

Abstract
Members of the α- and β-subfamily of herpesviridae encode glycoproteins that specifically bind to the Fc part of immunoglobulin (Ig)G. Plasma membrane resident herpesviral Fc receptors seem to prevent virus-specific IgG from activating antibody-dependent effector functions. We show that the mouse cytomegalovirus (MCMV) molecule fcr-1 promotes a rapid down-regulation of NKG2D ligands murine UL16-binding protein like transcript (MULT)-1 and H60 from the cell surface. Deletion of the m138/fcr-1 gene from the MCMV genome attenuates viral replication to natural killer (NK) cell response in an NKG2D-dependent manner in vivo. A distinct N-terminal module within the fcr-1 ectodomain in conjunction with the fcr-1 transmembrane domain was required to dispose MULT-1 to degradation in lysosomes. In contrast, down-modulation of H60 required the complete fcr-1 ectodomain, implying independent modes of fcr-1 interaction with the NKG2D ligands. The results establish a novel viral strategy for down-modulating NK cell responses and highlight the impressive diversity of Fc receptor functions.
NK cells play a crucial role in the control of many viruses (1, 2). The recognition of virus-infected cells by NK cells is regulated by the balance of signaling via inhibitory and stimulatory receptors (1, 3). NKG2D is a dominant activating NK cell receptor involved in immune responses to viruses (3). It is also expressed by activated CD8+ T cells, subsets of γδ T cells, and NK1.1+ T cells (4). Several mouse NKG2D ligands can be distinguished as follows: murine UL16-binding protein like transcript (MULT)-1 (5), the minor histocompatibility antigen H60 (6), and the retinoic acid early inducible (RAE)-1 isoforms (7). Both human cytomegalovirus (HCMV) and mouse cytomegalovirus (MCMV) encode proteins that negatively regulate the cell surface expression of NKG2D ligands and thus compromise the efficacy of NK and T cell responses (2). Among the members of the MCMV m145 gene family, the m152-encoded gp40 serves as a modulator of the RAE-1 family of NKG2D ligands (8, 9). The product of the MCMV m155 gene down-modulates H60 (10, 11), whereas the m145-encoded protein affects the expression of MULT-1 (12).
Members of the α- and β-subfamily of the herpesviruses encode transmembrane glycoproteins that selectively bind IgG via its Fc domain. The viral Fcγ receptors (vFcγRs) are expressed on the surface of infected cells (13, 14). According to the model of “antibody bipolar bridging,” the IgG molecule that recognizes an epitope on an infected cell is sequestered via its Fc part by vFcγR. Thus, the engagement of the IgG Fc domain may prevent antiviral effector activities such as the triggering of NK cells via cellular FcγRs and the activation of its complement. MCMV expresses a vFcγR encoded by the m138/fcr-1 gene (15). Deletion of this gene results in a dramatic virus attenuation in vivo irrespective of the presence of antibodies, suggesting that the observed phenotype is not only dependent on the fcr-1 property to bind IgG (16).
A detailed comparison of the effects of WT and mutant MCMV infections on cellular H60 expression level suggested the presence of an additional m155 independent inhibitory function encoded by MCMV genome (8, 10). Furthermore, the up-regulation of MULT-1 mRNA and only a modest up-regulation of surface MULT-1 on cells infected with Δm145 virus also opened the possibility for an additional viral inhibitor of MULT-1 (12). Systematic analysis of MCMV deletion mutants guided our search to a single gene, m138/fcr-1, as a causal principle, being able to down-modulate both NKG2D ligands. This finding provides an explanation for the IgG-independent attenuation of Δm138/fcr-1 MCMV and demonstrates novel immune-evasive functions of viral FcγR.
RESULTS AND DISCUSSION
MCMV down-modulation of NKG2D ligands requires fcr-1
The MCMV m155 and m145 gene products prevent the surface expression of H60 and MULT-1 on MCMV-infected cells, respectively (10, 12). However, the deletion of these genes from MCMV genome could not fully explain H60 and MULT-1 down-regulation. This prompted us to continue in vitro screening for additional inhibitors using MCMV mutants lacking different sets of nonessential genes. NIH3T3 cells were infected with mutant MCMVs and analyzed for surface density of NKG2D ligands using a NKG2D tetramer. As controls, WT MCMV and the mutant virus Δ6 lacking most of the m145 gene family members including m145, m152, and m155 were used. In line with previous results (8, 10), the infection with WT MCMV resulted in a strong down-modulation of NKG2D ligands, whereas cells infected with Δ6 virus remained positive (Fig. 1 a). Interestingly, the infection with ΔA1 MCMV mutant lacking the gene region m128 through m138 also preserved NKG2D ligand expression. Because the gene encoding the MCMV receptor for the Fc fragment of IgG, m138/fcr-1 is located in this region, we examined whether this protein might be involved. Indeed, two independent mutants possessing only the deletion of m138/fcr-1 gene were unable to down-regulate NKG2D ligands to the level of WT MCMV (Fig. 1 a). Next, we analyzed which of the NKG2D ligands are regulated by m138/fcr-1. The specific down-modulation of MULT-1 by fcr-1 was demonstrated because all three viral mutants lacking m138/fcr-1 were unable to affect its surface expression (Fig. 1 b). In contrast, the m138/fcr-1 revertant virus (RMS95.9) was able to down-modulate surface MULT-1. The possibility that fcr-1 caused down-modulation of surface MULT-1 relating to its ability to bind IgG via the Fc domain (15) was ruled out by the use of F(ab)2 fragments of anti–MULT-1 mAb (Fig. 1 c). Experiments performed with H60-3T3 transfectants revealed that the surface expression of H60 is also regulated by m138/fcr-1, although not as efficiently as in the case of MULT-1 (Fig. 1 d). To assess the specificity of the fcr-1 interaction with potential targets, surface expression of RAE-1αβγ ligands of the NKG2D receptor and the MHC class I allele H-2Kd was analyzed. The cells were infected with WT MCMV, Δm138/fcr-1, Δm152, and the virus strain lacking m06 in addition to m152. The down-modulation of RAE-1 (Fig. 1 e) and MHC class I H-2Kd (Fig. 1 f) turned out to be independent of the presence or absence of fcr-1. Altogether, the data established that fcr-1 selectively down-modulates expression of NKG2D ligands MULT-1 and H60 on the surface of infected cells.
