

Abstract
The transcription factor SOX9 is essential for multiple steps during skeletal development, including mesenchymal cell chondrogenesis and endochondral bone formation. We recently reported that the human SOX9 proximal promoter region is regulated by the CCAAT-binding factor through two CCAAT boxes located within 100 bp of the transcriptional start site. Here we report that the human SOX9 proximal promoter is also regulated by the cyclic-AMP response element binding protein (CREB) and Sp1. We show by DNaseI protection and EMSA analysis that CREB and Sp1 interact with specific sites within the SOX9 proximal promoter region. By transient transfection analysis we also demonstrate that mutations of the CREB and Sp1 binding sites result in a profound reduction of SOX9 promoter activity. Chromatin immunoprecipitation (ChIP) assay demonstrated that both Sp1 and CREB interact with the SOX9 promoter in vivo. Finally, we demonstrate that IL-1β treatment of chondrocytes isolated from human normal and osteoarthritic (OA) cartilage down-regulates SOX9 promoter activity, an effect accompanied by a reduction of Sp1 binding to the SOX9 proximal promoter.
Keywords: Transcription factor, CREB, Sp1, human chondrocytes, gene regulation, human SOX9 promoter, IL-1β, osteoarthritis
INTRODUCTION
The transcription factor SOX9 is expressed during skeletal development in mesenchymal cells, pre-chondrocytes, and differentiated chondrocytes but not in hypertrophic chondrocytes [1–4]. SOX9 is a potent transcriptional activator of the COL2A1 gene and also controls the expression of other genes encoding chondrocyte-specific extracellular matrix proteins such as aggrecan core protein, COL11A2 and COL9A1 [5–7]. Studies in mice have shown that SOX9 is required for multiple steps along the chondrocyte differentiation pathway. Sox9−/− cells are not able to participate in mesenchymal condensation and hence chondrogenesis, and do not express ECM genes known to be activated by SOX9 in chondrocytes [8]. SOX9 was found to be required for mesenchymal cells to undergo condensation and for chondrogenesis to proceed following condensation [9]. SOX9 was also implicated in regulating the conversion of prehypertrophic into hypertrophic chondrocytes during endochondral bone formation in the growth plate [9]. Based on the considerable evidence linking SOX9 to chondrocyte function and differentiation, SOX9 is considered to be a master regulator of chondrocyte-specific gene expression and chondrogenesis [3, 5–11].
Although there is substantial information about the tissue, and temporal-specific expression profiles of SOX9 and its target genes, information on the transcription factors that regulate the level of SOX9 gene transcription is scarce and only a few studies have performed a functional analysis of the human SOX9 promoter. In one study, the sequences of the mouse Sox9 and human SOX9 proximal promoter regions were compared and some functional analyses of the mouse Sox9 promoter were performed [12]. The sequence analysis revealed conserved transcription factor binding sites for GATA, CREB and two sites for CBF/NF-Y, along with a possible SOX/Sry binding site. A DNaseI hypersensitive site within the mouse Sox9 proximal promoter region (approximately between −200 to −70 bp) was observed in gonadal somatic cells, undifferentiated C3H10T1/2 cells and L-3T3 cells, but not in liver cells. The mouse Sox9 proximal promoter activity was also assessed in C3H10T1/2 and L-3T3 cells, and in primary testes, ovary and liver cells. The mouse Sox9 promoter was active in C3H10T1/2 and L-3T3 cells until a deletion past -193 bp relative to the transcriptional start site was made. These findings are similar to our recent results with the human SOX9 promoter in which activity decreased markedly when a deletion was made past position -172 bp [13]. More recently, Morishita, et al., [14] reported on the activity of the human SOX9 promoter region in several different cell types. Their work showed that promoter activity decreased as much as 6 fold when deletions were made past –1453 in rat articular chondrocytes (RACs) and approximately 3–5 fold in rat chondrosarcoma cells (RCS), ATDC5 cells and MC3T3-E1 cells. The highest activity was observed with a promoter fragment starting at position −734 bp relative to the transcriptional start site. Interestingly, in this work an enhancer element within the first intron was reported to function mainly in undifferentiated ATDC5 cells, in which it resulted in approximately an 8-fold increase in promoter activity. The enhancer element was less active in RACs and RCS cells, in which it resulted only in a 2-fold increase.
Other potential Sox9 gene regulatory pathways include the fibroblast growth factors, the cytokines IL-1β and TNF-α and certain members of the hedgehog family of proteins. Fibroblast growth factors have been shown to up-regulate Sox9 mRNA expression in mouse primary chondrocytes and in the mesenchymal C3H10T1/2 cell line through a MAP kinase mediated pathway [15]. In contrast, IL-1β and TNF-α cause a marked suppression of gene expression in primary mouse chondrocytes and MC615 cells (immortalized mouse chondrocytes) [16]. The suppression of Sox9 gene transcription appears to be mediated through the direct/indirect action of NF-κB [16]. In certain instances BMP-2 and -4 have been shown to up-regulate SOX9 expression while retinoic acid yielded mixed results [6, 15, 17–20]. Recent findings have shown that hedgehog protein family members can cause an up-regulation of Sox9 gene expression through the promoter region [21].
Despite the extensive studies reviewed above, transcription factors that interact with the SOX9 proximal promoter region and/or the upstream elements have yet to be identified and functionally characterized. Here, we describe our studies on the identification and functional analysis of transcription factors that interact with the SOX9 proximal promoter and on the modulation of its activity by the cytokine IL-1β. We demonstrate that two Sp1-binding sites within the human SOX9 proximal promoter region can specifically bind Sp1 and that mutation or deletion of these sites diminishes SOX9 promoter activity. We also show that CREB can interact with a CREB response element (CRE) ½-site located in the SOX9 promoter and that mutation of this site also results in down-regulation of SOX9 promoter activity. Chromatin immunoprecipitation (ChIP) assays demonstrated the in vivo occupancy of the SOX9 promoter by CREB and Sp1. Finally, we showed that IL-1β has a potent inhibitory effect on SOX9 promoter activity which is mediated, at least in part, by a reduction in Sp1 binding to the promoter.
