the nucleus in response to nuclear accumulation of mutant ataxin-1 at early stages of the disease.
Keywords: Transglutaminase, Ataxin-1, Cerebellum, Degeneration
Spinocerebellar ataxia 1 (SCA1) is one of a number of late-onset neurodegenerative diseases that are caused by the expansion of a CAG repeat encoding an endogenous polyglutamine (polyQ) tract within the respective disease protein [4]. The mutated protein, different in each disease, confers a dominant gain of function ultimately leading to the disease state [4]. However, the given reasoning for the pathological sequence of events observed as a result of aggregation of these mutated proteins is still debated. The formation of insoluble aggregates, largely confined to inclusions in neuronal nuclei are the one feature that stand out starkly and therefore, are the focal interest of most researchers. These inclusions contain a variety of proteins in addition to the mutant protein, which is a major portion of the conglomerate [18,19]. Such inclusions are and have been known to be intransigent to most solublization techniques [6,9]. Two general mechanisms of stabilization of such protein aggregates have been proposed [9] and much research has been focused on the highly aggregating nature of a polyglutamine repeat in a β-sheet conformation [17,21]. The second theory involves incorporation of non polyQ proteins into the aggregates by the formation of covalent bonds catalyzed by transglutaminases (TGs) [5].
Transglutaminases are a family of calcium dependent enzymes which catalyze the post translational modification of proteins through the exchange of primary amines for ammonia at the γ-carboxamide group of glutamine residues [2]. The well characterized function of most of the mammalian TGs involves stabilization of biological structure via cross linking of proteins [1]. Tissue transglutaminase 2 is expressed in mammalian nervous system and human brain, localizing mainly in the neurons [12,15], and is highly expressed in Purkinje cells of the cerebellum [16]. Though in vitro studies [5,10] have demonstrated the increased potential of expanded polyQ repeat proteins as substrates for TGs, they have recently been implicated in a number of polyglutamine neurodegenerative disorders [2,13]. Therefore, the aim of this study was to determine if TG2 crosslinks ataxin-1 and thus has any role in SCA1 pathogenesis.
SCA1 transgenic mice (homozygous and heterozygous) were obtained from Drs. Harry Orr and Huda Zoghbi [3] at the University of Minnesota. Wildtype FVB/N were obtained from Jackson Labs. The use of animals was approved by the Institutional Animal Care and Use Committee. Mouse anti-GFP was obtained from Roche (Roche Diagnostics Corp). Rabbit polyclonal anti-transglutaminase 2 was obtained from Lab Vision Corp. Mouse anti-polyglutamine (MAB1574) and rabbit anti-calbindin D-28K were obtained from CHEMICOM Int. (Serologicals Corporation). Tissue Transglutaminase type 2 was obtained from Sigma Chemical Co. (St. Louis, MO)
Three to four individual cerebella (whole cerebellum) from 4 and 6 weeks old SCA1 heterozygous transgenic and wild type (FVB/N background) mice were used for preparation of cytosolic, nuclear and membrane fractions. The procedure described in the cytosol-nuclear extract preparation kit of Bio-Vision Research Products, CA was followed. Fractions were divided into different aliquots and stored at −80 °C.
HeLa cells from the American Type Culture Collection (ATCC) were cultured, transfected with ataxin-1 (Q2, 30 or 82)-GFP constructs and processed as described [25]. Briefly, for obtaining whole cell lysates, transfected cells were lysed in 50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 2 mM EDTA plus 1 tablet of complete protease inhibitor cocktail (Roche, Mannheim, Germany) per 50 ml lysis solution. The lysates were then repeatedly passed through a 25-gauge needle to shear DNA.
The reaction was performed at 37 °C for various time intervals in a buffer containing 125 mM Tris (pH 8.5), 2.5 mM CaCl2, 10 mM DTT, plus an appropriate amount of nuclear extracts or HeLa cell lysate. TG2 was added to a final concentration of 0.01 U/mg protein. In some reactions 1 mM EGTA or 10–40 mM cystamine was used to inhibit TG2 activity.