Figure 1.

Selective down-modulation of NKG2D ligands MULT-1 and H60 by MCMV fcr-1. 12 h p.i., NIH3T3 cells were analyzed for the expression of NKG2D ligands using the NKG2D-PE tetramer (a), whereas SVEC4-10 cells were stained for the expression of MULT-1 with rat anti–MULT-1 mAb (b) or with F(ab)2 fragments of rat anti–MULT-1 mAb (c) followed by anti–rat-PE. H60-3T3 cells were stained 16 h p.i. with rat anti-H60 mAb followed by anti–rat-FITC (d). 12 h p.i. B12 cells were stained with anti-RAE–1αβγ mAb (e) or anti–H-2Kd mAb (f) followed by FITC-labeled secondary Ab. Irrelevant primary mAbs were used as a negative control (thin line). Infection was performed with 1 PFU per cell of indicated viruses.
Down-modulation of NKG2D ligands by fcr-1 is independent of other viral proteins
The MCMV m145- and m155-encoded glycoproteins have been demonstrated to act as autonomous inhibitors of MULT-1 and H60, respectively. To test whether fcr-1 can also inhibit MULT-1 and H60 on its own or if it requires MCMV coregulators, recombinant Vaccinia viruses (VV) expressing fcr-1, MULT-1, and H60 were used (Fig. 2 a). CV1 cells infected with MULT-1–VV showed a high expression of surface MULT-1, similar to the cells coinfected with MULT-1-VV and WT-VV. In contrast, coinfection with MULT-1–VV and VV expressing m145 or fcr-1 showed a similar extent of MULT-1 down-modulation. Analogous results confirmed that fcr-1 is sufficient to affect surface expression of H60. The specific and autonomous effect of fcr-1 on MULT-1 as well as on H60 was further confirmed after its transient coexpression with these NKG2D ligands in 293 HEK cells (Fig. 2 b). Although cotransfection with a HCMV US2 plasmid preserved high levels of MULT-1 and H60, cotransfection of the m138/fcr-1 gene resulted in much lower surface densities of both NKG2D ligands. We concluded that fcr-1 requires no other viral proteins for the down-modulation of MULT-1 and H60.
Figure 2.

fcr-1 is efficient both in vivo and in isolated in vitro conditions. (a) CV-1 cells infected with indicated Vaccinia viruses for 16 h or (b) HEK 293T cells cotransfected with indicated plasmids for 24 h were analyzed for the expression of MULT-1 or H60. Irrelevant primary mAbs were used as a negative control (thin line). NK cell–depleted or undepleted BALB/c mice were injected i.v. with (c) 2 × 105 PFU of indicated viruses or (d) 104 BALB/c MEF cells were infected for 12 h with 2 PFU of indicated viruses. (e) Untreated BALB/c mice or mice treated with anti-NKG2D mAb were i.v. injected with 2 × 105 PFU of indicated viruses. Titers in the lungs of individual mice (circles) 3 d p.i. and median values (horizontal bars) are shown. The differences between the groups of untreated mice infected with Δm138/fcr-1 and WT or m138/fcr-1 revertant virus were significant (P < 0.005). Depletion of NK cells and blockade of NKG2D resulted in a significant increase (P < 0.005 for c and d; P < 0.025 for e) of Δm138/fcr-1 virus titers.