Materials and methods
Cell Culture
Pluripotent mesenchymal C3H10T1/2 cells and human chondrosarcoma HTB cells were obtained from ATCC. Rat chondrosarcoma cells (RCS, early differentiated chondrocyte phenotype) were a kind gift from Dr. Benoit de Crombrugghe [22]. Prechondrogenic mesenchymal ATDC5 cells were obtained from the RIKEN BioResource Center Cell Bank (Ibaraki, Japan). C3H10T1/2, HTB and RCS cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% heat inactivated fetal bovine serum (FBS), supplemented with L-glutamine (2 mM), penicillin (100 units/ml) and streptomycin (100 μg/ml). For experiments involving treatment of C3H10T1/2 cells with interleukin-beta (IL-1β), the indicated concentration (0, 5 or 10 ng/ml) of recombinant human IL-1β (PeproTech, Inc., Rocky Hill, NJ) was added to the cells in complete media for 0–24 hrs. ATDC5 cells were propagated in DMEM/Ham’s F-12 (1:1) containing 5% FBS, 10 μg/ml human transferrin, 3x10−8M sodium selenite and supplements as above. Bovine fetal epiphyseal cartilage from the knee was obtained from Animal Technologies, Inc. (Tyler, TX). Normal human articular chondrocytes (HAC) were obtained from the knee joints of organ donors through the Cooperative Human Tissue Network. Osteoarthritic chondrocytes (OAC) were obtained with IRB approval from patients undergoing knee-joint replacement at Thomas Jefferson University Hospital. Chondrocytes were isolated as previously described [23]. Isolated chondrocytes were frozen in 90% FBS/10% DMSO until used. Upon thawing, chondrocytes were maintained in DMEM containing 10% FBS, supplemented with L-glutamine (2 mM), penicillin (100 units/ml), streptomycin (100 μg/ml), fungizone (2.5 μg/ml) and 50 μg/ml ascorbic acid (complete media). Adult human and fetal bovine chondrocytes (FBCs) were cultured in complete media at a density of 5 x 106 cells/well in 60 mm plastic petri dishes that had been previously coated with 0.9 ml of a 10% solution of poly-(2-hydroxyethyl) methacrylate (polyHEMA, Poly Sciences Inc., Malvern, PA) to prevent attachment as described previously [24]. Human chondrocytes were treated with 10 ng/ml of human recombinant IL-1β in complete media for 24 hrs.
Nuclear extracts
Nuclear extracts were prepared from sub-confluent cells according to the method of Dignam et al., [25] using the CellLytic NuCLEAR extraction kit (Sigma-Aldrich, St. Louis, MO). Briefly, cells were placed in hypotonic buffer (10 mM HEPES [pH 7.9], 1.5 mM MgCl2, 10 mM KCl, and 0.5 mM DTT) and incubated on ice for 15 min. Igeapal CA-630 detergent was added to a final concentration of 0.6% and the mixture was vortexed vigorously for 10 sec. Nuclei were recovered by centrifugation at 3,300xg for 30 sec at 4°C and extracted in buffer containing 20 mM HEPES pH 7.9, 0.42 M NaCl, 25% glycerol, 1.5 mM MgC12, 0.2 mM EDTA, and 0.5 mM DTT for 30 min at 4°C by gentle shaking. The extract was then centrifuged for 15 min at 25,000xg, and the supernatant was snap frozen at −70°C. All buffers contained a protease inhibitor cocktail (2 mM 4-(2-aminoethyl)benzenesulfonylfluoride, 1.4 pM trans-epoxysuccinyl-L-leucylamido [4-guanidinobutane], 130 pM bestatin, 1 μM leupeptin, and 0.3 pM aprotinin; Sigma-Aldrich).
DNaseI footprinting
DNaseI footprinting was carried out according the method of LeBlanc and Moss [26]. The following primers were used to amplify the region between position −41 and −315 (275 bp) of the SOX9 promoter: forward; AAGATTCGCGCGGAGAAGGCA, reverse; ACCTTAGAGCCACCCGCCAA. Before the PCR reaction, the reverse primer was 5-end labeled with 32P in order to generate an individual strand labeled probe. For DNA-protein binding reactions, purified-radiolabeled probe (50,000 cpm) was incubated with 30 μg of nuclear protein for 20 min on ice in a 50 μl volume containing 10 mM Hepes (pH 7.9), 100 mM NaCl, 0.1 mm EDTA, 0.5mM DTT, 1 μg of poly dA-dT or dI-dC and 10% glycerol. Following the binding reaction, 50 μl of 10 mM MgCl2/5 mM CaCl2 was added (final concentration of 5 and 2.5mM, respectively). Reactions were shifted to room temperature and then 0.001–0.005 units of DNaseI were added. The DNaseI reactions were incubated at room temperature for 2 min followed by the addition of 100 μl of stop solution (1% SDS, 200 mM NaCl 20 mM EDTA, pH 8.0, 40 μg/ml tRNA). The reactions were then phenol/chloroform extracted and ethanol precipitated. After washing twice in 70% ice-cold ethanol, the DNA was resuspended in 3.5 μl of formamide loading dye. The samples were then denatured at 90°C for 7 min and loaded onto 6.5% polyacrylamide-urea sequencing gels. Gels were pre-run at 2200v for 30 min and run for 1.5 hrs at 2200v. Gels were dried and exposed to x-ray film at −80°C.
Electromobility shift assays
EMSAs were carried out as previously described with minor modifications [7, 27, 28]. Briefly, binding reactions consisted of 12.5 mM Hepes, pH 7.9, 50–100 mM NaCl, 5 % glycerol, 2 mg/ml BSA, 1–2 μg poly-dIdC, 0.1 mM EDTA, 0.1 mM DTT, 0.5–1 ng of 32P-end labeled double stranded oligonucleotide probes and 10–15 μg of nuclear protein. Binding reactions were incubated for 30 min at room temperature and then loaded onto either 5% acrylamide-0.5X tris-borate-EDTA gels or 50mM Tris/0.38M glycine-2 mM EDTA gels and electrophoresed at 130 V for 2–3 hrs. For competition analyses, 100-fold excess of unlabeled-competitor probe was included in the binding reaction. For the supershift experiments, 1–2 μl of anti-Sp1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) or anti-CREB antibody (Chemicon International, Temecula, CA) was added to the binding reaction for 25 min at room temperature prior to the addition of labeled DNA probe. After addition of labeled DNA-probe, the binding reaction was incubated for additional 20 min at room temperature.
SOX9-luciferase constructs
The human SOX9 proximal promoter region from −1034 bp to +67 bp relative to the transcriptional start site was obtained by PCR using human genomic DNA as a template and the following primers: forward; 5’-GTGGAGCGTTTTGTCTGC-3’, reverse; 5’-TGAAACTGGCGAGTCTCC-3’. The PCR product was cloned into the pCR-Blunt vector (Invitrogen, Carlsbad, CA) and then transferred as a KpnI-EcoRV fragment into the pGL3-Basic vector (Promega, Madison, WI) that had been digested with KpnI-SmaI. Site directed mutagenesis of the SOX9-promoter construct was performed by a two-step PCR-based protocol [29]. Deletions were introduced into the SOX9-promoter construct using the Quick change mutagenesis kit (Stratagene, La Jolla, CA).
Transient transfection and luciferase assays
All cell types were transfected employing the FuGENE 6 transfection reagent (Roche, Indianapolis, IN). C3H10T1/2 cells were plated at 50,000 cells/well and ATDC5 cells at 100,000 cells/well in 6-well plates 24 hrs before transfection. Cell lines were transfected with 2 μg DNA/well (1.5 μg construct and 0.5 μg pCMVβ, a constitutive beta-galactosidase mammalian expression plasmid as a control for transfection efficiency, Clontech, Palo Alto, CA). FBCs were transfected utilizing the procedure of Madry and Trippel et al., [30] with modifications. FBCs were maintained in polyHEMA coated dishes in suspension culture in order to preserve the differentiated phenotype [24, 31]. FBCs in suspension culture were digested overnight with 0.5 mg/ml collagenase in DMEM containing 10% FBS and supplements. After digestion the cells were plated into 24-well plates at 75,000/well and grown to ~70% confluence. The FBCs were then treated with 10 U/ml hyaluronidase for 4 hrs before and during transfection. FBCs were transfected with 1 μg DNA/well (0.5 μg construct and 0.5 μg pCMVβ). Cells were harvested 36–42 hrs after transfection and luciferase assays were performed with a luciferase assay kit (Promega) and a Turner Designs TD 20/20 luminometer (Turner Designs, Sunnyvale CA). β-galactosidase assays were performed spectrophotometrically with a β-galactosidase enzyme assay system (Promega). Luciferase activity was normalized to β-galactosidase activity to correct for transfection efficiency. Transfection data represent at least three independent experiments each performed in triplicate.