SCA1 heterozygous and wildtype animals were sacrificed, brains removed and stored or processed for immunoblot analysis as described [23]. The cerebellar cytosolic and nuclear fractions prepared from animals in various groups were subjected to SDS–PAGE (4–20% or 15% acrylamide gels). For each post-natal age equal amounts of proteins from animals in the various groups were used. Protein estimations were carried out in the nuclear and cytosolic fractions, and the HeLa cell extracts using protein assay kit (Bio-Rad). Proteins were transferred to PVDF membrane (Bio-Rad), blocked for 1 h with blocking solution (Western Breeze, Invitrogen) and incubated for 1 h or overnight with appropriate concentration of primary antibodies (monoclonal anti-polyglutamine; 1:5000 and monoclonal or polyclonal calbindin D28k; 1:5000). Immunoreactive proteins were visualized by incubation (for 1 h) with the goat anti-mouse alkaline phosphatase-labeled secondary antibody followed by reaction with the luminescent substrates (Western Breeze, Invitrogen). The blots were then exposed to hyperfilm-ECL (Amersham). Some blots were stripped and reprobed with appropriate antibodies. Images were processed using Adobe Photoshop CS (version 8.0; San Jose, CA).
To determine if ataxin-1 is a substrate for TG2, we studied the effects of exogenous TG2 on HeLa cell lysates expressing GFP and GFP tagged ataxin-1 with normal and expanded polyglutamine repeats (Q2, Q30 and Q82) (Fig. 1). Fixed amount of enzyme was added to the HeLa cell lysates. Non-transfected HeLa cell lysates did not show any endogenous TG2 activity (data not shown). The control samples were incubated for 90 min in the absence of guinea pig TG2 (Fig. 1A). The reaction produced large multimers, which stayed on top of the gel (Fig. 1A). Time dependent progressive increase in aggregation was seen in Q2, Q30 through Q82 ataxin-1 (Fig. 1B). Control samples, however, did not show any aggregates. To determine if the crosslinking of ataxin-1 by exogenous TG2 is specific, gels were silver stained after 0 and 30 min incubation with TG2 (Fig. 1C). The silver staining revealed that not all proteins are cross-linked in vitro. Furthermore, HeLa cells lysates transfected with GFP with no extra Q tract, when probed with anti-GFP antibody showed no crosslinking as compared to polyQ transfected lysates (Fig. 1D). The ataxin-1 aggregate formation was inhibited by TG2 inhibitor cystamine (Fig. 1E) in a concentration dependent manner (Fig. 1E).
Fig. 1.
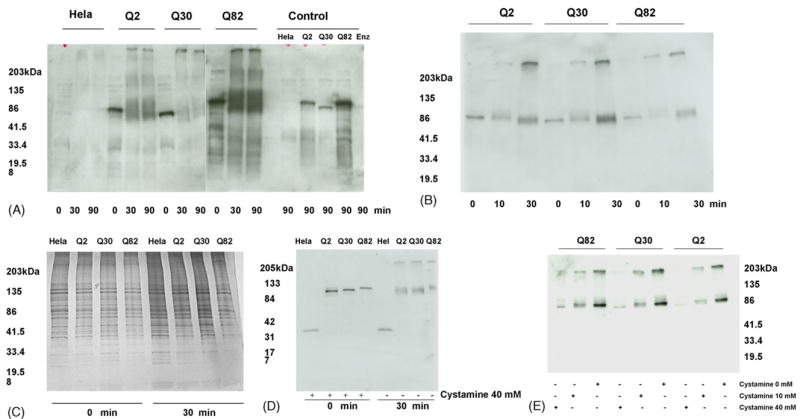
Western blots of HeLa cell lysates expressing ataxin-1 (Q2, 30 or 82)-GFP. (A and B) Time course crosslinking of ataxin -1-GFP by TG2. Immunoreactive bands were detected with anti-polyglutamine (A) and anti-GFP (B) antibodies. When indicated, 10 μl aliquots were removed and electrophoresed on a reducing 4–20% polyacrylamide gel. In controls (A) TG2 was omitted except ‘Enz’ (enzyme) lane. The reaction produced large multimers, which stayed on top of the gel. (C) Silver stained gel after 0 and 30 min incubations with TG2. (D and E) GFP immunoblots showing inhibition of ataxin-1 aggregate formation in the presence of various concentrations of TG2 inhibitor cystamine. In ‘E’ the reaction mixtures were incubated for 10 min with guinea pig TG2. Lysate prepared from HeLa cells transfected with GFP without extra Q tract (D) shows no crosslinking.