NKG2D-dependent early attenuation of Δm138/fcr-1 replication in vivo
We have previously found that MCMV lacking m138/fcr-1 is severely attenuated in vivo during the primary phase of infection (16) in which IgG does not contribute to the immune control (17). This already suggests an additional function of fcr-1, unrelated to its IgG-binding capacity. Therefore, we tested whether the attenuation of Δm138/fcr-1 mutant is as the result of its increased susceptibility to NK cells in vivo. BALB/c mice were infected with Δm138/fcr-1 virus or WT MCMV and the viral load in different organs was determined 3 d post-infection (p.i.). Injection of rabbit antiserum to asialo-GM1 (AGM1), which leads to the depletion of NK cells, restored the replication of Δm138/fcr-1 to the same level as in WT control virus on day 3 p.i. (Fig. 2 c). Similar results were obtained when mice received an i.v. injection of MCMV-infected cells instead of infectious virus (Fig. 2 d). The mice infected with Δm138/fcr-1 showed significantly lower viral titers as compared with mice that received cells infected with m138 revertant virus RMS95.9. Importantly, injection of NKG2D-specific blocking antibodies rescued the titers of Δm138/fcr-1 mutant to the WT level (Fig. 2 e). These data demonstrate that fcr-1 enhances MCMV replication in vivo in an NKG2D- and NK cell–dependent manner and reconcile the seemingly paradoxical finding of Δm138/fcr-1 MCMV attenuation in IgG-deficient mice.
Identification of Ig-like domains of the fcr-1 ectodomain down-modulating MULT-1 and H60
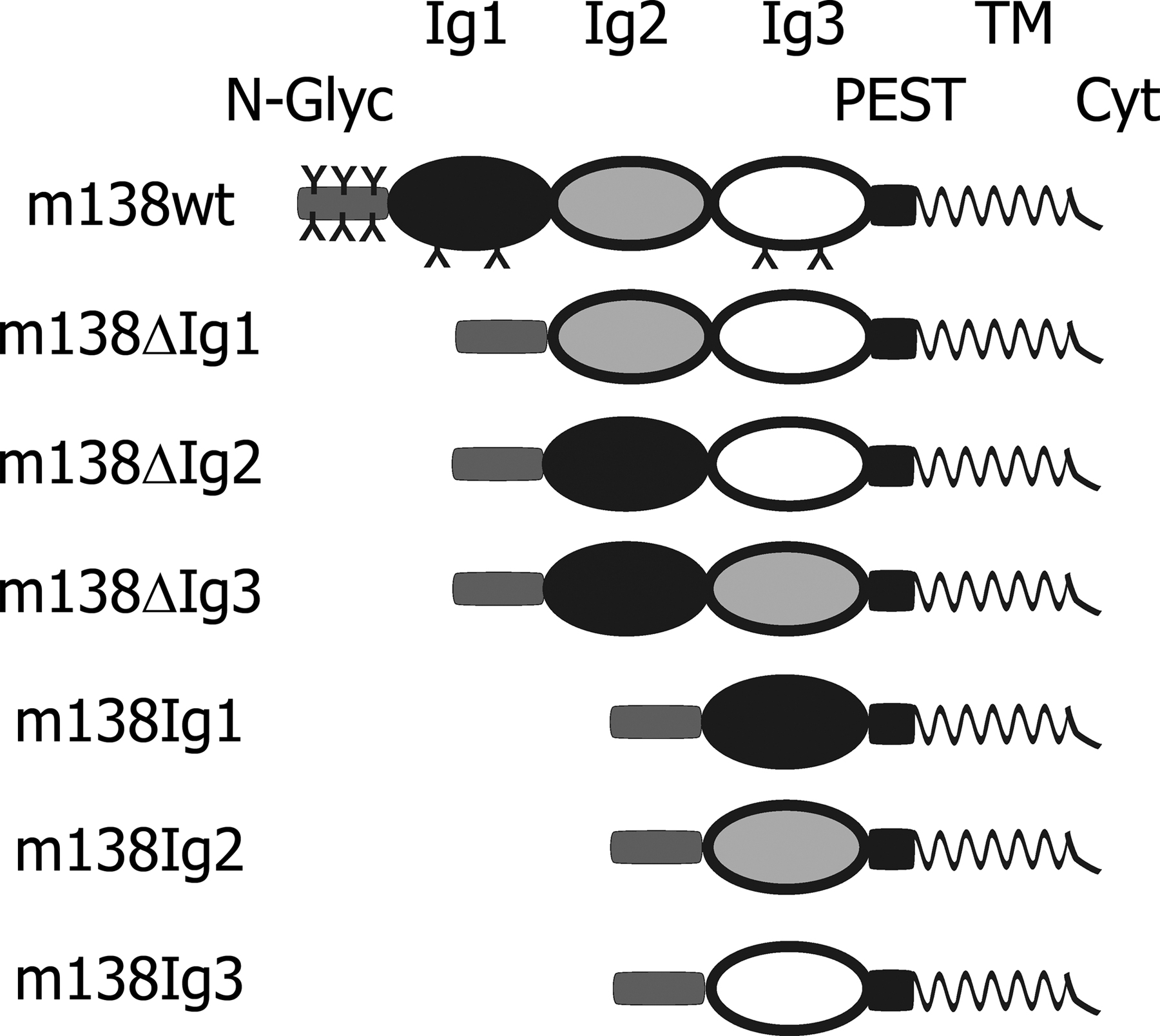
A detailed sequence alignment revealed the fcr-1 molecule to be phylogenetically congruent with host FcγR structures (unpublished data). fcr-1 is predicted to preserve three FcγR-related putative IgSF-like domains Ig1, Ig2, and Ig3, which exhibit a low but still significant sequence homology with IgSF domains of cellular FcγRs CD16/FcγRIII and CD32/FcγRII, reaching 17% identity and 24% similarity, and are adjacent to the top N-glycan–rich domain (13). To identify the domains involved in down-modulation of MULT-1 and H60, we constructed mutant viruses lacking either of three IgSF-like domains, as well as viruses possessing only one of them in conjunction with the stabilizing N-glycan–rich domain (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20060514/DC1). SVEC4-10 cells were infected with either MCMV lacking m138/fcr-1, with m138/fcr-1-revertant, WT, or with mutant MCMVs possessing truncated forms of Ig-like domains (Fig. 3). As expected, WT virus and the revertant caused the down-regulation of MULT-1 and H60, whereas the m138/fcr-1 negative mutant failed to inhibit their surface expression. The mutant lacking Ig1 domain also lost its ability to regulate both ligands. The