

Abstract
In autoimmune arthritis, traditionally classified as a T helper (Th) type 1 disease, the activation of T cells results in bone destruction mediated by osteoclasts, but how T cells enhance osteoclastogenesis despite the anti-osteoclastogenic effect of interferon (IFN)-γ remains to be elucidated. Here, we examine the effect of various Th cell subsets on osteoclastogenesis and identify Th17, a specialized inflammatory subset, as an osteoclastogenic Th cell subset that links T cell activation and bone resorption. The interleukin (IL)-23–IL-17 axis, rather than the IL-12–IFN-γ axis, is critical not only for the onset phase, but also for the bone destruction phase of autoimmune arthritis. Thus, Th17 is a powerful therapeutic target for the bone destruction associated with T cell activation.
Skeletal homeostasis is dynamically influenced by the immune system (1–3), and lymphocyte- or macrophage-derived cytokines are among the most potent mediators of osteoimmunological regulation (3–7). Therefore, the effect of individual cytokines on bone cells has been extensively studied (3–7), but the subset of immune cells with selective cytokine production that specifically affects bone cell differentiation has not been well characterized. Upon activation, CD4+ T cells undergo distinct developmental pathways to the specialized effector subsets: Th1 cells produce IFN-γ and regulate cellular immunity, whereas Th2 cells produce IL-4 and IL-5 and mediate humoral immunity (8). In addition, accumulating evidence suggests that newly recognized IL-17–producing T (Th17) cells have a crucial role in autoimmune inflammation (9, 10). CD4+CD25+Foxp3+ regulatory T (T reg) cells also constitute a distinct subset that prevents immune pathology through suppression of pathogenic T cells (11). Activation of CD4+ T cells is often linked to pathological bone resorption (3, 4), but the distinct CD4+ T cell subset that induces the differentiation of bone-resorbing osteoclasts has not been identified (2, 3).
Osteoclasts are multinucleated cells (MNCs) of monocyte/macrophage lineage that degrade bone matrix and dynamically remodel the skeleton (4–6). The generation of osteoclasts is physiologically supported by mesenchymal cells such as osteoblasts, which provide essential signals for differentiation of the osteoclast lineage: macrophage colony-stimulating factor (M-CSF), receptor activator of NF-κB ligand (RANKL), and costimulatory signals for RANKL (12). RANKL is the key osteoclastogenic cytokine expressed by osteoclastogenesis-supporting mesenchymal cells, but the same molecule has been shown to be expressed by T cells, indicating that RANKL is a molecule that bridges the skeletal and immune systems (4).
In autoimmune arthritis, bone destruction is attributable to excessive bone resorption by osteoclasts, the formation of which is directly and indirectly regulated by CD4+ T cells infiltrating into the lesion (2, 3, 13, 14). Indirect effects are mainly mediated by inflammatory cytokines produced by macrophage-like synovial cells such as TNF-α and IL-1 that induce RANKL on synovial fibroblasts (14–16), but it is poorly understood how T cells exert direct effects (3). Although T cells express RANKL, the T cell–mediated positive effect is not easily observed because T cells also produce IFN-γ, which counterbalances the effect of the RANKL, making the net effect on osteoclastogenesis inhibitory (3, 13, 14). Although autoimmune arthritis has traditionally been assumed to be a Th1 disease (17, 18), there is controversy over the role of Th1 cells in the onset phase of the disease based on the observations that typical Th1 cytokines, such as IFN-γ, are not always highly expressed in the lesion (19, 20), and that collagen-induced arthritis is exacerbated in mice lacking IFN-γ signaling (21, 22). Therefore, neither bone destruction nor inflammation may be attributable to Th1 cells. It is a critically important issue to determine the type of T cells linked to the activated osteoclastogenesis under such inflammatory conditions.
Recently, it has been reported that the IL-23–IL-17 axis, rather than the IL-12–IFN-γ axis, is critical for the onset of autoimmune arthritis (23, 24). It is also reported that IL-17 is detectable in the synovial fluid from rheumatoid arthritis (RA) patients and enhances osteoclastogenesis by inducing RANKL on mesenchymal cells (25). Here, we explored the effects of various CD4+ T cell subsets on osteoclast differentiation and identified Th17 cells as the exclusive osteoclastogenic T cell subset among the known CD4+ T cell lineages. The importance of the IL-23–IL-17 axis in the bone destruction phase was underscored by the observations in mice lacking either IL-17 or IL-23 (p19). We also found that the mRNA expression of RANKL correlates with that of IL-23 (IL23A), but not that of IL-12 (IL12A), in the synovial tissues of RA patients. Collectively, these results suggest that autoimmune arthritis can be deemed a Th17-type disease in terms of both the onset and destruction phases and provide a molecular basis for targeting the IL-23–IL-17 axis in the treatment of RA.
RESULTS
Effects of Th1, Th2, and T reg cells on osteoclastogenesis
Although the effects of activated T cells on osteoclastogenesis have been documented in previous reports (13, 14, 26), these T cells were only stimulated by anti-CD3 antibody or PMA and the characterization of the T cells was not strictly performed. In this study, to investigate the effects of effector Th cell subsets on osteoclastogenesis, we added Th subsets, which were strictly developed under Th1 or Th2 conditions. Purified CD4+ T cells were stimulated with anti-CD3/CD28 mAbs in the presence of either IL-12 (with anti–IL-4 mAb) or IL-4 (with anti–IFN-γ mAb) for Th1 or Th2 polarization. The Th cells were added to the two types of in vitro osteoclast differentiation systems: osteoclast precursor cells derived from BM cells (BMCs) were stimulated with recombinant RANKL and M-CSF (the RANKL–M-CSF system), or co-cultured with osteoblasts in the presence of 1,25 (OH)2 vitamin D3 (VitD3) and prostaglandin E2 (PGE2) (the co-culture system), and the formation of MNCs stained for tartrate-resistant acid phosphatase (TRAP), a marker enzyme for osteoclasts, was evaluated (Fig. 1 A). When Th1 or Th2 cells were added to the RANKL–M-CSF system at the same time as RANKL, both subsets had a marked inhibitory effect on the formation of TRAP+ MNCs and the inhibitory effects were dependent on the number of added T cells (Fig. 1, B and D). These inhibitory effects were significantly enhanced by restimulation with soluble anti-CD3 mAb, suggesting that restimulation of T cell receptor augments the polarized cytokine secretion and the inhibitory effects on osteoclastogenesis. If Th1 or Th2 cells were added to the co-culture system 2 d after BMC addition, the inhibitory effects of both subsets on osteoclastogenesis were exerted only by T cells restimulated with anti-CD3 mAb (Fig. 1, C and D). Th1 and Th2 cells both had less suppressive effects in the co-culture system, possibly because osteoblasts provide protection against the inhibitory effects through costimulatory signals (27) (see Discussion). As expected, the Th1 and Th2 subsets used in these experiments produced a significant amount of IFN-γ and IL-4, respectively (Fig. 1 E). The inhibitory effects of Th1 cells on osteoclastogenesis were completely abrogated on the BM-derived monocyte/macrophage precursor cells (BMMs) derived from IFN-γ receptor–deficient (Ifngr1 −/−) mice (28), indicating that IFN-γ is responsible for the Th1 cell–mediated inhibition of osteoclastogenesis (Fig. 1 F). We further analyzed the effects of CD4+CD25+ T reg cells on osteoclastogenesis in both systems, but they were found to have neither an enhancing nor an inhibitory effect (Fig. 1 G), suggesting that T reg cells are not directly related to the T cell–mediated regulation of bone resorption.
