

Abstract
Given the global impact of persistent infections on the human population, it is of the utmost importance to devise strategies to noncytopathically purge tissues of infectious agents. The central nervous system (CNS) poses a unique challenge when considering such strategies, as it is an immunologically specialized compartment that contains a nonreplicative cell population. Administration of exogenously derived pathogen-specific memory T cells (referred to as adoptive immunotherapy) to mice burdened with a persistent lymphocytic choriomeningitis virus (LCMV) infection from birth results in eradication of the pathogen from all tissues, including the CNS. In this study, we sought mechanistic insights into this highly successful therapeutic approach. By monitoring the migration of traceable LCMV-specific memory CD8+ T cells after immunotherapy, it was revealed that cytotoxic T lymphocytes (CTLs) distributed widely throughout the CNS compartment early after immunotherapy, which resulted in a dramatic elevation in the activity of CNS antigen-presenting cells (APCs). Immunotherapy induced microglia activation as well as the recruitment of macrophages and dendritic cells (DCs) into the brain parenchyma. However, DCs emerged as the only CNS APC population capable of inducing memory CTLs to preferentially produce the antiviral cytokine tumor necrosis factor-α, a cytokine demonstrated to be required for successful immunotherapeutic clearance. DCs were also found to be an essential element of the immunotherapeutic process because in their absence, memory T cells failed to undergo secondary expansion, and viral clearance was not attained in the CNS. These experiments underscore the importance of DCs in the immunotherapeutic clearance of a persistent viral infection and suggest that strategies to elevate the activation/migration of DCs (especially within the CNS) may facilitate pathogen clearance.
Given the challenges associated with the eradication of persistent viral infections, it is important to devise and fully understand therapeutic strategies that achieve systemic viral elimination without severe pathological consequences. Remarkably, a series of seminal studies conducted in the lymphocytic choriomeningitis virus (LCMV) model system (1) have revealed that total body elimination of a fully established persistent viral infection is attainable (2, 3). If mice are infected at birth or in utero with LCMV (referred to as carrier mice), the virus establishes lifelong persistence in nearly every tissue compartment (e.g., spleen, thymus, lymph nodes, liver, lung, heart, kidney, central nervous system [CNS], etc.; reference 4). T cells become tolerant to LCMV in carrier mice (5), and, thus, attempts to eliminate the pathogen through vaccination have been unsuccessful (6). To further complicate matters, neurons are the predominant LCMV-infected cell population residing in the brain parenchyma (2, 7). Neurons reside in an immunologically specialized compartment (8) and do not readily express MHC (9, 10). Consequently, they do not represent an optimal target for T cell interactions. Nevertheless, one study pioneered a therapeutic intervention more than four decades ago in the LCMV model system (referred to as immunocytotherapy) that relied on the adoptive transfer of LCMV-specific memory splenocytes into carrier mice (11). Administration of immunocytotherapy resulted in the complete eradication of the virus from all peripheral tissues as well as the CNS (7).
Analysis of immunocytotherapy in LCMV carrier mice provides an ideal model system to gain mechanistic insights into how to relieve a delicate, nonreplicative cell population (neurons) of a persistent viral infection. Studies in the LCMV model system have shown that successful immunotherapeutic clearance requires the cooperative efforts of as few as 350,000 CD8+ and 7,000 CD4+ memory T cells (12) as well as the cytokine IFN-γ (13). In addition, a unique pattern of clearance was observed after immunocytotherapy: most peripheral tissues are purged within 14 d, whereas 100 d are required to eliminate the virus from the CNS (7). Presently, the precise mechanism by which CNS neurons are purged of a persistent viral infection without obvious signs of damage (7, 9) remains unclear. Based on the supposition that neurons provide a suboptimal target for adoptively transferred CTLs in carrier mice as a result of their limited or negligible expression of MHC (9, 10), we theorized that professional APCs such as DCs might also be involved in the modus operandi of immunocytotherapy by promoting optimal interactions and antiviral cytokine release. Unlike most peripheral tissues, DCs cannot be found in the undisturbed brain parenchyma (14, 15). However, several studies have revealed that DCs are recruited into the brain parenchyma during a variety of inflammatory conditions (16–21). Moreover, recent studies have also demonstrated that DCs are involved in the optimization of secondary T cell responses to a variety of peripheral pathogens (22) and can exacerbate T cell responses in nonlymphoid tissues (23). Therefore, we set out to address several pertinent, unanswered questions in this study pertaining to the role of DCs in adoptive immunotherapy. First, does adoptive immunotherapy promote DC recruitment and antigen presentation within the CNS parenchyma? Second, what is the antiviral cytokine profile of CTLs that interact with CNS DCs during immunotherapy? Third, are DCs absolutely required for successful immunotherapeutic clearance?
RESULTS
Localization of virus in the brain of an LCMV carrier mouse
LCMV distributes evenly throughout the brains of LCMV carrier mice (Fig. 1 A), and higher resolution colabeling analyses confirmed previous studies (7, 9) indicating that the establishment of a persistent LCMV infection from birth results primarily in neuronal infection within the brain parenchyma. Greater than 99% of the parenchymal LCMV-positive cells were found to colocalize with neuronal (NeuN) staining (Fig. 1, B–D). Outside of the parenchyma, LCMV was also found to localize to the meninges, ependyma, and choroid plexus, which are the very same regions infected in intracerebrally inoculated adult mice that succumb to the fatal choriomeningitis for which LCMV is named (24).
Figure 1.

Localization of LCMV in the brain of a carrier mouse persistently infected from birth. (A) Six-μm sagittal brain sections from C57BL/6 LCMV carrier mice (n = 3) at >8 wk of age were stained with a polyclonal anti-LCMV antibody (green) and a nuclear dye (red). Brain reconstructions were performed to illustrate the anatomical distribution of LCMV. Note the equal distribution of LCMV throughout the brain parenchyma. (B) High resolution analyses of LCMV-infected cells (green; C and D) in the brain parenchyma confirmed that >99% of the cells colocalized with neuronal staining (red; B and D). Overlapping fluorescence appears in D.
LCMV-specific memory T cells migrate into the brain parenchyma early after adoptive immunotherapy
To gain unique mechanistic insights into adoptive immunotherapy, we developed an experimental approach to monitor a population of LCMV-specific memory CTLs during the immunocytotherapeutic process in LCMV carrier mice. We opted to monitor LCMV-specific CTLs because these cells are required for efficient clearance to occur (12). To generate a traceable population of memory CTLs, C57BL/6 donor mice were seeded with 104 GFP+ (or Thy1.1+) naive TCR-transgenic (tg) CD8+ precursors specific to amino acids 33–41 of the LCMV glycoprotein (GP) as described previously (25). 1 d after the injection of the traceable DbGP33–41- specific T cells, the memory donor mice were infected with 105 PFU of LCMV Armstrong, which is cleared acutely in 8–10 d, and, afterward, memory T cells specific to the virus emerge (26).