MULT-1, but not H60, regulating function was preserved in mutants lacking either the middle or the membrane-proximal third Ig domain. The absence of the Ig3 domain decreased the ability of fcr-1 to down-modulate MULT-1 to a certain extent. Because the Ig3 as well as the Ig2 domain had no effect on MULT-1 when expressed in isolation (Fig. 3), the intermediate MULT-1 phenotype may be as a result of steric constraints on the activity of the Ig1 domain caused by the alteration in the protein structure. The influence of the Ig1 domain on MULT-1 was confirmed by the expression of m138/fcr-1 mutants in HEK 293T cells cotransfected with MULT-1. In contrast, the Ig1 domain alone failed to decrease surface expression of H60. When fcr-1 mutants lacking the cytoplasmic or transmembrane part of the molecule were analyzed, membrane insertion, but not the cytoplasmic tail of fcr-1, was identified as being essential for MULT-1 modulation (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20060514/DC1). Collectively, the data identified the Ig1 domain of the fcr-1 ectodomain to be necessary to down-regulate MULT-1 and even sufficient when expressed in the absence of Ig2 and Ig3. In contrast, H60 down-regulation required the preserved composition of the complete fcr-1 ectodomain, indicating a different mode of interaction with fcr-1.
Figure 3.

The fcr-1 Ig1 domain is responsible for MULT-1 down-modulation. SVEC4-10 cells infected for 22 h with 5 PFU per cell of indicated viruses or left uninfected were stained with rat anti–MULT-1 mAb or anti-H60 mAb. Irrelevant primary antibody was used as a negative control (thin line).
m138/fcr-1 constitutes a surface resident FcγR
The finding that fcr-1 down-regulates NKG2D ligands from the cell surface prompted us to reevaluate the surface disposition of the molecule to qualify its FcγR function. Fibroblasts were infected with WT MCMV and Δm138/fcr-1 mutant for 14 h before staining with mouse IgG-Fc fragments on the cell surface and intracellularly (Fig. 4 a). Fc-binding activity was readily detectable both on the cell surface and inside of the WT-infected, but not in the Δm138/fcr-1–infected, cells. fcr-1 molecule thus specifically binds IgG-Fc on the cell surface, constituting a true vFcγR.
Figure 4.

fcr-1 is readily detected on the cell surface of MCMV-infected cells and surface resident MULT-1 is rapidly recycling. (a) (10)1 cells were mock infected or infected with 5 PFU per cell of WT MCMV or Δm138/fcr-1. 15 h p.i., the cells were surface stained and intracellularly stained with mouse IgG Fc fragment and Cy5-labeled anti–mouse IgG. (b) Confocal analysis was performed on MULT-1–3T3 cells Ab-tagged for surface MULT-1 (red) as described for the uptake assay. Transferrin-FITC (green) was added in parallel with anti–MULT-1 mAb.