Overexpression of CREB and Sp1
C3H10T1/2 cells were plated in monolayer cultures and transfected following the same protocol described above. Co-transfections of the SOX9-promoter construct along with constructs expressing either CREB (pCMV50ACREB, gift from Dr. Vinson) or Sp1 (pEGFP-C1-NLS-Sp1, gift from Dr. Folz) were performed. As a control the cells were co-transfected with the SOX9-promoter construct along with the empty pCMVβ vector.
DNA affinity precipitation assay
DNA affinity precipitation assay (DAPA) was performed according to the method described by Zhu et al. [32]with some modifications. The binding assay was performed by mixing 50 μg of nuclear extract proteins from ATDC5 cells cultured in chondrogenic differentiation media (DMEM/Ham’s F-12 1:1 containing 10% FBS supplemented with L-glutamine (2 mM), penicillin (100 units/ml) and streptomycin (100 μg/ml)) for 0, 3, 5, 7 and 13 days with Dynabeads M-208 Streptavidin (Invitrogen) which had been previously bound to 10 pmol of either biotinylated SOX9 promoter specific CRE sequence probe or the same probe with a mutated CREB site in the SOX9 promoter. The mixture was incubated at room temperature for 30 min with shaking and then the beads were separated and washed following the manufacturer’s standard protocol. Non- specifically bound proteins were removed by two washes in a 10 mM Tris buffer containing 50 mM KCl, 1 mM MgCl2, 1 mM EDTA, 5.5 mM DTT, 5% Glycerol and 0.03% Nondiet P-40. Finally the bound proteins were eluted by resuspending the beads in sample buffer and boiling them for 5 min and then separated by SDS-PAGE in 4–10% polyacrylamide gradient gels. Western Blot analysis was performed with a specific anti-CREB antibody (Chemicon). Appropriate secondary antibodies coupled to peroxidase and the SuperSignal detection system (Pierce) were employed for detection.
Chromatin immunoprecipitation assay
The chromatin immunoprecipitation assay was performed using the Chip-IT kit (Active Motif, Carlsbad, CA) following the manufacturer’s instructions. Briefly, three 100 cm2 plates of 70–80%-confluent HTB cells were treated with 1% formaldehyde in PBS for 10 min at room temperature. The formaldehyde was inactivated by the addition of 0.125 M glycine in PBS to the cells for 5 min at room temperature. The cells were then washed in ice-cold PBS and harvested by scraping. The cells were then resuspended in lysis buffer and the nuclei released by Dounce homogenization. The nuclei were pelleted by centrifugation at 2400xg for 10 min at 4°C. To shear genomic DNA, the nuclei were subjected to an enzymatic digestion with 5U of Enzymatic Shearing Cocktail solution (Active Motif) for 5 min at 37 °C. The sheared DNA was then centrifuged at 12000xg for 10 min at 4°C and the supernatant collected. For each immunoprecipitation, 50 μl of chromatin was pre-cleared with Protein G agarose beads for 1–2 hrs at 4°C. Six μg of the appropriate antibody was incubated with a pre-cleared chromatin aliquot overnight at 4°C with gentle rotation. Anti-Sp1 antibodies were obtained from Santa Cruz Biotechnology and anti-CREB antibody from Chemicon. The antibody-chromatin mixtures were incubated with Protein G beads for 1.5 hrs at 4°C. The antibody/chromatin/Protein G bead mixtures were then washed extensively and the DNA eluted. Crosslinks were reversed and the DNA was purified for semiquantitative PCR analysis. The sequences of the primers utilized to amplify a 150 bp segment approximately 3 kb upstream of the SOX9 transcriptional start site were: forward; 5′-ACACACTTGGAAGTCCCGGG-3′ and reverse; 5′-TTGGGAGGGAGGAGGATTG-3′. The initial denaturation was performed at 94 °C for 20 s and the annelation at 59 °C for 30 s with 36 cycles of amplification. The sequences of the PCR products were confirmed by sequencing.
RESULTS
DNaseI protection analysis of the human SOX9 proximal promoter region
Figure 1 shows the sequence of the human SOX9 proximal promoter region from −315 to +5 bp relative to the start site of transcription as mapped by Wagner et al. [33]. Within this region there are predicted to be several transcription factor-binding sites including two Sp1-like sites, a CRE ½-site and two CCAAT boxes [12]. We have recently shown that the two CCAAT boxes can interact with the CCAAT-binding factor and are important for SOX9 promoter activity [13]. In order to determine whether the Sp1 sites and the CRE ½-site are also functionally important for the activity of the SOX9 promoter, we undertook a DNaseI footprinting analysis of the region from −315 to −41 bp of the SOX9 proximal promoter (bracketed region, Fig. 1). Figure 2A shows the results of a DNaseI footprint analysis of the region from −166 to −112 bp that contains the CRE ½-site. The CRE was protected when incubated with nuclear extracts from prechondrogenic mesenchymal ATDC5 cells. Figure 2B shows protection of the Sp1-1 and Sp1–2 sites with nuclear extracts from ATDC5 cells. We conclude from these experiments that ATDC5 nuclear extracts contain specific factors that bind to the predicted CRE and Sp1 sites within the human SOX9 promoter.
Figure 1. Sequence of the human SOX9 proximal promoter region from −315 to +5 bp.

The Sp1-1 and Sp1–2 sites, the CRE ½-site, the CCAAT boxes, and the TATA box sequences are in bold and labeled above the corresponding sequence. The sequences of the transcription factor binding sites are underlined. The transcriptional start site is indicated by the open arrow. Brackets indicate the region that was analyzed by DNaseI protection (see Fig. 2).
Figure 2. DNaseI protection experiment.

(A) Lane IT; ATDC5 nuclear extract with poly dI-dC as non-specific competitor, lane AT; ATDC5 nuclear extract with poly dA-dT as non-specific competitor, lanes N; naked DNA; lane L; G+A DNA sequence ladder. The sequence shown is from −166 to −112 bp. (B) DNaseI footprinting of the Sp1 sites within the human SOX9 proximal promoter region with nuclear extracts from ATDC5 cells. L; G+A DNA sequence ladder, N, naked DNA; A; ATDC5 nuclear protein.