The distribution of memory GFP+DbGP33–41-specific CD8+ T cells in a whole spleen cross section of a representative donor mouse at day 45 after infection is shown in Fig. 2 A. At this time point, the GFP+DbGP33–41-specific memory CTLs localize throughout the spleen. It is important to note that the spleen at this time point also harbors a polyclonal repertoire of endogenous memory T cells (both CD8+ and CD4+) specific to LCMV (26), which can assist in viral clearance. Thus, GFP+DbGP33–41-specific CTLs represent a single traceable population of memory T cells in a diverse antiviral repertoire.
Figure 2.

Kinetics of DbGP33–41-specific T cell expansion and viral clearance in the blood after immunocytotherapy. C57BL/6 mice seeded with 104 GFP+ (or Thy1.1+) DbGP33–41-specific CD8+ T cells and i.p. infected 1 d later with LCMV served as memory donors for immunocytotherapy experiments. (A) The distribution of GFP+DbGP33–41-specific memory CD8+ T cells (green) on a splenic reconstruction is shown for a donor mouse 45 d after infection. The illustration is a representative example (n = 3) of an entire splenic cross section. Note the even distribution of GFP+ memory cells throughout the spleen. Cell nuclei are shown in blue for anatomical purposes. (B and C) 107 memory splenocytes containing GFP+DbGP33–41-specific memory CD8+ T cells were injected i.p. into LCMV carrier mice. The viral clearance kinetics in relation to the expansion of GFP+DbGP33–41-specific T cells in the blood is shown for two representative immunocytotherapy recipients (n = 3 mice). Note that a rapid decline in serum viral titers (gray dots) coincides perfectly with the expansion of GFP+DbGP33–41-specific T cells (black dots). DbGP33–41-specific T cells are represented as the percentage of CD8+ T cells that are GFP+.
To perform immunocytotherapy, 107 bulk memory splenocytes (which included the traceable population of CTLs) from donor mice >45 d after infection (Fig. 2 A) were adoptively transferred into adult LCMV carrier mice. After the administration of memory splenocytes, the traceable population of GFP+DbGP33–41-specific CTLs expanded rapidly in peripheral compartments. Elevated frequencies of GFP+DbGP33–41-specific CTLs (representing secondary effectors) were noted in the blood as early as 7–8 d after the therapeutic intervention, and this always coincided with a complete reduction in serum viral titers (Fig. 2, B and C). No expansion of GFP+DbGP33–41-specific CTLs was observed when immunotherapy was performed in naive C57BL/6 mice (unpublished data).
To evaluate the anatomical distribution of the traceable LCMV-specific CTL and to determine whether secondary effectors gained access to the CNS after immunocytotherapy, we performed three-color organ reconstructions of the brain and spleen (a representative peripheral compartment known to be heavily infected by LCMV in carrier mice; Fig. 3; reference 4). 8 d after the administration of immunocytotherapy, the spleen was completely inundated with GFP+DbGP33–41-specific CTLs (Fig. 3, D and F), and the virus in this compartment was reduced to an undetectable level (Fig. 3, E and F), indicating that the secondary effectors had successfully purged the pathogen. The clearance of virus from the spleen also mirrored the reduction in serum viral titers (Fig. 2, B and C). At this same time point, the virus continued to persist in the CNS (Fig. 3, B and C), which is consistent with the delayed immunocytotherapeutic clearance previously described in this compartment (7). Importantly, the failure to purge virus in a timely manner could not be explained by an inability of CTLs to access this compartment. GFP+DbGP33–41-specific CTLs were found throughout the brain parenchyma within 8 d of adoptive immunotherapy and were dispersed in a manner that overlapped with the distribution of the persisting virus (Fig. 3, A and C). Furthermore, the quantity of GFP+DbGP33–41-specific CTLs in the brain at this time point correlated negatively (r = −0.83) with the amount of virus (as determined by quantitative image analysis). These data suggested that CTLs exerted an immunological pressure on CNS virus in immunotherapy recipients.
Figure 3.

Distribution of DbGP33–41-specific CD8+ T cells in the brain and spleen of an immunocytotherapy recipient. Three-color coronal brain (A–C) and spleen (D–F) reconstructions were generated for immunocytotherapy recipients (n = 3) 8 d after the administration of immunocytotherapy to establish the localization of GFP+DbGP33–41-specific CD8+ T cells during the clearance of a persistent LCMV infection (red). Cell nuclei are shown in blue for anatomical purposes. The GFP+DbGP33–41-specific CD8+ T cells are widely spread throughout the brain parenchyma (A) 8 d after immunocytotherapy, and the distribution is analogous to that of the remaining persistent virus (B). A three-color overlay of the coronal brain section is shown in C. Note that the GFP+DbGP33–41-specific CD8+ T cells also distribute throughout the spleen (D) of the same animal; however, the virus (E) has dropped to an undetectable level at this time point. A three-color overlay of the spleen is shown in F.
Adoptive immunotherapy induces APC recruitment and antigen presentation in the brain parenchyma
Because neurons do not readily express antigen-presenting machinery (9, 10), we postulated that professional APCs might contribute to the process of CNS viral clearance by serving as an intermediary. According to this theory, interactions between peptide–MHC-presenting APCs and memory T cells would facilitate the release of antiviral cytokines that noncytopathically cleanse adjacently infected neurons. Therefore, we set out to address whether adoptive immunotherapy influenced APC activity within the CNS. This was accomplished initially by analyzing MHC class II expression on sagittal brain reconstructions at day 8 after immunocytotherapy. MHC class II expression is restricted primarily to cell populations with the capacity to present antigens and thus serves as an excellent marker for APCs (e.g., DCs, macrophages, and microglia). Day 8 after immunotherapy was selected because it represents a time point when GFP+DbGP33–41-specific CTLs were widely distributed throughout the brain parenchyma (Fig. 3, A and C). In contrast to the extremely low levels of MHC class II expression found in the brains of naive C57BL/6 and LCMV carrier mice, a marked increase in expression (14.4-fold; P < 0.001) was observed on sagittal brain sections from day 8 immunocytotherapy recipients (Fig. 4, A, C, E, and G). These data indicate that the LCMV carrier state itself did not influence CNS MHC class II expression. Rather, the substantial elevation in expression resulted from the adoptive immunotherapy.