fcr-1 interferes with the recycling of surface MULT-1 and leads to its subsequent degradation in lysosomes
MULT-1 maturation in MCMV-infected cells is not altered before it reaches the medial-Golgi compartment (12). Moreover, MULT-1 is able to reach the trans-Golgi compartment (TGN) in infected cells (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20060514/DC1). We therefore studied the fate of surface resident MULT-1 by a modified uptake assay (18). By tagging MULT-1 with specific mAb in uninfected cells, we observed that although MULT-1 colocalizes with internalized transferrin in vesicles (Fig. 4 b), it also persists on the cell surface for a prolonged period of time (Fig. 5 a, first column), implying vesicular recycling. Therefore, we tested whether fcr-1 could down-modulate surface MULT-1 by interfering with the recycling pathway. In contrast with stable expression of surface resident MULT-1 on uninfected cells, MCMV infection causes a rapid fcr-1–dependent down-modulation, whereas the virus lacking m138/fcr-1 gene cannot affect the stability of surface resident MULT-1 (Fig. 5 a). The lack of down-modulation of CD29 (β1 integrin) as detected by an isotype-matched mAb demonstrated the fcr-1 specificity for MULT-1 (Fig. 5 c). Because fcr-1 is known to bind the Fc fragment of the IgG, as a further control we used F(ab)2 fragments of anti–MULT-1 mAb in the uptake assay and obtained the same results of fcr-1–dependent MULT-1 down-regulation. Next, we determined the fate of MULT-1 molecules and observed that MULT-1 is not only down-modulated from the cell surface, but also degraded in the presence of fcr-1 (Fig. 5 b). The fcr-1 effect on MULT-1 was completely abolished by imipramine, which inhibits the AP2-mediated clathrin pathway known to be involved in the recycling of transferrin receptor. Ammonium chloride and leupeptin, inhibitors of the lysosomal degradation pathway, prevented MULT-1 degradation, whereas specific inhibitors of the proteasome, lactacistine, and epoxomicin, had no effect (unpublished data). The flow cytometry data provided the evidence of imipramine rescuing MULT-1 on the surface of infected cell (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20060514/DC1), whereas the downstream inhibitors of MULT-1 degradation could not prevent MULT-1 down-regulation from the plasma membrane. Altogether, the data confirmed that fcr-1 is responsible for the interference with the recycling of the surface portion of MULT-1, leading to its subsequent lysosomal degradation. We did not observe a similar recycling process of H60 (unpublished data), the fcr-1 effect upon which involves also other domains of fcr-1 and is apparently based on another mechanism that requires further studies.
Figure 5.

fcr-1 targets surface resident MULT-1 for lysosomal degradation. SVEC4-10 (a and c) or MULT-1–3T3 (b) cells either mock infected or infected with 4 PFU per cell of WT MCMV or Δm138/fcr-1 were Ab-tagged for surface proteins. Cells were treated as described for uptake assay with rat anti–MULT-1 mAb (IgG2a). In addition, F(ab)2fragments of anti–MULT-1 mAb or rat anti-CD29 mAb (IgG2a) were used (c). After incubation for the indicated time periods, the cells were analyzed for the surface proteins (a and c). MULT-1 expression was examined after treatment with different inhibitors as described in the confocal microscopy section (b).
Concluding remarks
This work identifies an MCMV antagonist of NKG2D-mediated NK cell responses, the existence of which was predicted by the studies on NKG2D inhibitors m145 (12) and m155 (8, 10). Surprisingly, the previously described MCMV FcγR, fcr-1, turned out to act as a strong and autonomous regulator of two NKG2D ligands, MULT-1 and H60. This underlines the enormous selective pressure of NK cell control on the virus to subvert NKG2D receptor triggering. The modular composition of the fcr-1 molecule may allow the protein to approach structurally diverse targets. Indeed, fcr-1 is able to down-modulate at least two additional immune receptor ligands involved in the activation of T cell responses (unpublished data and Mintern, J.D., personal communication). The same N-terminal part of the fcr-1 ectodomain, Ig1, which is sufficient for MULT-1 function, is also needed to complex with the Fc part of IgG. In fact, soluble Fc fragments of IgG are able to inhibit fcr-1–mediated down-modulation of MULT-1, presumably by competing for the binding to the same fcr-1 domain (unpublished data). Evidence is provided that fcr-1 interferes with the clathrin-dependent endocytosis of MULT-1, leading to its subsequent lysosomal degradation. Altogether, these remarkable properties of fcr-1 explain the prominent functional roles of this vFcγR in vivo as suggested by the dramatic, IgG-independent attenuation of MCMV mutants lacking this gene (16).
MATERIALS AND METHODS
Cells.
CV-1 (CCL-70; American Type Culture Collection); SVEC4-10 (CRL-2181; American Type Culture Collection); NIH3T3 (CRL1658; American Type Culture Collection); HEK 293T (CRL-11268; American Type Culture Collection); B12 fibroblasts (10); (10)1 fibroblasts (19); H60-3T3 (H60-transfected NIH3T3 cells; reference 10); and MULT-1–3T3 (MULT-1–transfected NIH3T3 cells; reference 12) were cultivated in DMEM supplemented with 10% FCS. Mouse embryonic fibroblasts (MEFs) prepared from BALB/c mice were cultivated in MEM supplemented with 3% FCS. HEK 293T cells were cotransfected with pcDNA3.1–MULT-1 and pIRES-US2-EGFP or pIRES2-m138-EGFP in a ration of 1:6, using Polyfect (QIAGEN) according to the manufacturer's instructions.
Viruses.
A bacterial artificial chromosome (BAC)-derived MCMV, MW97.01, has previously been shown to be biologically equivalent to the MCMV Smith strain (VR-194 [reaccessioned as VR-1399]; American Type Culture Collection) and is here referred as WT MCMV (20). The generation of the following recombinant MCMV mutants was described elsewhere: MCMV-Δm145 (12); MCMV-Δ6 (m144-m158), MCMV-Δ7 (m159-m170) (21); MCMV-Δm152, MCMV-Δm06Δm152 (22); and the Δm138/fcr-1 virus (ΔMC95.15) and its revertant MCMV-m138Rev (RMS95.9) (16). MCMV mutagenesis and the construction of MCMV ΔA1 and m138/fcr-1 mutants are described in detail in the online supplemental material.