EMSA analysis of the CRE ½-site within the human SOX9 proximal promoter
Sequence analysis of the CRE protected site in the human SOX9 promoter revealed that it was a CRE ½-site (consensus CGTCA) [34]. CREB has been shown to bind to and transactivate from both low affinity ½-sites and the higher affinity full palindromic sites (TGACGTCA) [34, 35]. In order to further define the specificity of the CRE ½-site within the SOX9 promoter, we performed EMSA competition and supershift analyses. Figure 3A shows strong binding activity to the wild-type (WT) SOX9 CREB site in nuclear extracts from several different cell lines. Figure 3B shows that the binding is specific for the CRE ½-site. Binding to the CRE ½-site was observed in nuclear extracts from ATDC5 cells, RCS cells, FBCs and C3H10T1/2 cells with a WT SOX9 CRE ½-site probe, but not a mutated version (Fig 3B). Figure 4 shows that either an unlabeled WT SOX9 CREB-½ site probe or an unlabeled CRE-consensus site probe could compete for binding with a labeled WT SOX9 CRE ½-site probe in nuclear extracts from ATDC5 cells, whereas an unlabeled mutant CRE-consensus probe was unable to efficiently compete. In Figure 5, we show that antibodies to CREB can specifically supershift the CREB-SOX9 probe complexes, as well as CREB-consensus probe complexes, in nuclear extracts from ATDC5 cells, RCS cells, FBCs and C3H10T1/2 cells. From these experiments we conclude that CREB can specifically interact with the WT SOX9 CRE ½-site probe.
Figure 3. EMSA of the CREB binding site in the human SOX9 promoter.
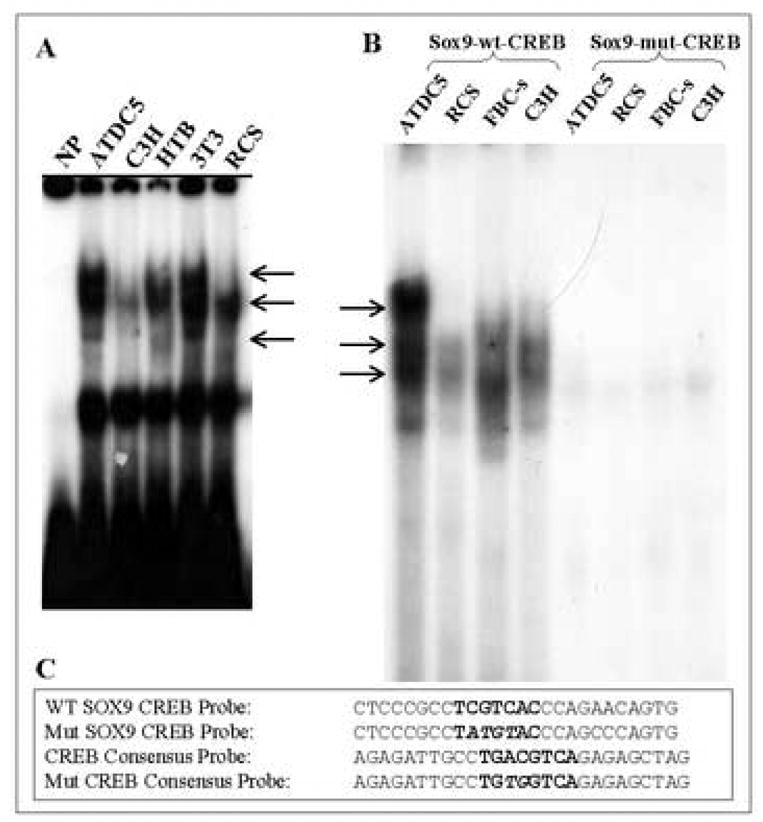
(A) Binding activity of the SOX9 promoter CRE ½-site in nuclear extracts from various cell lines. NP= no protein; (B) Binding of CREB from nuclear extracts from ATDC5, C3H10T1/2, RCS cells and from primary fetal bovine chondrocytes (FBC) to a WT or mutated (MUT) SOX9 CREB-½ site probe. (C) Sequences of the WT and mutated (Mut) EMSA probes. Arrows indicate putative DNA-CREB complexes.
Figure 4. EMSA competition analysis of CREB binding to the human SOX9 promoter.

Binding of CREB to the SOX9 CRE ½-site was competed with either the WT SOX9 CRE ½-site probe, a CREB-consensus site probe or a mutated CREB-consensus site probe. Sequences of the EMSA probes can be found in Figure 3C. Arrows indicate putative DNA-CREB complexes.
Figure 5. Supershift analysis of CREB binding to the human SOX9 promoter in nuclear extracts from various cell lines.

Supershift analysis of CREB binding to the SOX9 CRE ½-site or a CRE-consensus site probe with an anti-CREB antibody. A=ATDC5, R=RCS, C=C3H10T1/2 and F= fetal bovine chondrocyte nuclear extract. Sequences of the EMSA probes can be found in Figure 3C. Thick arrows indicate putative DNA-CREB complexes. Thin arrows point supershifted complexes.
EMSA analysis of the Sp1 sites within the SOX9 proximal promoter
In the DNaseI protection experiments described above (see Fig. 1), we demonstrated that the Sp1-1 and Sp1–2 sites were protected by nuclear extracts from several different cell lines. To investigate the specificity of binding of Sp1 to the SOX9 Sp1-1 and Sp1–2 sites within the proximal-promoter region we performed EMSA competition and supershift analyses. In Figure 6A it can be seen that the binding activity to the WT SOX9 Sp1-1 and Sp1–2 sites in nuclear extracts from ATDC5 cells can be efficiently competed away with either unlabeled WT SOX9 Sp1-1 or Sp1–2 probes or with an unlabeled Sp1 consensus probe. In Figure 6B we demonstrate that Sp1 binding to the WT Sp1-1 or Sp1–2 sites within the SOX9 proximal promoter can be inhibited by inclusion of an anti-Sp1 antibody. In the supershift experiment (Fig. 6B) we noted that not all of the Sp1 binding activity to the SOX9 Sp1 sites was inhibited by the Sp1 antibody, as was the case for the Sp1 consensus probe, an observation which suggests that other Sp-like proteins can also interact with these sites.
Figure 6. Competition and supershift EMSA of the human SOX9-promoter Sp1-1 and Sp1–2 sites.

(A) EMSA competition analysis of the human SOX9 promoter Sp1-1 and Sp1–2 sites (see Fig. 1) with ATDC5 cell nuclear extracts. In each case 100X of either an unlabeled SOX9 WT Sp1-1 or Sp1–2 probe or an unlabeled Sp1-consensus probe was used as a competitor. Sp1Con= Sp1 consensus probe. Note decreased binding (arrow). (B) EMSA super shift analysis of the human SOX9 promoter Sp1-1 and Sp1–2 sites with ATDC5 cell nuclear extracts and an anti-Sp1 antibody. Inclusion of the Sp1 antibody resulted in partial inhibition of binding to the SOX9 Sp1-1 site and almost complete inhibition of binding to the SOX9 Sp1–2 site (arrow). In contrast, the antibody fully inhibited binding to the Sp1 consensus probe (Cons).
Functional analysis of the CRE-1/2 and Sp1-1 sites within the human SOX9 promoter
The roles of the CRE ½ site and the Sp1-1 site in transcriptional control of the human SOX9 proximal promoter were investigated next. SOX9 promoter-reporter constructs containing the WT, mutated CRE-½ or deleted Sp1-1 sites (Figure 7A) were transfected into C3H10T1/2 cells, ATDC5 cells or FBCs. In all three-cell types, SOX9 proximal-promoter activity was reduced by 40–60% when either the CRE ½-site was mutated or the Sp1-1 site was deleted (Figs. 7B, C and D). We conclude from these experiments that human SOX9 proximal-promoter transcriptional activity is regulated by CREB and Sp1.