Figure 1.

Effects of Th1, Th2, and T reg cells on in vitro osteoclastogenesis. (A) Schematics of two culture systems for osteoclast differentiation and Th cell addition. In the RANKL–M-CSF system, mouse nonadherent BMCs were stimulated with M-CSF for 2 d and adherent cells were used as BMMs. After BMMs were stimulated with recombinant RANKL and M-CSF for 3 d, the formation of TRAP+ MNCs was analyzed. In the co-culture system, BMCs were co-cultured with osteoblasts stimulated with VitD3 and PGE2, and the formation of TRAP+ MNCs was observed 7 d after the addition of BMCs. (B) Inhibitory effects of Th1 and Th2 cells on TRAP+ MNC formation in the RANKL–M-CSF system. Th cells (4,000 or 20,000 cells/ml) were added at the same time as RANKL (day 0) with (black bars) or without (white bars) anti-CD3 mAb. n.d., not detected. (C) Inhibitory effects of Th1 and Th2 cells on TRAP+ MNC formation in the co-culture system. The same number of T cells as in B was added 2 d after BMC addition (day 2). (D) Microphotographs of the in vitro osteoclast formation systems in the presence of Th1 or Th2 cells (20,000 cells/ml) with anti-CD3 mAb (TRAP staining). (E) Cytokine profile of culture supernatants in the presence of Th cells and 1 μg/ml of soluble anti-CD3 mAb (the RANKL–M-CSF system on day 2). Without restimulation by anti-CD3 mAb, cytokine production was much less than this result and was difficult to detect after 2-d culture with osteoclast precursor cells (not depicted). (F) Effects of Th1 and Th2 cells (20,000 cells/ml plus anti-CD3 mAb) on WT or IFN-γ receptor–deficient (Ifngr1 −/−) osteoclast precursor cells. (G) Effects of isolated CD4+CD25+ T reg cells (4,000 or 20,000 cells/ml plus anti-CD3 mAb) on osteoclastogenesis in vitro. n.s., not significantly different. The survival of a considerable number of T reg cells after 3 d was confirmed by CFSE staining (not depicted).
Characterization of MNCs induced by Th2 cells and IL-4
It has been reported that the inhibitory effect of IFN-γ on osteoclastogenesis is reduced if the osteoclast precursor cells encounter RANKL before IFN-γ stimulation, suggesting that RANKL-prestimulated preosteoclasts are resistant to such inhibitory cytokines (29). To test whether the inhibitory effects of Th cells on osteoclastogenesis are also dependent on the differentiation stage of the osteoclast precursor cells, we added the Th cells to the osteoclast formation systems 1 d later than in the previous experiment. Interestingly, the inhibitory effects of Th cells were less (Fig. 2 A). Although Th1 cells inhibited the formation of TRAP+ MNCs under this condition, Th2 cells induced a normal number of TRAP+ MNCs in the RANKL–M-CSF system and even an increased number in the co-culture system (Fig. 2 A).
Figure 2.

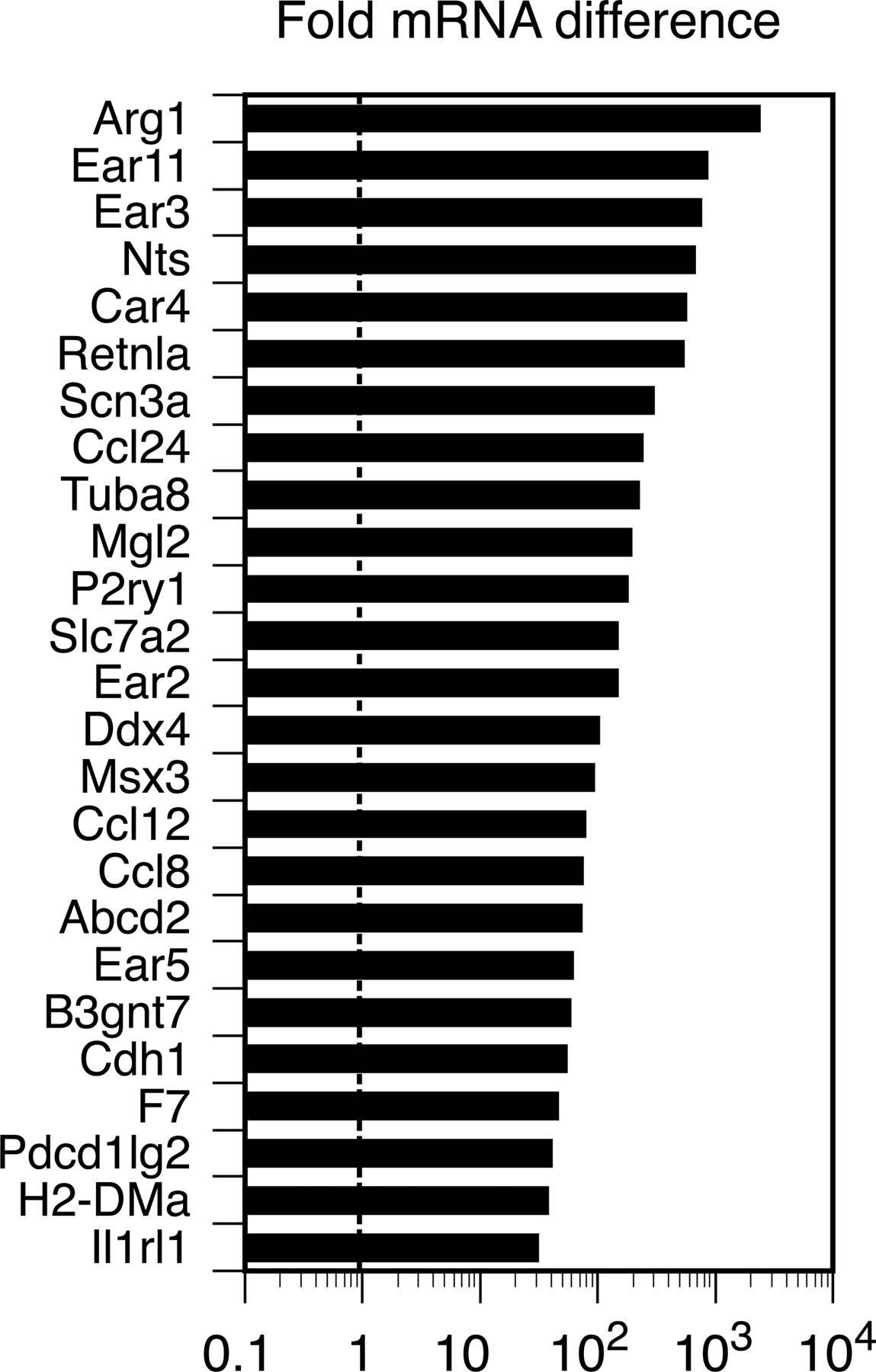
Formation of multinuclear cells with no bone-resorbing activity induced by Th2 cells and IL-4. (A) Inhibitory effects of Th1 and Th2 cells on osteoclastogenesis are reduced when T cells are added 1 d later. Th cells (20,000 cells/ml plus anti-CD3 mAb) were added on days 0 (at the same time as RANKL, gray bars) or 1 (black bars) to the RANKL–M-CSF system and on days 2 (2 d after BMC addition, gray bars) or 3 (black bars) to the co-culture system. (B) Microphotographs and (C) quantification of in vitro osteoclast formation (left, TRAP staining) and resorption pit formation (right). Th1 and Th2 cells (20,000 cells/ml plus anti-CD3 mAb) were added to WT or Stat6 −/− osteoclast precursor cells on day 1. (D) Effect of IL-4 on mRNA expression of osteoclast-related genes in osteoclast precursor cells (GeneChip analysis). Osteoclast precursor cells were stimulated by 10 ng/ml IL-4 from day 1 in the RANKL–M-CSF system and harvested on day 3. Fold mRNA difference was calculated by dividing the average difference of the IL-4–treated sample by that of the control sample. The expressions of most of the osteoclast-specific genes are down-regulated. (E) Reduced expression of NFATc1 protein in the cells treated with IL-4. Osteoclast precursor cells were stimulated by 10 ng/ml IL-4 from day 1 in the RANKL–M-CSF system, fixed on day 3, and stained with anti-NFATc1 antibody followed by Alexa Fluoro 488–labeled secondary antibody.