Figure 4.

Elevated MHC class I and II expression in the brains of immunocytotherapy recipients. Representative sagittal brain sections from naive (A and B; n = 4), LCMV carrier (C and D; n = 4), and LCMV carrier 8 d after immunocytotherapy (E and F; n = 4) were stained with an antibody directed against MHC class II (A, C, and E) or MHC class I (B, D, and F; green). Note the marked increase in the level of MHC class II (E; 14.4-fold increase) and class I (F; 27.9-fold increase) expression on sagittal brain reconstructions obtained from immunocytotherapy recipients. A modest increase in MHC I expression was also observed in LCMV carrier mice that did not receive immunotherapy (D; 4.4-fold). Cell nuclei are shown in blue. An image analysis program was used to quantify the amount of MHC class II (G) and I (H) staining on the sagittal brain sections shown in A–F. The data are represented as the mean + SD (error bars). Statistical differences from naive mice as determined by a one-way ANOVA (P < 0.05) are denoted by asterisks.
To determine the influence that adoptive immunotherapy had on MHC I expression in the brain parenchyma, we next performed analyses analogous to that described for MHC II expression. A modest increase in MHC I expression (4.4-fold; P = 0.009) was observed in the brains of untreated LCMV carrier mice when compared with naive C57BL/6 mice (Fig. 4, B, D, and H). This indicates that the carrier state alone influences MHC I expression in the CNS. Interestingly, the brains of day 8 immunotherapy recipients showed a marked elevation in MHC I that far exceeded the levels observed in both naive (27.9-fold; P = 0.008) and LCMV carrier (6.3-fold; P = 0.008) mice (Fig. 4, B, D, F, and H). These data indicate that adoptive immunotherapy promotes MHC I as well as MHC II expression in the brain parenchyma.
To establish the identity/source of the APCs that localized within the brains of immunocytotherapy recipients and to determine the kinetics of APC recruitment in relation to the infiltration of Thy1.1+DbGP33–41-specific CTLs, we performed seven-color digital flow cytometric analyses at various time points after immunocytotherapy. This methodology permitted the simultaneous analysis of microglia, macrophages, DCs, and DbGP33–41-specific CTLs in a single CNS sample obtained from individual immunocytotherapy recipients at the indicated time points (days 1, 3, 5, and 7). Seven-color analyses were used to ensure that the three APC populations (which are all potential sources of MHC class II) could be distinguished from one another and from CNS-infiltrating mononuclear cell populations that share common markers. For example, CD11c (a prototypic DC marker) is known to be expressed on activated T cells and NK cells (27, 28). CD11b (a macrophage/microglia/conventional DC marker) can also be expressed on activated T cells (28). To minimize errors resulting from marker overlap, we included a “dump channel” in our flow cytometric studies to exclude T cells (CD3), NK cells (NK1.1), and B cells (CD19) from our analyses of microglia, macrophages, and DCs. We also used the common leukocyte marker CD45 to distinguish resident microglia from transient blood-derived APCs such as macrophages and DCs. It was reported previously that CNS microglia express low to intermediate levels of CD45 and could always be distinguished from blood-derived APCs, which express high levels of CD45 (29, 30).
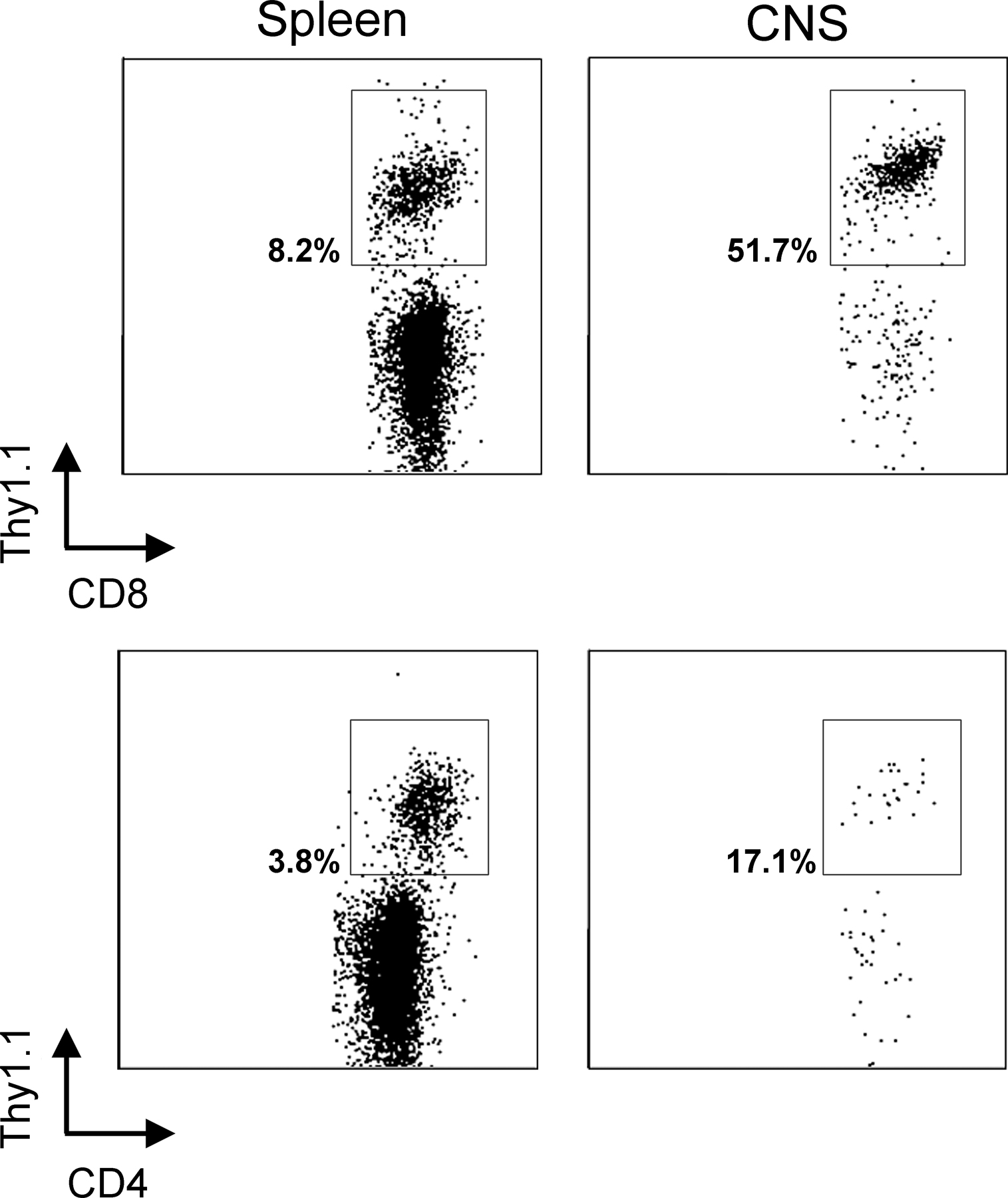
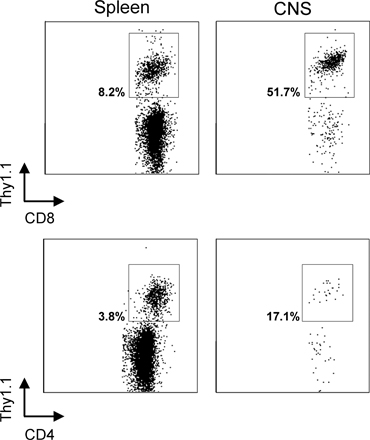
Using the aforementioned methodology, we simultaneously monitored DCs (CD45hiCD3−NK1.1−CD19−CD11c+), macrophages (CD45hiCD3−NK1.1−CD19−CD11c−CD11b+), microglia (CD45low/intCD3−NK1.1−CD19−CD11b+), and DbGP33–41-specific CTLs (CD45hiCD8+Thy1.1+) within the CNS of immunocytotherapy recipients over time. Because the naive brain parenchyma is normally devoid of DCs (14, 15), we were particularly interested in whether adoptive immunotherapy induced the recruitment of this potent APC into the CNS. Thy1.1+DbGP33–41-specific CTLs were observed in the CNS within 1 d of adoptive immunotherapy, and this early entry did not appreciably impact the CNS APC populations (Fig. 5 A). Nonactivated (CD45low) microglia represented the primary CNS APC population at this time, and the levels of macrophages and DCs were similar to those observed in LCMV carrier mice that did not receive immunocytotherapy (unpublished data). Over time, the frequency of Thy1.1+DbGP33–41-specific CTLs increased from 2.8% (day 1) to 38.3% (day 7), and this coincided with marked changes in the three APC populations. Thy1.1+ I-AbGP61–80-specific CD4+ helper T cells also trafficked into the CNS by day 7 after immunotherapy, although their frequency and numbers were reduced when compared with the traceable CTLs (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20060039/DC1).