Figure 7. Functional analysis of the CREB and Sp1-1 sites in the human SOX9 proximal promoter.
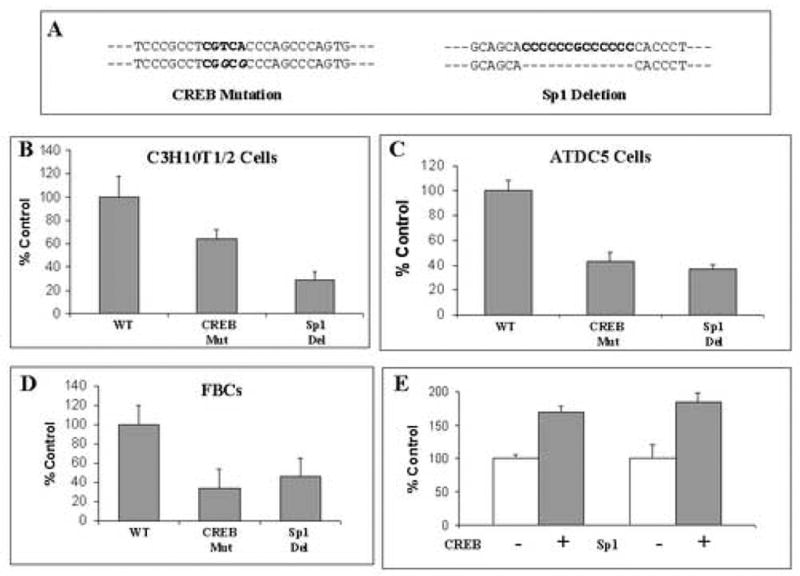
(A) Sequence of the CRE mutation and Sp1-1 deletion introduced into the WT human SOX9 promoter construct. (B–D) Relative promoter activity of the WT SOX9-luciferase promoter-reporter construct and constructs containing either a mutated CRE site or a deleted Sp1-1 site in C3H10T1/2 cells, ATDC5 cells or FBCs, respectively. Data are expressed as percent activity of the full length −1034/+67 bp SOX9-promoter-luciferase construct. (E) Relative promoter activity of the WT SOX9-luciferase promoter-reporter construct in C3H10T1/2 cells co-transfected with CREB and Sp1 expression constructs. Data are from three separate experiments each performed in triplicate and expressed as percent activity of the full length −1034/+67 bp SOX9-promoter-luciferase construct when co-transfected with either CREB or Sp1 expression constructs (+). Transfection efficiency was normalized by including the pCMVβ plasmid and then normalizing luciferase activity to β-galactosidase activity. The bars show S.D.
To confirm these observations we over-expressed CREB and Sp1 in C3H10T1/2 cells by co-transfection of CREB or Sp1 expression constructs. The results (Figure 7E) show that the activity of SOX9 promoter is increased 1.6 times when CREB is overexpressed. While Sp1 overexpression resulted in an increase in 1.8 times of SOX9 promoter activity.
DNA affinity precipitation analysis of CREB binding to the SOX9 promoter during chondrogenesis
To investigate the functional significance of CREB binding to the promoter region of the human SOX9 promoter analysis of CREB binding during the chondrogenesis process was performed by DAPA. ATDC5 cells were induced to undergo chondrogenic differentiation by culturing them in differentiation medium as described in Materials and Methods. After 0, 3, 5, 7 and 13 days cells were harvested and nuclear extracts were prepared. Biotinylated SOX9 promoter oligonucleotides which contained either the wild-type or a mutated CRE site were incubated with nuclear extracts obtained from the ATDC5 cells. Precipitation of the specific complex performed with streptavidin-agarose beads followed by electrophoresis and Western blot analysis indicated that chondrogenesis of ATDC5 cells was accompanied by an increase in CREB binding to the human SOX9 promoter with maximum binding at 7 days of differentiation (Figure 8).
Figure 8. DNA affinity precipitation of nuclear proteins from ATDC5 cells induced to undergo chondrogenic differentiation.

Nuclear extracts from ATDC5 cells during 0, 3, 5, 7 or 13 days of chondrogenic differentiation were incubated with biotinylated wild type (CREB wt) or mutated CREB site (CREB Mut) SOX9 promoter sequence oligonucleotides. Nuclear protein/oligonucleotide complexes were purified by precipitation with streptavidin-agarose beads, separated on SDS/PAGE, and subjected to a Western blot analysis using a specific anti-CREB antibody.
Association of CREB and Sp1 with the Sox9 promoter in vivo
In order to confirm the observations obtained with EMSA and to demonstrate that Sp1 and CBF interact with the SOX9 promoter in vivo, a chromatin immunoprecipitation (ChIP) assay was carried out. For this purpose HTB cells were subjected to cross-linking with formaldehyde, the cells were lysed and the chromatin was cleaved by nuclease digestion. The chromatin was then immunoprecipitated with anti-IgG, anti-Sp1 and anti-CREB antibodies and the DNA precipitated in the complexes was subjected to PCR amplification with primers flanking the region containing the two Sp1 sites and the CRE site (Fig. 9A). Figure 9B shows that both anti-CREB and anti-Sp1 antibodies precipitated proteins bound in vivo to the amplified sequence of the SOX9 proximal promoter whereas non-specific IgG antibody (control antibody) failed to precipitate proteins bound in vivo this sequence.
Figure 9. Chromatin immunoprecipitation analysis.

A chromatin immunoprecipitation (ChIP) analysis was performed to confirm the interaction of Sp1 and CREB with the SOX9 promoter in vivo. (A) Sequence of the region of the human SOX9 promoter and location of primers that was used in the PCR amplification after the ChIP assay. The Sp1 and CREB sites are underlined. (B) PCR products from the ChIP assay run on an agarose gel. Precipitation with an unrelated antibody was used as negative control. The input DNA represents total and 1/10 of the starting material.
IL-1β treatment reduces SOX9 promoter activity in C3H10T1/2 cells and Sp1 binding activity in normal and OA human articular chondrocytes
It has been well documented that pro-inflammatory cytokines, such as IL-1β and TNF-α down regulate the expression of chondrocyte-specific genes including SOX9 and COL2A1. Indeed, previous work has shown that IL-1β and TNF-α cause a potent suppression of SOX9 gene expression in primary chondrocytes and MC615 cells (immortalized mouse chondrocytes) [16]. In order to determine whether this suppression involves regulation of the SOX9 promoter, C3H10T1/2 cells were transiently transfected with a human SOX9 promoter (Figure 10A) and after 24 h were treated with IL-1β. Figure 10B shows that the activity of the SOX9 promoter is suppressed by approximately 50–60% upon treatment with either 5 or 10 ng/ml of IL-1β for 24 h. Figure 10C shows that the full effect of IL-1β on the SOX9 promoter occurs within 4 h of treatment. In order to determine whether this suppression involves modulation of Sp1 interaction with the SOX9 promoter we performed an EMSA analysis with nuclear extracts from human adult normal or OA articular chondrocytes that had been treated with IL-1β. As can be seen in Figure 9D, treatment of human normal (N) and OA (OA) chondrocytes with 10 ng/ml IL-1β for 24 h resulted in a significant reduction in binding to either the SOX9 Sp1-1 site (SX) or an Sp1 consensus site (CN).