These results appeared to suggest that Th2 cells increase osteoclastogenesis under certain conditions, but the MNCs induced in the presence of Th2 cells were only weakly stained for TRAP (TRAPdim) and were incapable of bone resorption (Fig. 2, B and C). Even in the presence of Th2 cells, TRAP+ MNCs with bone-resorbing activity were formed from BMMs derived from mice deficient in Stat6, which is an essential mediator of IL-4 signaling (30), suggesting that IL-4 is involved in the formation of TRAPdim MNCs. Consistent with this, the addition of IL-4 to the RANKL–M-CSF system at the same time as RANKL strongly inhibited TRAP+ MNC formation, and the addition of IL-4 1 d later induced TRAPdim MNCs (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20061775/DC1). The effects of IL-4 were abrogated if added to Stat6-deficient cells, which generated TRAP+ MNCs that were able to resorb bone.
To further characterize the TRAPdim MNCs induced by Th2 cells through IL-4, we performed a genome-wide microarray screening of the genes expressed in the TRAPdim MNCs (31). TRAPdim MNCs induced by IL-4 expressed a high level of genes characteristic of activated macrophages, including chemokine ligands and enzymes involved in allergic responses (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20061775/DC1). The expression of most of the genes important for osteoclast differentiation or functions was decreased (Fig. 2 D). The expression of NFATc1, an essential transcription factor for osteoclastogenesis (31, 32), was also revealed to be down-regulated by immunostaining (Fig. 2 E). Thus, the TRAPdim MNCs induced by Th2 cells are not authentic osteoclasts but rather should be classified as macrophage polykarions.
Th17 cells stimulate osteoclastogenesis through osteoclastogenesis-supporting cells
Because the above results show that neither Th1, Th2, nor T reg cells enhance osteoclastogenesis, we next focused on a newly identified CD4+ T cell subset producing IL-17 called Th17 (33, 34). We suspected the Th17 subset to be a good candidate for the osteoclastogenic Th subset because it has been reported that IL-17 induces RANKL on mesenchymal cells and promotes osteoclastogenesis in vitro (25). Moreover, Th17 cells, which produce IL-17 (IL-17A) and its related cytokines such as IL-17F, but not IFN-γ or IL-4, are responsible for a variety of autoimmune inflammatory effects (9, 10). Recent studies suggest that TGF-β and IL-6 are essential for the initiation of Th17 differentiation and IL-23 is critical for expanding the population (35, 36). IL-23 is one of the IL-12 family cytokines and is a heterodimer consisting of the subunits p40 and p19 (9, 10). Even though IL-23 shares a p40 subunit and one of its receptor subunits (IL-12β1) with IL-12, IL-23 and IL-12 selectively play critical roles in the regulation of Th1 and Th17 polarization, respectively.
To obtain the Th17 cells, we stimulated CD4+ T cells with anti-CD3/CD28 mAbs in the presence of IL-23, anti–IFN-γ mAb, and anti–IL-4 mAb. In the presence of Th17 cells, TRAP+ MNCs were efficiently formed in the RANKL–M-CSF system (Fig. 3 A) and possessed bone-resorbing activity (not depicted), although the efficiency is a little less than in the control culture without the T cells. Moreover, in the co-culture system, the Th17 cells significantly enhanced the formation of TRAP+ MNCs (Fig. 3 A). Consistent with the previous reports, Th17 cells used in the above experiments produced a large amount of IL-17 but little IFN-γ, but Th1 cells did the opposite (Fig. 3 B). When Th17 cells were added to the co-culture system even in the absence of VitD3 and PGE2, the formation of TRAP+ MNCs was observed (Fig. 3 C). The osteoclastogenic effects of Th17 cells in the co-culture system was greatly reduced when we used Th17 cells derived from Il17 −/− mice (37), indicating that the IL-17 produced from Th17 cells is mainly responsible for the osteoclastogenic effects of Th17 cells. IL-23 or IL-17 had no effect on osteoclastogenesis in the RANKL–M-CSF system, but IL-17 promoted osteoclastogenesis in the co-culture system, suggesting that IL-17 does not directly act on osteoclast precursor cells but rather on osteoclastogenesis-supporting cells (Fig. 3 D). This is consistent with the previous report that IL-17 promotes osteoclastogenesis through the induction of RANKL on osteoblastic cells (25). These results show that Th17 is the only osteoclastogenic Th subset according to the currently accepted categorization of CD4+ T cells, and that Th17 cells facilitate osteoclastogenesis, possibly through IL-17–mediated induction of RANKL on osteoblastic cells.
Figure 3.