Figure 5.

Kinetics of APC recruitment into the CNS after immunocytotherapy. (A) The recruitment kinetics of Thy1.1+DbGP33–41-specific CD8+ T cells (top), macrophages (middle), and DCs (bottom) into the CNS was evaluated in immunocytotherapy recipients (n = 3) at the indicated time points. The plots reflect a single animal and represent the mean of the group. The top panels are gated on CD45+CD8+ T cells, and the percentages denote the frequency of Thy1.1+DbGP33–41-specific CD8+ T cells. Note the substantial increase of antigen-specific T cells over time. The gates and corresponding percentages in the middle panels illustrate macrophages (CD45hiCD11b+) negatively gated on CD3 (to exclude T cells), CD19 (to exclude B cells), NK1.1 (to exclude NK cells), and CD11c (to exclude DCs). Note that the presence of macrophages in the CNS increases substantially over time. The bottom panels illustrate the percentages of activated microglia (CD45low/intCD11c+; left) and DCs (CD45hiCD11c+; right) negatively gated on CD3 and NK1.1. Note the increase in the frequency of DCs in the CNS over time and the fact that CD45int microglia also up-regulate CD11c at day 7 after immunocytotherapy. (B and C) The mathematical relationship between the frequency of Thy1.1+DbGP33–41-specific CD8+ T cells and macrophages over time (i.e., days 1, 3, 5, and 7) is shown in B. The relationship between Thy1.1+DbGP33–41-specific CD8+ T cells and DCs is shown in C. Note the near perfect direct linear relationship between the variables in both cases. Correlation coefficients (r) and P values were calculated using a Pearson Product Moment correlation test (P < 0.05). (D and E) The absolute number of macrophages (D) and DCs (E) were calculated for naive C57BL/6, LCMV carrier, and day 8 immunocytotherapy mice using the flow cytometric parameters described in A. Data are represented as the mean + SD (error bars). Asterisks denote statistical significance as determined by a one-way ANOVA (P < 0.05). The fold increase in the number of macrophages and DCs is also indicated on the graphs.
Microglia shifted from CD45low to CD45int between days 1 and 5 (not depicted) and up-regulated CD11c by day 7, indicating that these cells became activated as a consequence of the adoptive immunotherapy (Fig. 5 A, bottom). CD11c expression on activated microglia was reported previously (19, 31); nevertheless, microglia (CD45low/int) can be easily distinguished from DCs (CD45hi) based on the expression of CD45 (Fig. 5 A, bottom). Macrophages (CD45hiCD11c−CD11b+) and DCs (CD45hiCD11c+) increased significantly in the CNS of immunotherapy recipients over time, and this increase correlated almost perfectly (r = 1.000 for macrophages, and r = 0.997 for DCs) with the infiltration of Thy1.1+DbGP33–41-specific CTLs (Fig. 5, B and C). By day 7, 48,113 + 10,317 macrophages (94-fold increase; P = 0.029) and 4,063 + 1,588 DCs (10-fold increase; P = 0.004) could be extracted from the CNS of immunocytotherapy recipients. These two populations are normally represented at extremely low numbers in the CNS of naive C57BL/6 and LCMV carrier mice (Fig. 5, D and E).
Only CNS DCs stimulate memory CTLs
Because each of the aforementioned CNS APC populations has the potential to stimulate adoptively transferred memory T cells in situ, it is important to establish which populations (if any) are operational during the early phase of persistent viral clearance. To address this issue, we flow cytometrically sorted DCs, macrophages, and microglia from the CNS of day 8 immunocytotherapy recipients and cultured them ex vivo with purified Thy1.1+DbGP33–41-specific memory CD8+ T cells for 24 h. After the culture period, we evaluated whether the memory CTLs had produced two antiviral cytokines (IFN-γ and TNF-α) known to be rapidly expressed after reexposure to cognate antigen (32). Thus, this ex vivo assay enabled us to identify which CNS-derived APCs were presenting LCMV peptides in immunocytotherapy recipients.