To understand the plausible role of TG2 in mutant ataxin-1 aggregation in SCA1 we determined the levels of TG2 in the nuclear fractions prepared from the cerebella of 4 and 6 weeks old SCA1 and wildtype mice. Nuclear extracts from 4 and 6 weeks old SCA1 mice showed increased levels of TG2 as compared to wild type animals (Fig. 2A and B). Calcium binding protein calbindin D28k, which is down regulated in SCA1 ([23], H.Y. Zoghbi personal communication) was used as an internal control (Fig. 2A and B). In addition, the distribution of TG2 in cerebellar cytosolic and membrane fractions was assessed in 4 weeks old wildtype and SCA1 mice (Fig. 2A). We observed a decrease in cytosolic TG2 as compared to higher TG2 levels seen in the nuclear fractions suggesting altered TG2 distribution in SCA1 as compared to age-matched wildtype mice. However, no changes were seen in the membrane fractions. The guinea pig TG2 also crosslinked nuclear proteins in the nuclear extracts from 4 weeks old SCA1 and wildtype mice (Fig. 2C). However, the intensity of immunoreactive insoluble aggregates that remained on top the gel was higher in SCA1 mice as compared to wildtype animals. Aggregate formation was inhibited by the addition of 40 mM cystamine (Fig. 2C). We used anti-polyglutamine antibody (MAB1574, Chemicon) to detect ataxin-1 and/or polyglutamine containing proteins in insoluble aggregates. In addition, we measured endogenous TG2 activity in the nuclear fractions of SCA1 and wildtype mice as a measure of crosslinking of Hela Q82-GFP, and found that in SCA1 mice the intensity of GFP immunoreactive aggregates was markedly higher than the age-matched wildtype animals (Fig. 2D). These nuclear fractions were prepared from the cerebella of 5 weeks old SCA1 homozygous and wildtype mice. Equal amounts of proteins were used in all reactions.
Fig. 2.

Immunodetection of tissue transglutaminase type 2 (TG2) in cerebellar fractions in SCA1 transgenic and wildtype mice. (A and B) Western blots showing TG2 and calbindin D28k in cerebellar fractions in 4 (n = 4) and 6 (n = 3) weeks old wildtype and SCA1 heterozygous mice. Since calbindin D28k is expressed only in Purkinje cells of the cerebellum we used calbindin as an internal control. As reported previously [17], lower nuclear calbindin levels were observed in SCA1 mice as compared to wildtype animals. On the other hand, elevated levels of nuclear TG2 were seen in SCA1 mice. Figure A also shows altered nuclear and cytosolic distribution of TG2 in 4 weeks old SCA1 mice. TG2 appeared as a single band of about 75 kDa. (C) Immunoblot showing TG2 crosslinked nuclear proteins of 4 weeks old SCA1 and wildtype mice. The insoluble aggregates stayed on top of the gel. The crosslinked proteins were detected with anti-polyglutamine antibody. The reaction was catalyzed by exogenous guinea pig TG2 in the presence and absence of cystamine. (D) Endogenous TG2 in the nuclear fractions crosslinked Hela Q82-GFP (detected with anti-GFP antibody). The nuclear fractions were prepared from the cerebella of 5 weeks old SCA1 homozygous and wildtype mice. Equal amounts of proteins were used in all reactions. In SCA1 mice the intensity of immunoreactive aggregates was markedly higher than the age-matched wildtypes.
In the present study we were able to demonstrate that TG2 crosslinks full length ataxin-1 with normal and pathogenic polyQ domains. Recently, Zainelli et al. [26] reported that mutant huntingtin protein is also a substrate for TG2, and others transglutaminases. Further, in another report it was shown that TG2 crosslinked both normal and mutant huntingtin protein fragments. In addition, the degree of TG catalyzed aggregation was a function of the size of huntingtin fragments outside the polyQ domain [11].
We also observed elevated levels of TG2 in the SCA1 mouse cerebellar nuclei. These data are in agreement with the studies of Karpuj et al. [11], who saw increased TG activity in the nuclei isolated from the brains of patients with Huntington’s disease. Transglutaminases have a nuclear translocalization signal [22], and nuclear TG has been described in neuroblastoma cells in vitro [14]. However, their function in the nucleus is not fully understood. Using a live-cell nucleocytoplasmic transport assay, it was shown that wildtype, but not polyglutamine expanded mutant ataxin-1, has the ability to export from the nucleus [7,8]. Therefore, it is possible that in SCA1 mice TG2 is translocated to the nucleus due to retention of mutant ataxin-1 within Purkinje cell nuclei. Furthermore, increased calcium levels in cell culture medium have been shown to cause an increase in TG activity [10]. Moreover, in maitotoxin activated SH-SY5Y cells, tissue TG translocates to the nucleus in response to the elevation of intracellular calcium concentration [14]. Since calcium binding and signaling proteins are down regulated in SCA1 Purkinje cells [20,24], we speculate that elevated calcium levels due to alterations in calcium buffering could trigger activation of TG2 in SCA1 Purkinje cells. Recently, using Affymetrix gene arrays we performed expression profiling of 2 weeks old SCA1 and wild-type mouse cerebella, the data also shows significant increase in the expression of TG2 in SCA1 mice (unpublished observations) further supporting the role of TG2 in the etiology of SCA1.