Enhanced osteoclastogenesis by Th17 cells in the co-culture system but not in the RANKL–M-CSF system. (A) Effects of Th1 and Th17 cells on the osteoclast differentiation systems. T cells (4,000 or 20,000 cells/ml plus anti-CD3 mAb) were added on day 1 to the RANKL–M-CSF system and on day 3 to the co-culture system. When the Th17 cells were added 1 d earlier, or in the absence of soluble anti-CD3 mAb, enhancement of osteoclastogenesis was not observed even in the co-culture system (not depicted). (B) Cytokine profile of the culture supernatants obtained on day 3 from the RANKL–M-CSF system in the presence of Th1 and Th17 cells derived from either WT or Il17 −/− mice under the conditions described in A. (C) Effects of Th1 and Th17 cells derived from either WT or Il17 −/− mice on the formation of TRAP+ MNCs or TRAP+ cells in the co-culture system in the absence of VitD3 and PGE2. T cells (20,000 cells/ml plus anti-CD3 mAb) were added on day 3. (D) Effects of recombinant IL-17 and IL-23 (2 or 10 ng/ml) on osteoclastogenesis in vitro. (E) Expression of RANKL on Th subsets. CD4+ T cells cultured in each of the Th conditions for 3 d were restimulated with 1 μg/ml of plate-bound anti-CD3 mAb for 4 h and subjected to flow cytometric analysis using anti-RANKL mAb. Without the restimulation by anti-CD3 mAb, RANKL expression was barely detectable (not depicted).
We evaluated the expression level of RANKL on the surface of Th cells and found that Th17 cells express a significant amount of RANKL, but Th1 cells express only a minimal amount (Fig. 3 E). Neither subset, however, exhibited promotive effects on osteoclastogenesis in the RANKL–M-CSF system (Fig. 3 A) or induced any TRAP+ cells when added to the BMM culture in the absence of exogenous soluble RANKL (not depicted). Thus, it is evident that the RANKL expressed by Th cells alone is not sufficient to activate osteoclastogenesis (see Discussion).
The IL-23–IL-17 axis plays a critical role in inflammation-induced bone destruction in vivo
To clarify the role IL-17 and IL-23 play in bone metabolism in vivo, we investigated the phenotype of Il17 −/− and Il23a −/− (lacking p19) (38) mice. There was no significant difference in bone mineral density as evaluated by dual- energy x-ray absorptiometry (Fig. 4 A). Microradiography also revealed no obvious abnormality in skeletal development (Fig. 4 B). Bone morphometric analyses revealed the parameters of bone resorption and formation to be normal even in the mutant mice (Fig. 4 C), indicating that neither IL-17 nor IL-23 is involved in the physiological regulation of bone homeostasis.
Figure 4.

Contribution of IL-17 and IL-23 to the physiological and pathological bone resorption. (A) Bone mineral densities (measured in 20 longitudinal divisions of the femurs), (B) micro-computed tomography (at 10% length above the distal epiphyseal plate), and (C) bone morphometic analyses of WT, Il17 −/−, and Il23a −/− mice at the age of 12 wk. (D) Histological examination of calvarial bones of WT, Il17 −/−, and Il23a −/− mice treated with LPS (hematoxylin and TRAP staining). The degree of bone destruction was analyzed by the number of osteoclasts and the area of the eroded surface (%).
To further investigate the role of IL-17 and IL-23 in the disease conditions characterized by enhanced osteoclastogenesis associated with T cell activation, we used an LPS- induced model of inflammatory bone destruction, which is not induced by an autoantigen but is T cell dependent (14, 39). Because it is well documented that IL-17 and IL-23 play an important role in the development of autoimmune arthritis (23, 24), we used this inflammatory bone destruction model to evaluate their role in the osteoclast-mediated destruction phase. LPS injection into the calvarial bone results in severe bone destruction associated with aberrant formation of osteoclasts in WT mice, but the level of bone destruction was much less pronounced and the osteoclast formation was significantly reduced in both the Il17 −/− mice and Il23a −/− mice (Fig. 4 D). These results suggest that the Th17 cells expanded through IL-23 stimulation are involved in the T cell–mediated osteoclastogenesis in vivo. In contrast, the bone destruction was enhanced and a greater number of osteoclasts were formed in Ifngr1 −/− Stat6 −/− mice, which are deficient in the response to both IFN-γ and IL-4 (Fig. 4 D), suggesting that IFN-γ and IL-4 may play a protective role against bone destruction by suppressing osteoclastogenesis associated with inflammation.
The above results suggest that IL-23–stimulated proliferation of Th17 cells, a major osteoclastogenic Th subset, plays a pivotal role in inflammatory bone destruction by inducing RANKL through an IL-17 effect on mesenchymal cells. Consistent with this, it has been reported that RANKL is abundantly expressed in the synovial fibroblasts of RA patients (16, 40) and the IL-17 concentration is elevated in the synovial fluid of RA patients (25). To explore the role of IL-23 in the induction of RANKL in RA, we investigated whether IL-23 was detected in the synovium of RA patients. Quantitative RT-PCR analysis revealed the mRNA of the p19 subunit of IL-23 (IL23A) in all the samples of the synovium derived from RA patients, and the expression level of IL23A positively correlated with that of RANKL (Fig. 5 A). A similar correlation was observed between RANKL and the p40 subunit shared by IL-12 and IL-23 (IL12B), but the expression of the p35 subunit specific for IL-12 (IL12A) did not correlate with that of RANKL, suggesting that IL-23 is an important determinant of arthritic bone destruction through the induction of RANKL. These results lend further support to the notion that the IL-23–IL-17 axis, rather than the IL-12–IFN-γ axis, is critical for the bone destruction phase of autoimmune arthritis.
Figure 5.

Regulation of RANKL-mediated osteoclastogenesis by the IL-23–IL-17 axis in the RA synovial tissue. (A) Correlation of the mRNA expression level of RANKL with that of IL23A (p19), IL12A (p35), or IL12B (p40) in the synovium of RA patients. The relative expressions of RANKL, IL23A, IL12A, and IL12B were all standardized using that of GAPDH. (B) Model of Th17-mediated bone destruction in autoimmune arthritis. Th17 cells function as an osteoclastogenic Th cell subset by stimulating local inflammation, inducing RANKL on osteoclastogenesis-supporting cells, and expressing RANKL on themselves and stimulating local inflammation, all of which contribute to an acceleration of osteoclastogenesis. It is notable that RANKL on Th17 cells alone is not sufficient for the induction of osteoclast differentiation (a dotted line). See Discussion for the details. Op, osteoclast precursor cell.
DISCUSSION
Coordinated activation of the innate and adaptive immune systems is essential for the efficient eradication of pathogens, but aberrant or prolonged activation under certain pathological conditions, such as autoimmune inflammation, results in tissue damage through the activation of effector cells. In autoimmune arthritis, it has long been a challenging question as to how the abnormality of the immune system induces the skeletal damage, although the infiltration of CD4+ T cells in the RA synovium is a pathogenetic hallmark and is undoubtedly linked to the bone destruction that ensues (3, 13, 14, 20). After RANKL was cloned and the high RANKL expression in the synovium was brought to light (16, 40), the importance of bone-resorbing osteoclasts came into general acceptance (3). Based on recent reports using genetically modified mice, the crucial role of osteoclasts in the inflammatory bone loss has been established (41, 42), but which CD4+ T cells cause the induction of osteoclasts, and by what mechanism, has remained elusive.