As a positive control, memory CTLs were incubated with splenic DCs and macrophages obtained from LCMV carrier mice. Both APC populations are known to harbor LCMV antigen in carrier mice (33); however, only DCs possessed the capacity to induce antiviral cytokine production in memory CTLs (Fig. 6, middle row). When APCs were extracted from immunotherapy recipients, it was revealed that of the three CNS APC populations, only DCs (not macrophages or microglia) were capable of inducing cytokine production by memory CTLs (Fig. 6, bottom row). Moreover, the cytokine profile in the restimulated memory CTLs was heavily biased toward TNF-α production. A similar pattern emerged when APCs from immunotherapy spleens were analyzed (Fig. 6, fourth row). The bias toward TNF-α production is quite interesting given that the memory CTLs clearly have the capacity to simultaneously synthesize both IFN-γ and TNF-α (Fig. 6, middle row; reference 32).
Figure 6.

Stimulatory capacity of APCs extracted from the CNS of immunocytotherapy recipients. DCs (CD45hiCD3−NK1.1−CD19−CD11c+) and macrophages (CD45hiCD3−NK1.1−CD19−CD11c−CD11b+) were flow cytometrically sorted from the spleen and CNS of C57BL/6 naive, LCMV carrier, and day 8 immunocytotherapy recipients (see Materials and methods for details). Microglia (CD45low/intCD3−NK1.1−CD19−CD11b+) were only sorted from the CNS of day 8 immunocytotherapy recipients. The sources of the APCs are indicated on the left. Thy1.1+DbGP33–41-specific memory CD8+ T cells were incubated with the denoted APCs (using a 1:2 T cell/DC ratio) for a 24-h period and stained intracellularly with antibodies directed against IFN-γ (y axis) and TNF-α (x axis). Each FACs plot is gated on Thy1.1+CD8+ cells and denotes the frequency of IFN-γ+ (top left quadrant), IFN-γ+ TNF-α+ (top right quadrant), and TNF-α+ (bottom right quadrant) cells. No cytokine production was observed when CTLs were left unstimulated or combined with APCs from naive mice. APCs extracted from the spleens of LCMV carrier mice were used as a positive control. Note that DCs (but not macrophages) obtained from carrier spleens induced a marked number of IFN-γ+– and IFN-γ+ TNF-α+–producing CTLs, whereas DCs obtained from the spleens and CNS of immunotherapy recipients induced mainly TNF-α+ producers. Macrophages and microglia from immunotherapy recipients were unable to stimulate memory CTLs. These data are representative of three to five experiments.
Because CNS-infiltrating DCs were the only APC population capable of restimulating memory CTLs ex vivo, we next examined whether these cells differed from the nonstimulatory APC subsets (i.e., macrophage and microglia) in the expression of antigen-presenting machinery (Fig. 7, A and B) and costimulatory molecules (Fig. 7, C and D). To accomplish this objective, we examined the expression of MHC I (Fig. 7 A), MHC II (Fig. 7 B), B7.1 (Fig. 7 C), and B7.2 (Fig. 7 D) on the CNS APC subsets at day 8 after immunotherapy. DCs and macrophages did not differ from one another in MHC I (Db) or II expression but showed significantly (P < 0.05) higher levels than observed on microglia. Interestingly, the costimulatory molecules B7.1 and B7.2 were significantly (P < 0.05) increased only on DCs when compared with macrophages and microglia in the CNS. These data, combined with the results from the ex vivo cultures, strongly suggest that DCs are potent stimulators of memory CTLs in the brain parenchyma during the immunotherapeutic clearance of a persistent viral infection.
Figure 7.

MHC and costimulatory molecule expression on CNS APCs. The expression of MHC class I (A), MHC class II (B), B7.1 (C), and B7.2 (D) was analyzed flow cytometrically on DCs, macrophages, and microglia of day 8 immunocytotherapy recipients (n = 5). Note the elevated expression of B7.1 and B7.2 on CNS-infiltrating DCs. Data are represented as the mean + SD (error bars) geometric mean fluorescent intensity (GMFI). Asterisks denote statistical significance as determined by a one-way ANOVA (P < 0.05). MHC I, MHC II, and B7.2 were detected with antibodies conjugated to PE, and B7.1 was detected with an antibody conjugated to FITC. The geometric mean fluorescent intensities for a PE-isotype control antibody were 4.4 + 0.8, 4.3 + 0.7, and 4.5 + 0.1 on DCs, macrophages, and microglia, respectively. The geometric mean fluorescent intensities for a FITC-isotype control antibody were 3.5 + 0.4, 3.3 + 0.3, and 13.1 + 1.0 on DCs, macrophages, and microglia, respectively. The geometric mean fluorescent intensities for all molecules analyzed on the three APC populations were statistically higher (P < 0.05) than the corresponding isotype control antibody.
MHC II–expressing APCs interact with memory CTLs in the CNS of immunocytotherapy recipients
A marked increase in MHC II–bearing APCs was observed in the CNS after immunotherapy (Figs. 4 and 7), of which only DCs were found to stimulate DbGP33–41-specific memory CTLs ex vivo. To determine whether direct interactions between MHC II+ APCs and DbGP33–41-specific memory CTLs occurred within the brains of immunotherapy recipients, we conducted in situ analyses of immunological synapse formation (25). We demonstrated previously that in vivo immunological synapse formation between CTLs and their targets was associated with LFA-1 (an adhesion molecule) polarization toward the interface (25). Therefore, we set out to determine whether LFA-1 polarization could be observed between GFP+DbGP33–41-specific memory CTLs and MHC II+ APCs in the brains of LCMV carrier mice at day 8 after immunotherapy. Interestingly, we found examples of such interactions in the meninges, choroid plexus, and brain parenchyma of immunotherapy recipients (Fig. 8). Because DCs were the only MHC II–bearing APC found to stimulate cytokine production in DbGP33–41-specific CTLs ex vivo, these data suggest that DC–CTL interactions occur within the brains of immunotherapy recipients.
Figure 8.

Interactions between Thy1.1+DbGP33–41-specific T cells and MHC II+ APCs in the CNS of immunotherapy recipients. Three-color confocal microscopy was used to demonstrate immunological synapse formation between GFP+DbGP33–41-specific T cells (blue) and MHC II+ APCs (red) in the meninges, choroid plexus, and brain parenchyma of immunotherapy recipients. Immunological synapses were indicated by the polarization of the adhesion molecule LFA-1 (green) between the CTL and APC (25). Asterisks denote the engaged APC, and arrows denote the contact point between the two cells. LFA-1 is expressed on both CTLs and APCs, but note that all of the CTL-associated LFA-1 is focused toward a contact point at the CTL–APC interface. Two representative examples are shown for day 8 immunotherapy recipients (n = 3).