Recombinant VV expressing either MULT-1 (MULT-1–VV), H60 (H60-VV), MCMV m145 (m145-VV), MCMV m155 (m155-VV), or MCMV m138/fcr-1 (m138-VV) were described previously (12, 15) (8, 10).
Uptake assay.
To chase the surface resident proteins, a modified uptake assay was used (18). The surface portion of referred proteins was tagged with specific mAb in PBS for 30 min on ice. MULT-1 was tagged with rat anti–MULT-1 mAb (12) and in some experiments, F(ab)2 fragments of anti–MULT-1 mAb were used that were prepared by pepsin digestion and separated from Fc fragments and IgG by the use of HiTrap Protein G HP column (GE Healthcare). CD29 was tagged with rat anti–mouse CD29 mAb (BD Biosciences) of the same rat IgG2a isotype as determined for the rat anti–MULT-1 mAb. The excess of mAb was washed, the cells were returned to 37°C, and the antibody-tagged protein was chased for a given period of time. For flow cytometry analysis, cells were harvested, washed in PBS, and PE-labeled goat anti–rat IgG was added for 25 min on ice. For confocal analysis, cells were washed in PBS, fixed, and permeabilized as described in section confocal analysis. The TRITC-labeled goat anti–rat IgG was added for an additional 40 min at room temperature. Cells were uninfected or infected with 4 PFU of indicated viruses. The inhibitors of different cellular pathways were used at the following concentrations: imipramine (40 μg/ml, Sigma-Aldrich), NH4Cl (40 mM; Sigma-Aldrich), and leupeptin (75 μg/ml, Sigma-Aldrich). The cells were stained in parallel with irrelevant primary mAb followed by fluorescence-labeled secondary Ab.
Flow cytometry.
SVEC4-10, H60-3T3, MULT-1-3T3, (10)1, NIH3T3, and B12 cells were mock treated or infected with MCMV for indicated periods of time, harvested by trypsinization or ethylenediaminetetraacetic acid (EDTA), and washed in PBS supplemented with 1% BSA and 0.1% NaN3. The cells were incubated for 25 min on ice with rat anti–MULT-1 mAb (12), F(ab)2 fragments of anti–MULT-1 mAb, rat anti–RAE-1αβγ mAb CX1 (9), rat anti-H60 mAb (R&D Systems), and mouse anti-Kd mAb MA-215. After washing the cells, the binding of first antibodies was visualized by the addition of PE-labeled goat anti–rat IgG (Caltag Laboratories) or biotinylated anti–rat IgG (Jackson ImmunoResearch Laboratories) followed by PE-labeled streptavidin (Becton Dickinson). The first antibodies of mouse origin were visualized with FITC-labeled goat anti–mouse Ig (BD Biosciences). The binding of mouse IgG-Fc fragments (Rockland Immunochemicals) to infected and uninfected cells was visualized with anti–mouse IgG-Cy5 (Dianova). The detection of NKG2D ligands on the surface of uninfected and infected cells was performed by PE-NKG2D tetramers (8). For surface staining, the cells were labeled directly after harvesting; for intracellular staining, cells were fixed in paraformaldehyde and permeabilized in 0.1% saponin before incubation with antibodies. Cells infected with indicated viruses were discriminated by intracellular staining with mouse mAb CROMA 229, which recognizes MCMV gp48 antigen; mouse mAb CROMA 101, which recognizes MCMV pp89 antigen; or by surface staining with mouse mAb 38–14-8s (anti-H-2Lq). In some experiments, GFP viruses were used or an infection dose of 4 PFU per cell was used to achieve an infection rate of >95% of cells.
CV-1 cells were mock treated or infected with VV by a multiplicity of infection PFU/cell of 4, harvested 16 h after infection, and stained with rat anti–MULT-1 mAb (12), rat anti-H60 mAb clone 205326 (provided by J.P. Houchins, R&D Systems, Minneapolis, MN) or rat IgG2a isotype control, followed by PE-labeled goat anti–rat IgG (Caltag Laboratories).
HEK 293T cell transfectants were harvested 24 h after transfection, washed in PBS supplemented with 2.5% (vol/vol) FCS and 0.05% (wt/vol) NaN3, (FACS-PBS), and subsequently incubated with rat anti–MULT-1 mAb, followed by biotinylated anti–rat IgG and PE-labeled streptavidin, each for 25 min on ice with three washing steps after incubations. Transfected cells were identified by gating on EGFP-positive cells. The cells were stained in parallel with irrelevant primary mAb followed by fluorescence-labeled secondary Ab. All samples were analyzed with a Becton Dickinson FACScan or Becton Dickinson FACSCalibur.
Confocal analysis.