As RANKL is expressed in activated T cells, T cells may have the capacity to induce osteoclast differentiation by directly acting on osteoclast precursor cells (13, 26). However, because T cells also secrete a variety of cytokines and express membrane-bound factors other than RANKL, the effects of T cells on osteoclastogenesis should be dependent on the balance of positive and negative factors expressed by the T cells. As summarized in Fig. 5 B, the results in this study show that Th1 and Th2 cells inhibit osteoclastogenesis by acting on the precursor cells, mainly through IFN-γ and IL-4, respectively. The inhibitory effects of these cytokines were less observed in the co-culture system than in the RANKL–M-CSF system (Figs. 1, B and C, and 2 A). We infer that osteoblasts may provide membrane-bound RANKL and stimulate costimulatory signals for RANKL simultaneously, enabling the strong cell–cell contact between osteoblasts and osteoclast precursor cells and preventing the access of T cells or inhibitory cytokines to osteoclast precursor cells.
Previous observations that IL-12 and IL-18, which drive Th1 differentiation, both inhibit osteoclastogenesis via IFN-γ or GM-CSF (43, 44), and that IL-10, which is released from Th2 cells, also negatively regulates osteoclastogenesis (45) further support the negative role of Th1 and Th2 cells on osteoclastogenesis. In contrast, Th17 cells stimulated by IL-23 promote osteoclastogenesis mostly through production of IL-17 (Fig. 3, A and C). Therefore, the osteoclastogenic ability of Th17 cells does not require cell–cell contact with osteoclast precursor cells, but additional membrane-bound mediators such as RANKL and CD40L may also contribute (46, 47). IL-17 is known to act on the osteoclastogenesis-supporting cells to induce RANKL (25). It should be noted that the effect of IL-17 is not limited to this direct effect on the osteoclastogenesis-supporting cells. IL-17 facilitates local inflammation by recruiting and activating immune cells, which leads to an abundance of inflammatory cytokines such as TNF-α and IL-1 (9, 10). The inflammatory cytokines enhance RANKL expression on osteoclastogenesis-supporting cells and activate osteoclast precursor cells by synergizing with RANKL signaling. A relatively high expression of RANKL on Th17 cells may also participate in the enhanced osteoclastogenesis (Fig. 3 E). Collectively, Th17 cells can be called an osteoclastogenic Th subset not only because Th17 cells have positive effects on osteoclastogenesis in vitro, but also because they tip the balance of the microenvironments in favor of osteoclast differentiation.
It is worth noting that Th17 cells do not induce osteoclastogenesis in the absence of osteoblasts. This strongly suggests that RANKL expressed on Th17 cells alone is not sufficient to induce osteoclastogenesis, although this is partly because even Th17 cells produce a small amount of IFN-γ, which counterbalances the RANKL action. To understand the role of RANKL on T cells in more detail, we need mice of T cell–specific ablation of the RANKL gene, which are currently unavailable. But it is conceivable that RANKL expressed on adherent cells such as osteoblasts has more potent effects than that expressed on T cells. This mechanism may also explain why osteoclasts are formed only in the bone microenvironments, but it currently remains to be clarified. We consider the following explanations: (a) T cell expression of membrane-bound RANKL, which is more osteoclastogenic than the soluble form (48), is very low compared with that on osteoblasts; (b) costimulatory signals provided specifically by osteoblasts (12, 27) are missing in T cells; and (c) cell adhesion induces specific signals including those mediated by integrins, which are also important for osteoclastogenesis (49).
In our study, T reg cells had no apparent effect on osteoclastogenesis in vitro (Fig. 1 G). However, their function in the regulation of bone metabolism should be investigated in vivo considering the recent finding that the development of Th17 cells and T reg cells is coordinately regulated (10, 35, 36).
The importance of the IL-23–IL-17 axis in the autoimmune inflammation has been demonstrated in a variety of models of autoimmune diseases such as arthritis and encephalomyelitis (23, 24, 38). In arthritis models, IL-17 −/− mice were protected from the development of destructive arthritis (24), whereas collagen-induced arthritis is exacerbated in IFN-γ receptor–deficient mice (21, 22). The specific role of IL-23 compared with IL-12 in the development of arthritis has been clearly demonstrated by a genetic study using mice deficient in p19 and p35 (23). Based on these observations, the IL-23–IL-17 axis inducing Th17 cells, rather than the IL-12–IFN-γ axis inducing Th1 cells, is critical for the development of autoimmune arthritis. Our study also provides evidence that the IL-23–IL-17 axis plays a critical role even in a model of bone loss induced by local inflammation that is independent of autoimmunity (Fig. 4 D), suggesting that the IL-23–IL-17 axis is not only essential for the onset phase, but also for the destruction phase of autoimmune arthritis characterized by the T cell–mediated activation of osteoclastogenesis. Thus, Th17 cells, an osteoclastogenic subset, have profound relevance in the bone damage that takes place in autoimmune arthritis. The identification of T cell subsets in the synovium of arthritis is a challenging issue of great importance that should be pursued in a future study. Considering the strong inhibitory effects of Th1 cells on osteoclastogenesis, Th17 cells may be overwhelmingly dominant and the colocalization of Th1 cells is unlikely, at least under the microenvironments in which osteoclastogenesis efficiently occurs. The positive correlation between IL-23 and RANKL expression in the synovium of RA patients further suggests the importance of IL-23 in the regulation of local osteoclastogenesis through IL-17 (Fig. 5 A). Despite the importance of TGF-β and IL-6 in the initiation of Th17 development (10, 35, 36), Th17 cells can be obtained in an IL-23–stimulated culture system without adding exogenous TGF-β/IL-6, suggesting that the endogenous level of TGF-β/IL-6 may suffice for the initiation and that osteoclastogenic activity of Th17 cells is mainly determined by IL-23 under certain pathological conditions.
For the treatment of RA, there are several drugs available, most of which were developed to modulate immune reactions. The antirheumatic drugs are effective in treating pain and inflammation, but patients still fairly frequently have to undergo joint replacement surgery because of the progressive bone damage despite long-term treatment with antirheumatic drugs. Therefore, it is clinically an urgent issue to establish a method to prevent such persistent bone destruction (3). Although rheumatologists are now aware of the great impact that anti-TNF therapy has had on the management of RA (50), it is still not determined whether all patients respond to the therapy or, indeed, whether bone destruction will be completely prevented by it. Recent progress in understanding the mechanism of bone loss in RA has provided promising new strategies, one of which is an anti-RANKL antibody directly suppressing RANKL-mediated osteoclastogenesis (51). As we have demonstrated a new role of Th17 in the context of bone damage in RA, the significance of the IL-23– IL-17 axis extends beyond the simple initiation or development of the autoimmunity. Because osteoclastogenic Th17 cells link the autoimmune inflammation to bone damage, inhibition of this axis has the potential of a doubly beneficial impact on RA, i.e., in the context of both the immune and skeletal systems, and thus appears to be an ideal therapeutic strategy for ameliorating the bone destruction associated with T cell activation.