In sum, these data indicate that both normal and mutant ataxin-1 may be substrates of TG2. The increased levels of nuclear TG2 in SCA1 suggests that TG2 may be translocating to the nucleus in response to nuclear accumulation of the mutant ataxin-1. Whether cerebellar TG2 is associated with normal ataxin-1 in vivo awaits further investigation.
Acknowledgments
This work was partly supported by grants from the National Institute of Neurological Disorders and Stroke and Lucky Day Foundation, USA.
References
- 1.Aeschlimann D, Tomazy V. Protein crosslinking in assembly and remodeling of extracellular matrices: the role of transglutaminases. Connect Tissue Res. 2000;41:1–27. doi: 10.3109/03008200009005638. [DOI] [PubMed] [Google Scholar]
- 2.Bailey CD, Tucholski J, Johnson GV. Transglutaminases in neurodegenerative disorders. Prog Exp Tumor Res. 2005;38:139–157. doi: 10.1159/000084238. [DOI] [PubMed] [Google Scholar]
- 3.Burright EN, Clark HB, Servadio A, Matilla T, Feddersen RM, Yunis WS, Duvick LA, Zoghbi HY, Orr HT. SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell. 1995;82:937–948. doi: 10.1016/0092-8674(95)90273-2. [DOI] [PubMed] [Google Scholar]
- 4.Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- 5.Gentile V, Sepe C, Calvani M, Melone MA, Cotrufo R, Cooper AJ, Blass JP, Peluso G. Tissue transglutaminase-catalyzed formation of high-molecular-weight aggregates in vitro is favored with long polyglutamine domains: a possible mechanism contributing to CAG-triplet diseases. Arch Biochem Biophys. 1998;352:314–321. doi: 10.1006/abbi.1998.0592. [DOI] [PubMed] [Google Scholar]
- 6.Hazeki N, Tukamoto T, Goto J, Kanazawa I. Formic acid dissolves aggregates of an N-terminal huntingtin fragment containing an expanded polyglutamine tract: applying to quantification of protein components of the aggregates. Biochem Biophys Res Commun. 2000;277:386–393. doi: 10.1006/bbrc.2000.3682. [DOI] [PubMed] [Google Scholar]
- 7.Howell JL, Truant R. Live-cell nucleocytoplasmic protein shuttle assay utilizing laser confocal microscopy and FRAP. Biotechniques. 2002;32:80–87. doi: 10.2144/02321st04. [DOI] [PubMed] [Google Scholar]
- 8.Irwin S, Vandelft M, Pinchev D, Howell JL, Graczyk J, Orr HT, Truant R. RNA association and nucleocytoplasmic shuttling by ataxin-1. J Cell Sci. 2005;118:233–242. doi: 10.1242/jcs.01611. [DOI] [PubMed] [Google Scholar]
- 9.Iuchi S, Hoffner G, Verbeke P, Djian P, Green N. Oligomeric and polymeric aggregates formed by proteins containing expanded polyglutamine. PNAS. 2003;100:2409–2414. doi: 10.1073/pnas.0437660100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Junn E, Ronchetti RD, Quezado MM, Kim SY, Mouradian MM. Tissue transglutaminase-induced aggregation of α-synuclein: implications for Lewy body formation in Parkinson’s disease and dementia with Lewy bodies. PNAS. 2003;100:2047–2052. doi: 10.1073/pnas.0438021100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karpuj MV, Garren H, Slunt H, Price DL, Gusella J, Becher MW, Steinman L. Transglutaminase aggregates huntingtin into nonamyloidogenic polymers, and its enzymatic activity increases in Huntington’s disease brain nuclei. PNAS. 1999;96:7388–7393. doi: 10.1073/pnas.96.13.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim SY, Grant P, Lee JH, Pant HC, Steinert PM. Differential expression of multiple transglutaminases in human brain. Increased expression and cross-linking by transglutaminases 1 and 2 in Alzheimer’s disease. J Biol Chem. 1999;274:30715–30721. doi: 10.1074/jbc.274.43.30715. [DOI] [PubMed] [Google Scholar]