DCs and TNF-α are required for successful viral clearance after immunocytotherapy
We next addressed whether DCs were required for successful viral clearance in LCMV carrier mice. To accomplish this aim, we used a well-established tg strain of mice that expresses the diphtheria toxin (DT) receptor (DTR) under the CD11c promoter (34). Administration of a single injection of DT to CD11c-DTR mice results in DC depletion within 10–15 h that lasts for ∼1 wk (34). To evaluate the importance of DCs in the restimulation of adoptively transferred memory T cells, we established a colony of CD11c-DTR tg mice persistently infected with LCMV. The persistently infected colony, which consisted of CD11c-DTR mice and wild-type littermate controls, was first injected with 2 ng/g DT to initiate DC depletion, and, 10 h later, the mice received immunotherapy. The success of the immunotherapy in CD11c-DTR and control mice was evaluated by measuring traceable Thy1.1+DbGP33–41-specific CTLs (Fig. 9 A) and viral titers (Fig. 9 B) in the blood over time. The advantage of using this approach is that individual mice can be monitored serially. After the administration of DT and immunotherapy in wild-type carrier mice, a robust expansion of Thy1.1+DbGP33–41-specific CTLs was observed in the blood within 8 d (Fig. 9 A), which coincided with a reduction in serum viral titers (Fig. 9 B). In stark contrast, CD11c-DTR mice receiving the same regimen showed minimal expansion of Thy1.1+DbGP33–41-specific CTLs (Fig. 9 A) and no reduction in serum viral titers by day 18 after immunotherapy (Fig. 9 B).
Figure 9.

Role of DCs and TNF-α in adoptive immunotherapy. (A and B) CD11c-DTR tg mice (closed circles; n = 5) and wild-type littermate controls (open circles; n = 6) persistently infected with LCMV received an i.p. injection of 2 ng/g DT followed 10 h later by an injection of 107 memory cells that included traceable Thy1.1+DbGP33–41-specific CD8+ T cells. Expansion of Thy1.1+DbGP33–41-specific T cells (A) and viral titers (B) were monitored serially in the blood of mice over time. Note that Thy1.1+DbGP33–41-specific T cells failed to expand, and serum viral titers remained elevated only in CD11c-DTR tg mice. The data in A are represented as the mean + SD (error bars) of the percentage of CD8+ T cells that are Thy1.1+. (C and D) When examined at day 8 after immunotherapy, a substantial reduction in the number of Thy1.1+DbGP33–41-specific T cells (C; 23.8-fold decrease) and DCs (D; 7.1-fold decrease) was observed in the CNS of CD11c-DTR tg mice. (E) CNS viral clearance was also impaired at day 125 after immunotherapy in CD11c-DTR tg mice. The mean viral titer (PFU/gram) for each group is indicated on the bar graph. Asterisks denote statistical significance as determined by a Student's t test (P < 0.05). (F) B6 × 129 LCMV carrier mice deficient in TNF-α received 2 × 107 wild-type (closed circles) or TNF-α KO (closed triangles) memory splenocytes. Carrier mice receiving no cells (open circles) served as a negative control group. Serum viral titers (represented as PFU/milliliter on the y axis) were evaluated at the time points denoted on the x axis. Note the failure of TNF-α–KO memory splenocytes to reduce serum viral titers. The data illustrated on all graphs represent the mean of three to seven mice per group.
To determine whether DC depletion also interfered with the CTL and APC recruitment into the CNS, we quantified the number of Thy1.1+DbGP33–41-specific CTLs (Fig. 9 C) and APCs (Fig. 9 D) in the CNS of tg and littermate controls at day 8 after immunotherapy, a time point when DCs were shown to return to baseline levels in the CD11c-DTR system (34). At day 8 after immunotherapy, a 24-fold decrease in the number of Thy1.1+DbGP33–41-specific CTLs was observed in the CD11c-DTR carrier mice. Furthermore, the CD11c-DTR mice did not show the characteristic APC changes associated with immunotherapy (Figs. 4 and 5). Specifically, a reduced number of DCs (Fig. 9 D), macrophages (not depicted), and activated microglia (not depicted) was observed in the CNS of CD11c-DTR mice at day 8. Finally, to determine the long-term impact of DC depletion on the immunotherapeutic process, we measured CNS viral titers at day 125 after immunotherapy, a time point past the 100-d time window required for CNS viral clearance in carrier mice (7). At this time, serum viral titers were reduced to undetectable levels in both CD11c-DTR and control mice (unpublished data). This can be explained by the fact that DCs eventually reemerge in CD11c-DTR mice within 1 wk after a single injection of DT (34). However, this single injection of DT before immunotherapy had a long-term impact on CNS viral clearance (Fig. 9 E). At 125 d after immunotherapy, four out of five wild-type carrier mice had no detectable virus in the CNS. In contrast, none of the CD11c-DTR mice had cleared the virus by this time point. Collectively, these data indicate that DCs are not only capable of restimulating memory CTLs ex vivo (Fig. 7) but are also required for the success of immunocytotherapy in vivo.
Our ex vivo restimulation cultures also demonstrated that DCs extracted from immunotherapy recipients preferentially induced the production of TNF-α (rather than IFN-γ) by memory CTLs. Therefore, we set out to evaluate the importance of this antiviral cytokine by establishing a colony of LCMV carrier mice as well as memory donors deficient in TNF-α. Generation of TNF-α–deficient memory donors was possible because LCMV can be cleared in the absence of TNF-α (35). This experimental paradigm enabled us to identify the source of TNF-α (endogenous vs. adoptively transferred immune cells) if the cytokine was deemed important in viral clearance. When wild-type memory cells were administered to TNF-α–deficient carrier mice, viral clearance was observed within 2 wk. However, when TNF-α–deficient memory cells were injected into mice from the same carrier colony, no reductions in serum viral titers were observed. These data demonstrate that TNF-α is required for successful immunotherapy and must be produced by adoptively transferred rather than endogenous immune cells for clearance to occur.
DISCUSSION
In this study, we sought unique mechanistic insights into immunocytotherapy in an attempt to better understand this remarkable process. Our studies led to several important observations that link professional APCs to the successful therapeutic clearance of a persistent viral infection. First, we demonstrated that LCMV-specific CTLs arrive in the CNS early after adoptive immunotherapy, and this correlates almost perfectly with the activation/recruitment of CNS APCs (i.e., DCs, macrophages, and microglia) in the brain parenchyma. Second, of the three CNS APC populations, only DCs possessed the ability to restimulate memory CTLs directly ex vivo. Third, DCs extracted from immunotherapy recipients preferentially stimulated memory CTLs to produce the antiviral cytokine TNF-α, a cytokine shown in this study to be required in donor memory cells for successful immunotherapy. Finally, we demonstrated an absolute dependence of adoptively transferred memory T cells on DCs. In the absence of DCs, secondary effector T cells failed to significantly expand in carrier mice after adoptive immunotherapy, CTL/APC migration into the CNS was impaired, and CNS viral clearance was not attained.