MATERIALS AND METHODS
Mice.
Ifngr1 −/− (28), Stat6 −/− (30), Il17 −/− (37), and Il23a −/− mice (38) were described previously. All the mice were maintained under specific pathogen-free conditions and were backcrossed to C57BL/6 mice. All animal experiments were performed with the approval of the Animal Study Committee of Tokyo Medical and Dental University and conformed to relevant guidelines and laws.
Analysis of bone phenotype and LPS-induced bone destruction.
The mice were subjected to histomorphometric and microradiographic examinations as described previously (27). 8-wk-old mice were injected with 25 mg/kg body weight LPS (Sigma-Aldrich) subperiosteally in the calvarial bone. After 5 d, calvarial bones were analyzed as described previously using decalcified paraffin sections (14).
In vitro assays for osteoclast differentiation and function.
In vitro osteoclast differentiation was described previously (27, 52). For the RANKL–M-CSF system, we cultured BMCs with 10 ng/ml M-CSF (R&D Systems) for 2 d and used them as BMMs. The cells were cultured with 50 ng/ml RANKL (PeproTech) and 10 ng/ml M-CSF for 3 d, and TRAP+ multinucleated (more than three nuclei) cells were counted. The co-culture of osteoblasts derived from mouse calvarial cells and BMCs was performed in the presence of 10−8 M VitD3 (Wako) and 10−6 M PGE2 (Wako) for 7 d. For the assessment of the bone-resorbing function of osteoclasts, we cultured osteoclast precursors on a hydroxy appatite–coated disc (Osteologic; BD Biosciences). After the culture period, the cells were washed away as described in the manufacturer's protocol by 6% NaOCl and 5.2% NaCl.
Th cell differentiation.
CD4+ T cells were purified from the spleen using a magnetic sorter and anti-CD4 microbeads (MACS; Miltenyi Biotec). The purity of the CD4+ T cells was >95%. These CD4+ T cells were stimulated with a plate-bound anti-CD3 mAb and anti-CD28 mAb (1 μg/ml each) for 3 d in the presence of (a) 10 ng/ml IL-12 and 10 μg/ml anti–IL-4 mAb for the Th1 cells, (b) 10 ng/ml IL-4 and 10 μg/ml anti–IFN-γ mAb for the Th2 cells, and (c) 10 ng/ml IL-23 along with 10 μg/ml each of anti–IFN-γ and anti–IL-4 mAbs for the Th17 cells. When indicated, the T cells were added to the culture system with 1 μg/ml anti-CD3 mAb for restimulation. All the antibodies were purchased from BD Biosciences except for the anti-RANKL mAb (provided by H. Yagita, Juntendo University School of Medicine, Tokyo, Japan). Recombinant IL-17 and the other cytokines were purchased from Genzyme and R&D Systems, respectively. T reg cells were purified using a MACS CD4+CD25+ Regulatory T Cell Isolation kit.
Analysis of mRNAs expressed in RA synovial tissues.
Synovial tissues were obtained at the time of total knee arthroplasty from five patients (age range, 55–70 yr) who fulfilled the American College of Rheumatology criteria and gave informed consent (16). The experiments were performed with the approval of the institutional ethical committee. The tissues were minced and homogenized in Sepasol-RNA (Nacalai Tesque), and total RNA was extracted and purified according to the manufacturer's protocol.
GeneChip analysis and quantitative RT-PCR.
Total RNA (15 μg) was used for cDNA synthesis by reverse transcription followed by the synthesis of biotinylated cRNA through in vitro transcription. After cRNA fragmentation, we performed hybridization with a mouse A430 GeneChip (Affymetrix, Inc.) (31). We performed quantitative RT-PCR using a LightCycler (Roche), as described previously (52). The following primers were used: IL23A: 5′-CTGCTTGCAAAGGATCCACC-3′ (sense), 5′-TTGAAGCGGAGAAGGAGACG-3′ (antisense); IL12A: 5′-AGCCTCCTCCTTGTGGCTA-3′ (sense), 5′-TGTGCTGGTTTTATCTTTTGTG-3′ (antisense); IL12B: 5′-TCACAAAGGAGGCGAGGTT-3′ (sense), 5′-ATGACCTCAATGGGCAGACTC-3′ (antisense); and RANKL: 5′-AACCAGATGGGATGTCGGTGGCATTA-3′ (sense), 5′-AGCGATGGTGGATGGCTCATGGTTAG-3′ (antisense). The level of mRNA expression was normalized with that of GAPDH expression in Fig. 5 A.
Statistical analyses.
All data were expressed as the mean ± SEM (n = 4, unless otherwise indicated). Mann-Whitney U test was used for statistical analyses (*, P < 0.05; **, P < 0.01), and comparisons were made between each sample and the control (not treated with T cells/cytokines or WT mice).
Online supplemental material.
Fig. S1 shows the effect of recombinant IL-4 on osteoclast precursor cells derived from WT or Stat6 −/− mice in the RANKL–M-CSF system. Fig. S2 shows the list of genes whose expression was increased by IL-4 in osteoclast precursor cells (GeneChip analysis). Figs. S1 and S2 are available at http://www.jem.org/cgi/content/full/jem.20061775/DC1.
Supplemental Material
Acknowledgments
We are grateful to H. Yagita for providing anti-RANKL mAb. We also thank T. Taniguchi, S. Hida, S. Taki, H. Murayama, J. Taka, M. Asagiri, M. Shinohara, T. Nakashima, H.J. Gober, T. Koga, Y. Sato, and I. Takayanagi for fruitful discussion and assistance.
This work was supported in part by Grant-in-Aid for Creative Scientific Research from Japan Society for the Promotion of Science (JSPS), SORST program of JST, grants for Genome Network Project from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT), grants for the 21st century COE program from MEXT, Grants-in-Aid for Scientific Research from MEXT, Health Sciences Research Grants from the Ministry of Health, Labor and Welfare of Japan, and grants from the Naito Foundation, Suzuken Memorial Foundation, Uehara Memorial Foundation, Kato Memorial Bioscience Foundation, Cell Science Research Foundation, Inamori Foundation, and the Nakatomi Foundation.
The authors have no conflicting financial interests.
Abbreviations used: BMC, BM cell; BMM, BM-derived monocyte/macrophage precursor cell; M-CSF, macrophage colony-stimulating factor; MNC; multinucleated cell; PGE2, prostaglandin E2; RA, rheumatoid arthritis; RANKL, receptor activator of NF-κB ligand; TRAP, tartrate-resistant acid phosphatase; T reg, regulatory T; VitD3, 1,25 (OH)2 vitamin D3.