Figure 10. Suppression of SOX9 promoter activity and Sp1 binding by IL-1β.

(A) Schematic depiction of the −1034 bp human SOX9 promoter construct showing relevant transcription factor binding sites. (B) Dose-response of the full-length human SOX9 promoter activity to IL-1β treatment in C3H10T1/2 cells. C3H10T1/2 cells transiently transfected with the −1034 SOX9 promoter construct were treated with either 0, 5 or 10 ng/ml IL-1β for 24 h. (C) Time-course of IL-1β treatment. C3H10T1/2 cells transiently transfected with the −1034 bp SOX9 promoter construct were treated with 10 ng/ml IL-1β for 0, 1, 2, 4, 6 or 8 h. (D) Effect of IL-1β treatment on Sp1 binding to the SOX9 promoter. Primary adult human normal and OA chondrocytes were cultured in suspension and treated with 10 ng/ml IL-1β for 24 h followed by nuclear extract preparation. The SOX9 Sp1-1 probe or an Sp1 consensus probe was then utilized in an EMSA. CN, Sp1 consensus probe; SX, SOX9 Sp1-1 probe; NP, no protein; N, normal chondrocyte nuclear extract; OA, osteoarthritis chondrocyte nuclear extract.
DISCUSSION
In this work, we have documented the specific interaction of CREB with the SOX9 promoter through a CRE ½-site located at position −147. We have also demonstrated that mutation of the CRE ½-site reduces SOX9 promoter activity. In response to several signaling cascades, including cAMP levels, FGFs, IGFs, hypoxia and parathyroid hormone-related peptide (PTHrP), the dimeric, basic-leucine zipper transcription factor CREB is phosphorylated at a specific serine residue leading to the induction of genes containing a cyclic AMP response element [34]. Signaling through CREB has been shown to be important for cell survival, proliferation and neural cell function, and CRE-containing target genes are found in many different functional categories [34]. The CRE consists of the palindromic sequence TGACGTCA but CREB can also bind to a ½-site, CGTCA, with relatively high affinity (5 nM) [34, 36]. CREs are generally found 50–150 bp upstream from the start site of transcription although one third of CRE sites are located further away than 150 bp [34, 37]. Interestingly, CREB can stimulate both basal transcription and inducible transcription through its bipartite transcriptional activation domain [34, 38, 39]. The domain contains a constitutive activation domain that can interact with components of the general transcriptional machinery and a kinase-inducible domain that when phosphorylated at a serine residue promotes the recruitment of transcriptional co-activators such as CREB-binding protein and p300 [38–43]. A recent study of CREB binding has shown that the protein has the ability to bind its cognate site in responsive gene promotes either as a monomer or as a homodimer or a heterodimer with other transcription factors such as NFIL3 [44]. These observations might explain the presence of multiple shifted bands in our EMSA analyses of CREB binding to the SOX9 promoter. Another alternative to explain the presence of multiple bands on the EMSA experiments is the previously demonstrated molecular heterogeneity of CREB with several species of varying molecular sizes (see Figure 2, frame A, in reference [45]).
CREB has been implicated in the process of chondrocyte differentiation during endochondral bone formation based on the observations that the expression of a dominant negative form of CREB (A-CREB) in transgenic mice [46] resulted in a reduction in the proliferative zone of the growth plate along with a delay in chondrocyte hypertrophy, presumably owing to the loss of the pool of precursor chondrocytes. Furthermore, a recent study which examined the process of chondrogenic differentiation induced in human dermal fibroblasts cultured in collagen sponges containing demineralized bone powder found a remarkable increase in SOX9 gene expression which was accompanied by a two fold increase in CREB protein, and a three-fold in CREB phosphorylation [47]
In other studies, CREB has been implicated in PTHrP signaling pathway in chondrocytes [48]. In these studies it was demonstrated that inhibition of CREB function during PTHrP signaling led to a decrease in chondrocyte proliferation and an increase in maturation, similar to that observed in PTHrP-KO mice [49]. More recent results have demonstrated that forced expression of sonic hedgehog (Shh) in mice can up-regulate the Sox9 gene and Shh over-expression in cells can stimulate the Sox9 promoter [21]. Interestingly, PTHrP is known to be downstream of hedgehog signaling in the regulation of chondrocyte differentiation in the growth plate [50]. Based on our results, we postulate that CREB influences the basal transcription of the SOX9 promoter through its constitutive activation domain and that regulation of the SOX9 gene by agents such as the hedgehog proteins may proceed through PTHrP to the SOX9 promoter via the CREB binding site and activation of CREB.
We have also demonstrated in this work that the human SOX9 promoter contains two Sp1-like sites, designated Sp1-1 and Sp1–2, and that deletion of the Sp1-1 site results in decreased promoter activity. Interestingly, the Sp1–2 site is located adjacent to the CRE ½-site. Sp1 is a member of a family of transcription factors containing a conserved set of Cys-His zinc-fingers within their DNA-binding domain that recognize both GC and GT boxes, which are found in the proximal-promoter regions of many genes [51]. To date, there are at least 8 related Sp-proteins (Sp1–Sp8) found in humans. Some members, such as Sp1 and Sp3, are expressed in most tissues whereas some show a more tissue-restricted pattern of expression (i.e., Sp4 and Sp7) [51]. Related Sp-transcription factors can act both as activators or repressors. Some authors have shown that Sox9 gene expression is repressed by IL-1β treatment and we have shown that SOX9 gene expression is down-regulated during chondrocyte de-differentiation [16, 28]. In this work we confirm that the human SOX9 promoter is down-regulated by IL-1β treatment of transiently transfected C3H10T1/2 cells and that the binding activity of Sp1 to the SOX9 promoter is decreased in IL-1β treated normal and OA human articular chondrocytes. Other studies have shown that IL-1β treatment of rabbit articular chondrocytes resulted in decreased Sp1 protein expression[14, 52] thus, it is likely that IL-1β induced reduction of Sp1 is responsible for the reduced DNA binding activity of the SOX9 promoter observed in our study.
Elucidating the identity and function of transcription factors that regulate the expression of the SOX9 gene, a master regulator of chondrogenesis, will ultimately provide insight into the pathways that control mesenchymal cell and chondrocyte differentiation and maturation during development as well as changes in chondrocyte function occurring in disease states such as OA. Indeed, evidence suggests that SOX9 is up-regulated in OA articular cartilage, perhaps in an effort to repair damaged tissue [19]. Unfortunately, the repair process is not efficient and eventually declines, leading to cartilage degeneration and failure. An in depth understanding of pathways and factors that govern SOX9 gene expression, a gene that promotes the expression of many cartilage extracellular matrix protein genes, would provide novel molecular targets for developing therapeutic agents that could promote the repair of damaged or diseased cartilage.