- 13.Kim S, Nollen EA, Kitagawa K, Bindokas VP, Morimoto RI. Polyglutamine protein aggregates are dynamic. Nat Cell Biol. 2002;4:826–831. doi: 10.1038/ncb863. [DOI] [PubMed] [Google Scholar]
- 14.Lesort M, Attanavanich K, Zhang J, Johnson GV. Distinct nuclear localization and activity of tissue transglutaminase. J Biol Chem. 1998;273:11991–11994. doi: 10.1074/jbc.273.20.11991. [DOI] [PubMed] [Google Scholar]
- 15.Lesort M, Chun W, Johnson GV, Ferrante RJ. Tissue transglutaminase is increased in Huntington’s disease brain. J Neurochem. 1999;73:2018–2027. [PubMed] [Google Scholar]
- 16.Maggio N, Sellitti S, Capano CP, Papa M. Tissue-transglutaminase in rat and human brain: light and electron immunocytochemical analysis and in situ hybridization study. Brain Res Bull. 2001;56:173–182. doi: 10.1016/s0361-9230(01)00649-9. [DOI] [PubMed] [Google Scholar]
- 17.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. PNAS. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perez MK, Paulson HL, Pendse SJ, Saionz SJ, Bonini NM, Pittman RN. Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J Cell Biol. 1998;143:1457–1470. doi: 10.1083/jcb.143.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Preisinger E, Jordan BM, Kazantsev A, Housman D. Evidence for a recruitment and sequestration mechanism in Huntington’s disease. Philos Trans R Soc Lond B Biol Sci. 1999;354:1029–1034. doi: 10.1098/rstb.1999.0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serra HG, Byam CE, Lande JD, Tousey SK, Zoghbi HY, Orr HT. Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum Mol Genet. 2004;13:2535–2543. doi: 10.1093/hmg/ddh268. [DOI] [PubMed] [Google Scholar]
- 21.Sharma D, Sharma S, Pasha S, Brahmachari SK. Peptide models for inherited neurodegenerative disorders: conformation and aggregation properties of long polyglutamine peptides with and without interruptions. FEBS Lett. 1999;456:181–185. doi: 10.1016/s0014-5793(99)00933-3. [DOI] [PubMed] [Google Scholar]
- 22.Takeuchi Y, Ohashi H, Birckbichler PJ, Ikejima T. Nuclear translocation of tissue type transglutaminase during sphingosine-induced cell death: a novel aspect of the enzyme with DNA hydrolytic activity. Z Naturforsch [C] 1998;53:352–358. doi: 10.1515/znc-1998-5-609. [DOI] [PubMed] [Google Scholar]
- 23.Vig PJS, Subramony SH, Burright EN, Fratkin JD, McDaniel DO, Desaiah D, Qin Z. Reduced immunoreactivity to calcium-binding proteins in Purkinje cells precedes onset of ataxia in spinocerebellar ataxia-1. Neurology. 1998;50:106–113. doi: 10.1212/wnl.50.1.106. [DOI] [PubMed] [Google Scholar]
- 24.Vig PJS, Subramony SH, Qin Z, McDaniel DO, Fratkin J. Relationship between ataxin-1 nuclear inclusions and Purkinje cell specific proteins in SCA-1 transgenic mice. J Neurol Sci. 2000;174:100–110. doi: 10.1016/s0022-510x(00)00262-8. [DOI] [PubMed] [Google Scholar]
- 25.Xu H, Somers ZB, Robinson ML, 2nd, Hebert MD. Tim50a, a nuclear isoform of the mitochondrial Tim50, interacts with proteins involved in snRNP biogenesis. BMC Cell Biol. 2005;6:29. doi: 10.1186/1471-2121-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zainelli GM, Dudek NL, Ross CA, Kim SY, Muma NA. Mutant huntingtin protein: a substrate for transglutaminase 1, 2, and 3. J Neuropathol Exp Neurol. 2005;64:58–65. doi: 10.1093/jnen/64.1.58. [DOI] [PubMed] [Google Scholar]