References
- 1.Walsh, M.C., N. Kim, Y. Kadono, J. Rho, S.Y. Lee, J. Lorenzo, and Y. Choi. 2006. Osteoimmunology: interplay between the immune system and bone metabolism. Annu. Rev. Immunol. 24:33–63. [DOI] [PubMed] [Google Scholar]
- 2.Takayanagi, H. 2005. Inflammatory bone destruction and osteoimmunology. J. Periodontal Res. 40:287–293. [DOI] [PubMed] [Google Scholar]
- 3.Sato, K., and H. Takayanagi. 2006. Osteoclasts, rheumatoid arthritis, and osteoimmunology. Curr. Opin. Rheumatol. 18:419–426. [DOI] [PubMed] [Google Scholar]
- 4.Theill, L.E., W.J. Boyle, and J.M. Penninger. 2002. RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu. Rev. Immunol. 20:795–823. [DOI] [PubMed] [Google Scholar]
- 5.Teitelbaum, S.L., and F.P. Ross. 2003. Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 4:638–649. [DOI] [PubMed] [Google Scholar]
- 6.Boyle, W.J., W.S. Simonet, and D.L. Lacey. 2003. Osteoclast differentiation and activation. Nature. 423:337–342. [DOI] [PubMed] [Google Scholar]
- 7.Suda, T., N. Takahashi, N. Udagawa, E. Jimi, M.T. Gillespie, and T.J. Martin. 1999. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr. Rev. 20:345–357. [DOI] [PubMed] [Google Scholar]
- 8.Mosmann, T.R., H. Cherwinski, M.W. Bond, M.A. Giedlin, and R.L. Coffman. 1986. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 136:2348–2357. [PubMed] [Google Scholar]
- 9.Dong, C. 2006. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat. Rev. Immunol. 6:329–333. [DOI] [PubMed] [Google Scholar]
- 10.Weaver, C.T., L.E. Harrington, P.R. Mangan, M. Gavrieli, and K.M. Murphy. 2006. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 24:677–688. [DOI] [PubMed] [Google Scholar]
- 11.Sakaguchi, S. 2005. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat. Immunol. 6:345–352. [DOI] [PubMed] [Google Scholar]
- 12.Takayanagi, H. 2005. Mechanistic insight into osteoclast differentiation in osteoimmunology. J. Mol. Med. 83:170–179. [DOI] [PubMed] [Google Scholar]
- 13.Kong, Y.Y., U. Feige, I. Sarosi, B. Bolon, A. Tafuri, S. Morony, C. Capparelli, J. Li, R. Elliott, S. McCabe, et al. 1999. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 402:304–309. [DOI] [PubMed] [Google Scholar]
- 14.Takayanagi, H., K. Ogasawara, S. Hida, T. Chiba, S. Murata, K. Sato, A. Takaoka, T. Yokochi, H. Oda, K. Tanaka, et al. 2000. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-γ. Nature. 408:600–605. [DOI] [PubMed] [Google Scholar]
- 15.Hofbauer, L.C., D.L. Lacey, C.R. Dunstan, T.C. Spelsberg, B.L. Riggs, and S. Khosla. 1999. Interleukin-1β and tumor necrosis factor-α, but not interleukin-6, stimulate osteoprotegerin ligand gene expression in human osteoblastic cells. Bone. 25:255–259. [DOI] [PubMed] [Google Scholar]
- 16.Takayanagi, H., H. Iizuka, T. Juji, T. Nakagawa, A. Yamamoto, T. Miyazaki, Y. Koshihara, H. Oda, K. Nakamura, and S. Tanaka. 2000. Involvement of receptor activator of nuclear factor κB ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis. Arthritis Rheum. 43:259–269. [DOI] [PubMed] [Google Scholar]
- 17.Dolhain, R.J., A.N. van der Heiden, N.T. ter Haar, F.C. Breedveld, and A.M. Miltenburg. 1996. Shift toward T lymphocytes with a T helper 1 cytokine-secretion profile in the joints of patients with rheumatoid arthritis. Arthritis Rheum. 39:1961–1969. [DOI] [PubMed] [Google Scholar]
- 18.Smolen, J.S., M. Tohidast-Akrad, A. Gal, M. Kunaver, G. Eberl, P. Zenz, A. Falus, and G. Steiner. 1996. The role of T-lymphocytes and cytokines in rheumatoid arthritis. Scand. J. Rheumatol. 25:1–4. [DOI] [PubMed] [Google Scholar]
- 19.Husby, G., and R.C. Williams Jr. 1985. Immunohistochemical studies of interleukin-2 and γ-interferon in rheumatoid arthritis. Arthritis Rheum. 28:174–181. [DOI] [PubMed] [Google Scholar]
- 20.Kinne, R.W., E. Palombo-Kinne, and F. Emmrich. 1997. T-cells in the pathogenesis of rheumatoid arthritis villains or accomplices? Biochim. Biophys. Acta. 1360:109–141. [DOI] [PubMed] [Google Scholar]
- 21.Manoury-Schwartz, B., G. Chiocchia, N. Bessis, O. Abehsira-Amar, F. Batteux, S. Muller, S. Huang, M.C. Boissier, and C. Fournier. 1997. High susceptibility to collagen-induced arthritis in mice lacking IFN-γ receptors. J. Immunol. 158:5501–5506. [PubMed] [Google Scholar]
- 22.Vermeire, K., H. Heremans, M. Vandeputte, S. Huang, A. Billiau, and P. Matthys. 1997. Accelerated collagen-induced arthritis in IFN-γ receptor-deficient mice. J. Immunol. 158:5507–5513. [PubMed] [Google Scholar]
- 23.Murphy, C.A., C.L. Langrish, Y. Chen, W. Blumenschein, T. McClanahan, R.A. Kastelein, J.D. Sedgwick, and D.J. Cua. 2003. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 198:1951–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakae, S., A. Nambu, K. Sudo, and Y. Iwakura. 2003. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J. Immunol. 171:6173–6177. [DOI] [PubMed] [Google Scholar]
- 25.Kotake, S., N. Udagawa, N. Takahashi, K. Matsuzaki, K. Itoh, S. Ishiyama, S. Saito, K. Inoue, N. Kamatani, M.T. Gillespie, et al. 1999. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Invest. 103:1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horwood, N.J., V. Kartsogiannis, J.M. Quinn, E. Romas, T.J. Martin, and M.T. Gillespie. 1999. Activated T lymphocytes support osteoclast formation in vitro. Biochem. Biophys. Res. Commun. 265:144–150. [DOI] [PubMed] [Google Scholar]
- 27.Koga, T., M. Inui, K. Inoue, S. Kim, A. Suematsu, E. Kobayashi, T. Iwata, H. Ohnishi, T. Matozaki, T. Kodama, et al. 2004. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 428:758–763. [DOI] [PubMed] [Google Scholar]
- 28.Huang, S., W. Hendriks, A. Althage, S. Hemmi, H. Bluethmann, R. Kamijo, J. Vilcek, R.M. Zinkernagel, and M. Aguet. 1993. Immune response in mice that lack the interferon-γ receptor. Science. 259:1742–1745. [DOI] [PubMed] [Google Scholar]