Acknowledgments
Supported by NIH Program Project grant AR-39740 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Healy C, Uwanogho D, Sharpe PT. Expression of the chicken Sox9 gene marks the onset of cartilage differentiation. Ann N Y Acad Sci. 1996;785:261–2. doi: 10.1111/j.1749-6632.1996.tb56278.x. [DOI] [PubMed] [Google Scholar]
- 2.Wright E, Hargrave MR, Christiansen J, Cooper L, Kun J, Evans T, Gangadharan U, Greenfield A, Koopman P. The Sry-related gene Sox9 is expressed during chondrogenesis in mouse embryos. Nat Genet. 1995;9:15–20. doi: 10.1038/ng0195-15. [DOI] [PubMed] [Google Scholar]
- 3.Ng LJ, Wheatley S, Muscat GE, Conway-Campbell J, Bowles J, Wright E, Bell DM, Tam PP, Cheah KS, Koopman P. SOX9 binds DNA, activates transcription, and coexpresses with type II collagen during chondrogenesis in the mouse. Dev Biol. 1997;183:108–21. doi: 10.1006/dbio.1996.8487. [DOI] [PubMed] [Google Scholar]
- 4.Mertin S, McDowall SG, Harley VR. The DNA-binding specificity of SOX9 and other SOX proteins. Nucleic Acids Res. 1999;27:1359–64. doi: 10.1093/nar/27.5.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bridgewater LC, Lefebvre V, de Crombrugghe B. Chondrocyte-specific enhancer elements in the Col11a2 gene resemble the Col2a1 tissue-specific enhancer. J Biol Chem. 1998;273:14998–5006. doi: 10.1074/jbc.273.24.14998. [DOI] [PubMed] [Google Scholar]
- 6.Sekiya I, Tsuji K, Koopman P, Watanabe H, Yamada Y, Shinomiya K, Nifuji A, Noda M. SOX9 enhances aggrecan gene promoter/enhancer activity and is up-regulated by retinoic acid in a cartilage-derived cell line, TC6. J Biol Chem. 2000;275:10738–44. doi: 10.1074/jbc.275.15.10738. [DOI] [PubMed] [Google Scholar]
- 7.Zhang P, Jimenez SA, Stokes DG. Regulation of human COL9A1 gene expression. Activation of the proximal promoter region by SOX9. J Biol Chem. 2003;278:117–23. doi: 10.1074/jbc.M208049200. [DOI] [PubMed] [Google Scholar]
- 8.Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation. Nat Genet. 1999;22:85–9. doi: 10.1038/8792. [DOI] [PubMed] [Google Scholar]
- 9.Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002;16:2813–28. doi: 10.1101/gad.1017802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lefebvre V, Huang W, Harley VR, Goodfellow PN, de Crombrugghe B. SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol Cell Biol. 1997;17:2336–46. doi: 10.1128/mcb.17.4.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bell DM, Leung KK, Wheatley SC, Ng LJ, Zhou S, Ling KW, Sham MH, Koopman P, Tam PP, Cheah KS. SOX9 directly regulates the type-II collagen gene. Nat Genet. 1997;16:174–8. doi: 10.1038/ng0697-174. [DOI] [PubMed] [Google Scholar]
- 12.Kanai Y, Koopman P. Structural and functional characterization of the mouse Sox9 promoter: implications for campomelic dysplasia. Hum Mol Genet. 1999;8:691–6. doi: 10.1093/hmg/8.4.691. [DOI] [PubMed] [Google Scholar]
- 13.Colter DC, Piera-Velazquez S, Hawkins DF, Whitecavage MK, Jimenez SA, Stokes DG. Regulation of the human Sox9 promoter by the CCAAT-binding factor. Matrix Biol. 2005;24:185–97. doi: 10.1016/j.matbio.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 14.Morishita M, Kishino T, Furukawa K, Yonekura A, Miyazaki Y, Kanematsu T, Yamashita S, Tsukazaki T. A 30-base-pair element in the first intron of SOX9 acts as an enhancer in ATDC5. Biochem Biophys Res Commun. 2001;288:347–55. doi: 10.1006/bbrc.2001.5778. [DOI] [PubMed] [Google Scholar]
- 15.Murakami S, Kan M, McKeehan WL, de Crombrugghe B. Up-regulation of the chondrogenic Sox9 gene by fibroblast growth factors is mediated by the mitogen-activated protein kinase pathway. Proc Natl Acad Sci U S A. 2000;97:1113–8. doi: 10.1073/pnas.97.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murakami S, Lefebvre V, de Crombrugghe B. Potent inhibition of the master chondrogenic factor Sox9 gene by interleukin-1 and tumor necrosis factor-alpha. J Biol Chem. 2000;275:3687–92. doi: 10.1074/jbc.275.5.3687. [DOI] [PubMed] [Google Scholar]
- 17.Zehentner BK, Dony C, Burtscher H. The transcription factor Sox9 is involved in BMP-2 signaling. J Bone Miner Res. 1999;14:1734–41. doi: 10.1359/jbmr.1999.14.10.1734. [DOI] [PubMed] [Google Scholar]
- 18.Semba I, Nonaka K, Takahashi I, Takahashi K, Dashner R, Shum L, Nuckolls GH, Slavkin HC. Positionally-dependent chondrogenesis induced by BMP4 is co-regulated by Sox9 and Msx2. Dev Dyn. 2000;217:401–14. doi: 10.1002/(SICI)1097-0177(200004)217:4<401::AID-DVDY7>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 19.Uusitalo H, Salminen H, Vuorio E. Activation of chondrogenesis in response to injury in normal and transgenic mice with cartilage collagen mutations. Osteoarthritis Cartilage. 2001;9(Suppl A):S174–9. [PubMed] [Google Scholar]
- 20.Sekiya I, Koopman P, Tsuji K, Mertin S, Harley V, Yamada Y, Shinomiya K, Niguji A, Noda M. Transcriptional suppression of Sox9 expression in chondrocytes by retinoic acid. J Cell Biochem. 2001;81:71–78. doi: 10.1002/jcb.1077. [DOI] [PubMed] [Google Scholar]
- 21.Tavella S, Biticchi R, Schito A, Minina E, Di Martino D, Pagano A, Vortkamp A, Horton WA, Cancedda R, Garofalo S. Targeted Expression of SHH Affects Chondrocyte Differentiation, Growth Plate Organization, and Sox9 Expression. J Bone Miner Res. 2004;19:1678–88. doi: 10.1359/JBMR.040706. [DOI] [PubMed] [Google Scholar]
- 22.Mukhopadhyay K, Lefebvre V, Zhou G, Garofalo S, Kimura JH, de Crombrugghe B. Use of a new rat chondrosarcoma cell line to delineate a 119-base pair chondrocyte-specific enhancer element and to define active promoter segments in the mouse pro-alpha 1(II) collagen gene. J Biol Chem. 1995;270:27711–9. doi: 10.1074/jbc.270.46.27711. [DOI] [PubMed] [Google Scholar]
- 23.Coimbra IB, Jimenez SA, Hawkins DF, Piera-Velazquez S, Stokes DG. Hypoxia inducible factor-1 alpha expression in human normal and osteoarthritic chondrocytes. Osteoarthritis Cartilage. 2004;12:336–45. doi: 10.1016/j.joca.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 24.Reginato AM, Iozzo RV, Jimenez SA. Formation of nodular structures resembling mature articular cartilage in long-term primary cultures of human fetal epiphyseal chondrocytes on a hydrogel substrate. Arthritis Rheum. 1994;37:1338–49. doi: 10.1002/art.1780370912. [DOI] [PubMed] [Google Scholar]
- 25.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–89. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leblanc B, Moss T. DNase I footprinting. Methods Mol Biol. 2001;148:31–8. doi: 10.1385/1-59259-208-2:031. [DOI] [PubMed] [Google Scholar]