MULT-1–3T3 transfectants grown on glass coverslips were infected with 4 PFU per cell of WT-MCMV, Δm138/fcr-1, or left uninfected. The cells were washed in PBS, fixed with 2.5% (wt/vol) paraformaldehyde for 20 min at room temperature, and permeabilized using a 5-min incubation in 0.1% (wt/vol) Triton X-100. For visualization of MULT-1, rat anti–MULT-1 mAb (12) was used either as described for the uptake assay, or for 40 min in PBS at room temperature after the permeabilization, both followed by goat anti–rat IgG F(ab)2 TRITC-labeled secondary antibody (Santa Cruz Biotechnology, Inc.). The unspecific binding was blocked with 0.2% (wt/vol) fish skin gelatin. Cells incubated with secondary antibody upon binding to irrelevant rat IgG2a antibodies were used as a negative control. FITC-labeled transferrin (Sigma-Aldrich) was used in the uptake assay in parallel with anti–MULT-1 mAb as a marker of recycling pathway. The rabbit anti-Rab6 (Santa Cruz Biotechnology, Inc.) was used for the visualization of the medial- and trans-Golgi apparatus, followed by FITC-labeled secondary antibodies obtained from Santa Cruz Biotechnology, Inc. All samples were analyzed with Olympus FV300 confocal laser scanning microscope.
Mice.
BALB/c mice (H-2d) were housed and bred under specific pathogen-free conditions at the Central Animal Facility of the Faculty of Medicine University of Rijeka, in accordance with the general principles contained in the Guide for the Care and Use of Laboratory Animals (National Academies Press). The Ethical Committee at the Faculty of Medicine of the University of Rijeka approved all animal experiments. 6–8-wk-old female mice were used.
Infection conditions, detection of infectious MCMV in tissues, and statistical evaluation.
Mice were injected i.v. with 2 × 105 PFU of tissue culture–grown WT MCMV or recombinant virus in 500 μl of diluent. The mice were also infected by i.v. inoculation of 104 BALB/c MEF cells infected for 12 h with 2 PFU of indicated viruses (8). Organs were collected at 3 d p.i. and viral titers were determined by a standard plaque-forming assay (23). The statistical significance of the difference between experimental groups was determined by the Mann-Whitney exact rank test. Viral titers (from groups x and y) were considered significantly different for p-values (x versus y) of <0.05 (one-sided).
Depletion of NK cells.
The depletion of NK cells was performed by i.p. injection of rabbit antibodies to asialo-GM1 (Wako Chemicals) at the dose of 25 μl in 500 μl of diluent, 24 h before infection. NKG2D was blocked by i.p. injection of blocking anti-NKG2D mAb (R&D Systems) at a dose of 100 μg/mouse in 500 μl of diluent, 24 h before infection.
Online supplemental material.
MCMV mutagenesis and the construction of MCMV m138/fcr-1 mutants are described in detail in Fig. S1. Fig. S2 demonstrates the transmembrane domain of fcr-1, but not the cytoplasmic tail to be essential for MULT-1 regulation. Fig. S3 presents the colocalization of MULT-1 intracellular fraction with the Rab6 as the marker of medial- and trans-Golgi apparatus in the uninfected and WT MCMV-infected cells. The flow cytometry data (Fig. S4) show that the inhibitors of lysosomal degradation in contrast with the inhibitor of clathrin endocytosis cannot rescue the surface expression of MULT-1 in MCMV-infected cells. Online supplemental figures are available at http://www.jem.org/cgi/content/full/jem.20060514/DC1.
Supplemental Material
Acknowledgments
We are grateful to D.H. Busch for providing the NKG2D tetramer, L.L. Lanier for providing the CX1 mAb, and D. Margulies for critical comments.
This work was supported by Croatian Ministry of Science grant nos. 0062004 and 0062007; the Deutsche Forschungsgemeinschaft through grant nos. He 2526/6-2, ME1102/2-1, and SFB 455; NGFN grant 01GS0405; and FP6 Marie Curie Research Training grant no. 019248. A. Krmpotic is supported by the Howard Hughes Medical Institute International Research Scholars grant.
The authors have no conflicting financial interests.
T. Lenac and M. Budt contributed equally to this work.
References
- 1.French, A.R., and W.M. Yokoyama. 2003. Natural killer cells and viral infections. Curr. Opin. Immunol. 15:45–51. [DOI] [PubMed] [Google Scholar]
- 2.Jonjic, S., I. Bubic, and A. Krmpotic. 2006. Innate immunity to cytomegaloviruses. In Cytomegaloviruses: Molecular Biology and Immunology. M.J. Reddehase, editor. Caister Academic Press, Wymondham, Norfolk. 285–319 pp.