It was previously surmised that the clearance of virus from neurons after adoptive immunotherapy did not depend on the activities of memory T cells within the CNS (7, 36). This theory was based, in part, on the failure to detect cellular infiltrates in the brain parenchyma of immunotherapy recipients (7). By using more sensitive approaches, we demonstrated in this study that a requisite population of traceable LCMV-specific memory CTLs can indeed be found in the CNS as early as 1 d after immunotherapy, and, at the peak of expansion (day 8), these CTLs were spatially distributed throughout the brain parenchyma. Neuronal viral clearance was not achieved at this time; however, the quantity of CTLs on sagittal brain reconstructions was found to correlate negatively with the viral load in the CNS, suggesting that the CTLs were already exerting an immunological pressure at day 8. These data indicate that the failure to purge CNS virus in a timely manner cannot be explained by an inability of CTLs to migrate into the brain parenchyma. Rather, it is more likely that the delay in clearance is related to the constraints imposed on CTLs by the immunologically specialized CNS.
Although LCMV-specific CTLs were not able to achieve neuronal viral clearance with kinetics mirroring that observed in the periphery, their arrival in the CNS coincided perfectly with a dramatic elevation in MHC I and II expression. MHC expression was barely detectable in untreated carrier mice; however, by day 8 after immunotherapy, MHC I– and MHC II–bearing cells were distributed evenly throughout the parenchyma as well as the lining of the brain. CNS microglia and macrophages are APCs known to express MHC II (especially upon activation), and flow cytometric studies revealed that both cell populations were sources of MHC II in immunotherapy recipients. Although microglia/macrophages have the potential to reactivate memory T cells, we became intrigued by the possibility of DC involvement in the immunotherapeutic process given that this APC population is a potent stimulator of both naive (37) and memory (22) T cells. DCs are not normally found in the brain parenchyma (14, 15), but studies indicate that Toxoplasma gondii infection (16), experimental autoimmune encephalomyelitis (15, 20, 21), and acute brain injury (19) all promote the appearance of parenchymal DCs. In fact, one study demonstrated that DCs were capable of activating naive myelin-specific CD4+ T cells directly within the CNS (20). CNS DCs also appear capable of reactivating primed myelin-reactive CD4+ T cells during adoptive experimental autoimmune encephalomyelitis (21). Collectively, these data indicate that CNS DCs can inadvertently promote autoimmune pathogenesis.
Although CNS DCs can facilitate undesirable pathogenesis (20, 21), we demonstrate that a diametrically opposed outcome does exist upon DC migration into the CNS. As a consequence of immunotherapy, a substantial number of DCs were recruited into the brain parenchyma. These DCs showed an elevated expression of costimulatory molecules (B7.1/B7.2) as well as antigen-presenting machinery (MHC I/II) and were the only APC population capable of restimulating memory CTLs ex vivo. The selective stimulatory capacity of DCs could not be explained by a failure of other APC populations to become activated or migrate into the CNS. Macrophages were observed in the CNS after immunotherapy, yet these cells failed to restimulate memory CTLs. Microglia were also unable to restimulate memory CTLs despite up-regulating CD11c, a marker thought to signify microglial transformation into a DC-like cell population (31). Thus, DCs alone can harbor the potential to stimulate memory T cells in the CNS without initiating a pathogenic cascade.
Given that stimulatory DCs are recruited into the brain parenchyma as a consequence of immunotherapy, we propose that this professional APC population may participate in the noncytopathic clearance of neurons by sustaining the activities of the antiviral CTL or by promoting antiviral cytokine release within the brain parenchyma. T cells have the capacity to purge viruses noncytopathically through cytokine release (38), and engagement of peptide–MHC-bearing DCs in the brain parenchyma may favor viral clearance from adjacent neurons through this mechanism. In support of this theory, we demonstrated that CTLs engaged MHC II–bearing APCs in the CNS of immunotherapy recipients. We further demonstrated that DCs were the only MHC II+ APC extracted from the CNS of immunotherapy recipients that induced memory CTLs to produce TNF-α and, to a limited degree, IFN-γ. Although DCs from immunotherapy recipients preferentially induced TNF-α production through an as yet unknown mechanism, both TNF-α and IFN-γ (13) are required to achieve total body clearance in LCMV carrier mice.
One of the most important observations from this study was the dependence of memory T cells on DCs for successful immunotherapeutic clearance. In DC-depleted carrier mice, adoptively transferred memory CTLs demonstrated a dramatic reduction in secondary expansion and were impaired in their ability to purge LCMV systemically as well as from the CNS. DC depletion also significantly reduced CTL and APC migration/activation in the CNS of immunotherapy recipients. It is possible that memory CTLs also depend on macrophages for restimulation, as a recent study demonstrated that certain macrophage populations are depleted in CD11c-DTR tg mice (39). However, we consider this possibility unlikely given that macrophages derived from the CNS or spleen of immunotherapy recipients did not stimulate memory CTLs ex vivo. DCs likely represent the main APC population required for the optimization of secondary T cell responses to pathogens (22), and we postulate that this reliance on DCs becomes even greater when memory T cells are transferred into a host heavily burdened with a systemic viral infection. Our results demonstrate clearly that memory T cells require DCs for efficient secondary expansion in carrier mice; however, it remains to be determined whether DCs are continually required to sustain the activities of memory T cells in the CNS during the therapeutic clearance of a persistent viral infection. This latter scenario seems likely given the near perfect correlation between CTLs and stimulatory DC recruitment into the CNS after immunotherapy, and studies to evaluate this possibility are presently ongoing.
In conclusion, immunocytotherapy is a remarkably efficient means to achieve systemic clearance of a persistent viral infection. The precision and efficacy of this process is best exemplified by the ability of memory T cells to operate within the confines of the immunologically specialized CNS and noncytopathically purge virus from a cell population (neurons) that does not readily express MHC (9, 10). Thus, clearance of a persistent viral infection from the CNS is attainable. In this study, we demonstrated that adoptive immunotherapy influenced DC activity within the CNS, and the success of this therapeutic intervention was dependent on this potent APC subset. Therefore, we propose that a therapeutic enhancement of DC recruitment into the CNS may provide an excellent strategy to promote (or accelerate) the clearance of a persistent viral infection.
MATERIALS AND METHODS
Mice.