- 29.Huang, W., R.J. O'Keefe, and E.M. Schwarz. 2003. Exposure to receptor- activator of NFκB ligand renders pre-osteoclasts resistant to IFN-γ by inducing terminal differentiation. Arthritis Res. Ther. 5:R49–R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takeda, K., T. Tanaka, W. Shi, M. Matsumoto, M. Minami, S. Kashiwamura, K. Nakanishi, N. Yoshida, T. Kishimoto, and S. Akira. 1996. Essential role of Stat6 in IL-4 signalling. Nature. 380:627–630. [DOI] [PubMed] [Google Scholar]
- 31.Takayanagi, H., S. Kim, T. Koga, H. Nishina, M. Isshiki, H. Yoshida, A. Saiura, M. Isobe, T. Yokochi, J. Inoue, et al. 2002. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell. 3:889–901. [DOI] [PubMed] [Google Scholar]
- 32.Asagiri, M., K. Sato, T. Usami, S. Ochi, H. Nishina, H. Yoshida, I. Morita, E.F. Wagner, T.W. Mak, E. Serfling, and H. Takayanagi. 2005. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 202:1261–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harrington, L.E., R.D. Hatton, P.R. Mangan, H. Turner, T.L. Murphy, K.M. Murphy, and C.T. Weaver. 2005. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6:1123–1132. [DOI] [PubMed] [Google Scholar]
- 34.Park, H., Z. Li, X.O. Yang, S.H. Chang, R. Nurieva, Y.H. Wang, Y. Wang, L. Hood, Z. Zhu, Q. Tian, and C. Dong. 2005. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 6:1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mangan, P.R., L.E. Harrington, D.B. O'Quinn, W.S. Helms, D.C. Bullard, C.O. Elson, R.D. Hatton, S.M. Wahl, T.R. Schoeb, and C.T. Weaver. 2006. Transforming growth factor-β induces development of the TH17 lineage. Nature. 441:231–234. [DOI] [PubMed] [Google Scholar]
- 36.Bettelli, E., Y. Carrier, W. Gao, T. Korn, T.B. Strom, M. Oukka, H.L. Weiner, and V.K. Kuchroo. 2006. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 441:235–238. [DOI] [PubMed] [Google Scholar]
- 37.Nakae, S., Y. Komiyama, A. Nambu, K. Sudo, M. Iwase, I. Homma, K. Sekikawa, M. Asano, and Y. Iwakura. 2002. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 17:375–387. [DOI] [PubMed] [Google Scholar]
- 38.Cua, D.J., J. Sherlock, Y. Chen, C.A. Murphy, B. Joyce, B. Seymour, L. Lucian, W. To, S. Kwan, T. Churakova, et al. 2003. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 421:744–748. [DOI] [PubMed] [Google Scholar]
- 39.Ukai, T., Y. Hara, and I. Kato. 1996. Effects of T cell adoptive transfer into nude mice on alveolar bone resorption induced by endotoxin. J. Periodontal Res. 31:414–422. [DOI] [PubMed] [Google Scholar]
- 40.Gravallese, E.M., C. Manning, A. Tsay, A. Naito, C. Pan, E. Amento, and S.R. Goldring. 2000. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 43:250–258. [DOI] [PubMed] [Google Scholar]
- 41.Pettit, A.R., H. Ji, D. von Stechow, R. Muller, S.R. Goldring, Y. Choi, C. Benoist, and E.M. Gravallese. 2001. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am. J. Pathol. 159:1689–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Redlich, K., S. Hayer, R. Ricci, J.P. David, M. Tohidast-Akrad, G. Kollias, G. Steiner, J.S. Smolen, E.F. Wagner, and G. Schett. 2002. Osteoclasts are essential for TNF-α-mediated joint destruction. J. Clin. Invest. 110:1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horwood, N.J., J. Elliott, T.J. Martin, and M.T. Gillespie. 2001. IL-12 alone and in synergy with IL-18 inhibits osteoclast formation in vitro. J. Immunol. 166:4915–4921. [DOI] [PubMed] [Google Scholar]
- 44.Udagawa, N., N.J. Horwood, J. Elliott, A. Mackay, J. Owens, H. Okamura, M. Kurimoto, T.J. Chambers, T.J. Martin, and M.T. Gillespie. 1997. Interleukin-18 (interferon-γ–inducing factor) is produced by osteoblasts and acts via granulocyte/macrophage colony-stimulating factor and not via interferon-γ to inhibit osteoclast formation. J. Exp. Med. 185:1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hong, M.H., H. Williams, C.H. Jin, and J.W. Pike. 2000. The inhibitory effect of interleukin-10 on mouse osteoclast formation involves novel tyrosine-phosphorylated proteins. J. Bone Miner. Res. 15:911–918. [DOI] [PubMed] [Google Scholar]
- 46.Kadono, Y., F. Okada, C. Perchonock, H.D. Jang, S.Y. Lee, N. Kim, and Y. Choi. 2005. Strength of TRAF6 signalling determines osteoclastogenesis. EMBO Rep. 6:171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gohda, J., T. Akiyama, T. Koga, H. Takayanagi, S. Tanaka, and J. Inoue. 2005. RANK-mediated amplification of TRAF6 signaling leads to NFATc1 induction during osteoclastogenesis. EMBO J. 24:790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hikita, A., Y. Kadono, H. Chikuda, A. Fukuda, H. Wakeyama, H. Yasuda, K. Nakamura, H. Oda, T. Miyazaki, and S. Tanaka. 2005. Identification of an alternatively spliced variant of Ca2+-promoted Ras inactivator as a possible regulator of RANKL shedding. J. Biol. Chem. 280:41700–41706. [DOI] [PubMed] [Google Scholar]
- 49.Ross, F.P., and S.L. Teitelbaum. 2005. αvβ3 and macrophage colony- stimulating factor: partners in osteoclast biology. Immunol. Rev. 208:88–105. [DOI] [PubMed] [Google Scholar]
- 50.Palladino, M.A., F.R. Bahjat, E.A. Theodorakis, and L.L. Moldawer. 2003. Anti-TNF-α therapies: the next generation. Nat. Rev. Drug Discov. 2:736–746. [DOI] [PubMed] [Google Scholar]
- 51.McClung, M.R., E.M. Lewiecki, S.B. Cohen, M.A. Bolognese, G.C. Woodson, A.H. Moffett, M. Peacock, P.D. Miller, S.N. Lederman, C.H. Chesnut, et al. 2006. Denosumab in postmenopausal women with low bone mineral density. N. Engl. J. Med. 354:821–831. [DOI] [PubMed] [Google Scholar]
- 52.Kim, Y., K. Sato, M. Asagiri, I. Morita, K. Soma, and H. Takayanagi. 2005. Contribution of nuclear factor of activated T cells c1 to the transcriptional control of immunoreceptor osteoclast-associated receptor but not triggering receptor expressed by myeloid cells-2 during osteoclastogenesis. J. Biol. Chem. 280:32905–32913. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}