- 3.Cerwenka, A., and L.L. Lanier. 2001. Natural killer cells, viruses and cancer. Nat. Rev. Immunol. 1:41–49. [DOI] [PubMed] [Google Scholar]
- 4.Raulet, D.H. 2003. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 3:781–790. [DOI] [PubMed] [Google Scholar]
- 5.Carayannopoulos, L.N., O.V. Naidenko, D.H. Fremont, and W.M. Yokoyama. 2002. Cutting edge: murine UL16-binding protein-like transcript 1: a newly described transcript encoding a high-affinity ligand for murine NKG2D. J. Immunol. 169:4079–4083. [DOI] [PubMed] [Google Scholar]
- 6.Malarkannan, S., P.P. Shih, P.A. Eden, T. Horng, A.R. Zuberi, G. Christianson, D. Roopenian, and N. Shastri. 1998. The molecular and functional characterization of a dominant minor H antigen, H60. J. Immunol. 161:3501–3509. [PubMed] [Google Scholar]
- 7.Cerwenka, A., A.B. Bakker, T. McClanahan, J. Wagner, J. Wu, J.H. Phillips, and L.L. Lanier. 2000. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. 12:721–727. [DOI] [PubMed] [Google Scholar]
- 8.Krmpotic, A., D.H. Busch, I. Bubic, F. Gebhardt, H. Hengel, M. Hasan, A.A. Scalzo, U.H. Koszinowski, and S. Jonjic. 2002. MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat. Immunol. 3:529–535. [DOI] [PubMed] [Google Scholar]
- 9.Lodoen, M., K. Ogasawara, J.A. Hamerman, H. Arase, J.P. Houchins, E.S. Mocarski, and L.L. Lanier. 2003. NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J. Exp. Med. 197:1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hasan, M., A. Krmpotic, Z. Ruzsics, I. Bubic, T. Lenac, A. Halenius, A. Loewendorf, M. Messerle, H. Hengel, S. Jonjic, and U.H. Koszinowski. 2005. Selective down-regulation of the NKG2D ligand H60 by mouse cytomegalovirus m155 glycoprotein. J. Virol. 79:2920–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lodoen, M.B., G. Abenes, S. Umamoto, J.P. Houchins, F. Liu, and L.L. Lanier. 2004. The cytomegalovirus m155 gene product subverts natural killer cell antiviral protection by disruption of H60-NKG2D interactions. J. Exp. Med. 200:1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krmpotic, A., M. Hasan, A. Loewendorf, T. Saulig, A. Halenius, T. Lenac, B. Polic, I. Bubic, A. Kriegeskorte, E. Pernjak-Pugel, et al. 2005. NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J. Exp. Med. 201:211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Budt, M., H. Reinhard, A. Bigl, and H. Hengel. 2004. Herpesviral Fcγ receptors: culprits attenuating antiviral IgG? Int. Immunopharmacol. 4:1135–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dubin, G., E. Socolof, I. Frank, and H.M. Friedman. 1991. Herpes simplex virus type 1 Fc receptor protects infected cells from antibody-dependent cellular cytotoxicity. J. Virol. 65:7046–7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thäle, R., P. Lucin, K. Schneider, M. Eggers, and U.H. Koszinowski. 1994. Identification and expression of a murine cytomegalovirus early gene coding for an Fc receptor. J. Virol. 68:7757–7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crnkovic-Mertens, I., M. Messerle, I. Milotic, U. Szepan, N. Kucic, A. Krmpotic, S. Jonjic, and U.H. Koszinowski. 1998. Virus attenuation after deletion of the cytomegalovirus Fc receptor gene is not due to antibody control. J. Virol. 72:1377–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jonjic, S., I. Pavic, B. Polic, I. Crnkovic, P. Lucin, and U.H. Koszinowski. 1994. Antibodies are not essential for the resolution of primary cytomegalovirus infection but limit dissemination of recurrent virus. J. Exp. Med. 179:1713–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mansouri, M., E. Bartee, K. Gouveia, B.T. Hovey Nerenberg, J. Barrett, L. Thomas, G. Thomas, G. McFadden, and K. Fruh. 2003. The PHD/LAP-domain protein M153R of myxomavirus is a ubiquitin ligase that induces the rapid internalization and lysosomal destruction of CD4. J. Virol. 77:1427–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harvey, D.M., and A.J. Levine. 1991. p53 alteration is a common event in the spontaneous immortalization of primary BALB/c murine embryo fibroblasts. Genes Dev. 5:2375–2385. [DOI] [PubMed] [Google Scholar]
- 20.Wagner, M., S. Jonjic, U.H. Koszinowski, and M. Messerle. 1999. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J. Virol. 73:7056–7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brune, W., M. Wagner, and M. Messerle. 2006. Manipulating cytomegalovirus genomes by BAC mutagenesis: strategies and applications. In Cytomegaloviruses: Molecular Biology and Immunology. M.J. Reddehase, editor. Caister Academic Press, Wymondham, Norfolk. 63–89 pp.
- 22.Wagner, M., A. Gutermann, J. Podlech, M.J. Reddehase, and U.H. Koszinowski. 2002. Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J. Exp. Med. 196:805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brune, W., H. Hengel, and U.H. Koszinowski. 1999. A mouse model for cytomegalovirus infection. In Current Protocols in Immunology. John Wiley & Sons, New York. 19.17.11–19.17.13. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}