- 27.Stokes DG, Perry RP. DNA-binding and chromatin localization properties of CHD1. Mol Cell Biol. 1995;15:2745–53. doi: 10.1128/mcb.15.5.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stokes DG, Liu G, Dharmavaram R, Hawkins D, Piera-Velazquez S, Jimenez SA. Regulation of type-II collagen gene expression during human chondrocyte de-differentiation and recovery of chondrocyte-specific phenotype in culture involves Sry-type high-mobility-group box (SOX) transcription factors. Biochem J. 2001;360:461–70. doi: 10.1042/0264-6021:3600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Genuario RR, Kelley DE, Perry RP. Comparative utilization of transcription factor GABP by the promoters of ribosomal protein genes rpL30 and rpL32. Gene Expr. 1993;3:279–88. [PMC free article] [PubMed] [Google Scholar]
- 30.Madry H, Trippel SB. Efficient lipid-mediated gene transfer to articular chondrocytes. Gene Ther. 2000;7:286–91. doi: 10.1038/sj.gt.3301086. [DOI] [PubMed] [Google Scholar]
- 31.Stokes DG, Liu G, Coimbra IB, Piera-Velazquez S, Crowl RM, Jimenez SA. Assessment of the gene expression profile of differentiated and dedifferentiated human fetal chondrocytes by microarray analysis. Arthritis Rheum. 2002;46:404–19. doi: 10.1002/art.10106. [DOI] [PubMed] [Google Scholar]
- 32.Zhu Y, Saunders MA, Yeh H, Deng WG, Wu KK. Dynamic regulation of cyclooxygenase-2 promoter activity by isoforms of CCAAT/enhancer-binding proteins. J Biol Chem. 2002;277:6923–8. doi: 10.1074/jbc.M108075200. [DOI] [PubMed] [Google Scholar]
- 33.Wagner T, Wirth J, Meyer J, Zabel B, Held M, Zimmer J, Pasantes J, Bricarelli FD, Keutel J, Hustert E, et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell. 1994;79:1111–20. doi: 10.1016/0092-8674(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 34.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 35.Nichols M, Weih F, Schmid W, DeVack C, Kowenz-Leutz E, Luckow B, Boshart M, Schutz G. Phosphorylation of CREB affects its binding to high and low affinity sites: implications for cAMP induced gene transcription. Embo J. 1992;11:3337–46. doi: 10.1002/j.1460-2075.1992.tb05412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Craig JC, Schumacher MA, Mansoor SE, Farrens DL, Brennan RG, Goodman RH. Consensus and variant cAMP-regulated enhancers have distinct CREB-binding properties. J Biol Chem. 2001;276:11719–28. doi: 10.1074/jbc.M010263200. [DOI] [PubMed] [Google Scholar]
- 37.Tinti C, Yang C, Seo H, Conti B, Kim C, Joh TH, Kim KS. Structure/function relationship of the cAMP response element in tyrosine hydroxylase gene transcription. J Biol Chem. 1997;272:19158–64. doi: 10.1074/jbc.272.31.19158. [DOI] [PubMed] [Google Scholar]
- 38.Quinn PG. Distinct activation domains within cAMP response element-binding protein (CREB) mediate basal and cAMP-stimulated transcription. J Biol Chem. 1993;268:16999–7009. [PubMed] [Google Scholar]
- 39.Felinski EA, Quinn PG. The CREB constitutive activation domain interacts with TATA-binding protein-associated factor 110 (TAF110) through specific hydrophobic residues in one of the three subdomains required for both activation and TAF110 binding. J Biol Chem. 1999;274:11672–8. doi: 10.1074/jbc.274.17.11672. [DOI] [PubMed] [Google Scholar]
- 40.Quinn PG. Mechanisms of basal and kinase-inducible transcription activation by CREB. Prog Nucleic Acid Res Mol Biol. 2002;72:269–305. doi: 10.1016/s0079-6603(02)72072-2. [DOI] [PubMed] [Google Scholar]
- 41.Kim J, Lu J, Quinn PG. Distinct cAMP response element-binding protein (CREB) domains stimulate different steps in a concerted mechanism of transcription activation. Proc Natl Acad Sci U S A. 2000;97:11292–6. doi: 10.1073/pnas.97.21.11292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Felinski EA, Kim J, Lu J, Quinn PG. Recruitment of an RNA polymerase II complex is mediated by the constitutive activation domain in CREB, independently of CREB phosphorylation. Mol Cell Biol. 2001;21:1001–10. doi: 10.1128/MCB.21.4.1001-1010.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Felinski EA, Quinn PG. The coactivator dTAF(II)110/hTAF(II)135 is sufficient to recruit a polymerase complex and activate basal transcription mediated by CREB. Proc Natl Acad Sci U S A. 2001;98:13078–83. doi: 10.1073/pnas.241337698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Acharya A, Rishi V, Moll J, Vinson C. Experimental identification of homodimerizing B-ZIP families in Homo sapiens. J Struct Biol. 2006 doi: 10.1016/j.jsb.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 45.Kwast-Welfeld J, de Belle I, Walker PR, Whitfield JF, Sikorska M. Identification of a new cAMP response element-binding factor by southwestern blotting. J Biol Chem. 1993;268:19581–5. [PubMed] [Google Scholar]
- 46.Long F, Schipani E, Asahara H, Kronenberg H, Montminy M. The CREB family of activators is required for endochondral bone development. Development. 2001;128:541–50. doi: 10.1242/dev.128.4.541. [DOI] [PubMed] [Google Scholar]
- 47.Yates KE. Identification of cis and trans-acting transcriptional regulators in chondroinduced fibroblasts from the pre-phenotypic gene expression profile. Gene. 2006;377:77–87. doi: 10.1016/j.gene.2006.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ionescu AM, Schwarz EM, Vinson C, Puzas JE, Rosier R, Reynolds PR, O'Keefe RJ. PTHrP modulates chondrocyte differentiation through AP-1 and CREB signaling. J Biol Chem. 2001;276:11639–47. doi: 10.1074/jbc.M006564200. [DOI] [PubMed] [Google Scholar]
- 49.Karaplis AC, Luz A, Glowacki J, Bronson RT, Tybulewicz VL, Kronenberg HM, Mulligan RC. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994;8:277–89. doi: 10.1101/gad.8.3.277. [DOI] [PubMed] [Google Scholar]
- 50.Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–22. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 51.Bouwman P, Philipsen S. Regulation of the activity of Sp1-related transcription factors. Mol Cell Endocrinol. 2002;195:27–38. doi: 10.1016/s0303-7207(02)00221-6. [DOI] [PubMed] [Google Scholar]
- 52.Chadjichristos C, Ghayor C, Kypriotou M, Martin G, Renard E, Ala-Kokko L, Suske G, de Crombrugghe B, Pujol JP, Galera P. Sp1 and Sp3 transcription factors mediate interleukin-1 beta down-regulation of human type II collagen gene expression in articular chondrocytes. J Biol Chem. 2003;278:39762–72. doi: 10.1074/jbc.M303541200. [DOI] [PubMed] [Google Scholar]