C57BL/6, C57BL/6 Thy1.1+DbGP33–41 TCR-tg, C57BL/6 Thy1.1+I-AbGP61–80 TCR-tg, C57BL/6 GFP+DbGP33–41 TCR-tg, C57BL/6 CD11c-DTR tg, and C57BL/6 × 129 TNF-α–deficient mice were bred and maintained in a closed breeding facility at The Scripps Research Institute. C57BL/6 Thy1.1+I-AbGP61–80 TCR-tg mice were a gift of H. Hengartner (Institute of Experimental Immunology, University Hospital Zurich, Zurich, Switzerland). The handling of all mice conformed to the requirements of the National Institutes of Health and The Scripps Research Institute animal research committee.
Virus.
The parental Armstrong 53b of LCMV, which is a triple-plaque purified clone from Armstrong CD 1371 (40), was used for all experiments. 1-d-old C57BL/6 and C57BL/6 × 129 TNF-α–deficient mice were infected with 103 PFU of LCMV Armstrong to generate persistently infected adult mice as described previously (7). Because LCMV is known to spread through vertical transmission (i.e., mother to offspring), the resultant mice were bred to establish a persistently infected colony (referred to as LCMV carrier mice). Mice obtained from this LCMV carrier colony were used for all lines of experimentation. To establish a carrier colony of CD11c-DTR tg mice, persistently infected female C57BL/6 mice were crossed with uninfected male CD11c-DTR mice. All mice derived from this cross were persistently infected with LCMV. Viral titers were determined by plaque assay on Vero cells as described previously (41).
Immunocytotherapy.
To generate LCMV memory donors for immunocytotherapy experiments, C57BL/6 mice were seeded with 104 naive GFP+ (or Thy1.1+) DbGP33–41-specific CD8+ T cells (25, 42). For cotransfer experiments, C57BL/6 mice were seeded with 104 naive Thy1.1+ DbGP33–41-specific CD8+ T cells and 104 naive Thy1.1+ I-AbGP61–80-specific CD4+ T cells. All naive cells were purified from TCR-tg mice by negative selection (StemCell Technologies Inc.). The enrichment procedure resulted in a purity of >98%. 1 d after the adoptive transfer, mice were infected i.p. with 105 PFU of LCMV Armstrong. For the TNF-α KO experiments shown in Fig. 9, C57BL/6 × 129 TNF-α KO mice were infected with the same dose of virus to generate cytokine-deficient memory cells. Because TNF-α KO mice infected i.p. with LCMV Armstrong mounted a robust anti-LCMV CTL response and cleared the virus, they could be used as memory donors. These donor mice were not seeded with DbGP33–41-specific CD8+ T cells. Splenocytes were harvested from all mice at time points >45 d after infection, when LCMV-specific memory T lymphocytes are known to be present in the spleen (26, 43). For all immunocytotherapy experiments, 1–2 × 107 memory splenocytes either unseeded (TNF-α KO experiments) or seeded with traceable DbGP33–41-specific CD8+ T cells (see Fig. 2 A for an example) were injected i.p. into LCMV carrier mice.
DC depletion.
To deplete DCs, CD11c-DTR tg mice persistently infected with LCMV were injected i.p. with 2 ng/g DT in 100 μl PBS. This dose of DT was selected from a titration experiment in which CD11c-DTR carrier mice were injected with 4, 2, and 1 ng/g of toxin. All doses of DT resulted in a similar level of splenic DC depletion 1 d after injection. The 2-ng/g (instead of the traditional 4 ng/g; reference 34) dose of DT was ultimately used for all experiments because no evidence of toxicity was observed with this dosage.
Mononuclear cell isolations and tissue processing.
To obtain cell suspensions for flow cytometric analyses/stimulation cultures, the spleens and CNS were harvested from mice after an intracardiac perfusion with a 0.9% saline solution to remove the contaminating blood lymphocytes. Splenocytes and brain-infiltrating leukocytes were incubated with 1 ml collagenase D (1 mg/ml; Roche) at 37°C for 20 min and mechanically disrupted through a 70-μm filter. Splenocytes were treated with RBC lysis buffer (0.14 M NH4Cl and 0.017 M Tris-HCl, pH 7.2), washed twice, and analyzed. To extract brain-infiltrating leukocytes, homogenates were resuspended in 90% Percoll (4 ml), which was overlaid with 60% Percoll (3 ml), 40% Percoll (4 ml), and finally 1× HBSS (3 ml). The Percoll gradients were then centrifuged at 1,500 rpm for 15 min, after which the band corresponding to mononuclear cells was carefully extracted, washed, and ultimately analyzed. The number of mononuclear cells was determined from each organ preparation and used to calculate the absolute number of specific cell populations. PBMCs were obtained by centrifugation after incubating (for 1 h on ice) a capillary of blood (∼210 μl) with 10 ml RBC lysis buffer. To visualize GFP+DbGP33–41-specific T cells in situ, mice received an intracardiac perfusion of 4% paraformaldehyde. Brains and spleens were then removed and incubated for 24 h in 4% paraformaldehyde and for an additional 24 h in 30% sucrose. Afterward, tissues were submerged in optimal cutting temperature (Tissue-Tek) and frozen on dry ice. For all other immunohistochemical analyses, fresh, unfixed tissues were frozen in optimal cutting temperature.
Flow cytometry.
The following antibodies purchased from BD Biosciences were used to stain splenocytes and brain-infiltrating lymphocytes: anti-B7.1-FITC, anti-B7.2-PE, CD11b-PE-CY7, anti-CD11b-PerCP-Cy5.5, anti-CD11c-APC, anti-CD19-PE, anti-CD3-PE, anti-CD3-PerCP, anti-CD45.2-FITC, anti-CD45.2-biotin, anti-CD8-PE, anti-CD8-APC, anti-CD8-APC-CY7, anti-Db-PE, anti-I-Ab-PE, anti-IFNγ-APC, anti-NK1.1-PE, anti-NK1.1-PerCP, anti-Thy1.1-PerCP, and anti-TNFα-PE. Anti-CD8–Pacific blue was purchased from Caltag, and streptavidin–Pacific blue was purchased from Invitrogen. Before staining, all cell preparations were blocked with 3.3 μg/ml anti–mouse CD16/CD32 (Fc block; BD Biosciences) in PBS containing 1% FBS for 10 min. The Fc block was also included in all 20-min surface stains. Cells were acquired using a flow cytometer (Digital LSR II; Becton Dickinson), which allows up to 10-color detection by using four different excitation lasers. Flow cytometric data were analyzed with FlowJo software (Tree Star, Inc.).
CNS APC stimulation cultures.
Mononuclear cells were extracted from the CNS/spleens of LCMV carrier mice 8 d after immunocytotherapy as described in Mononuclear cell isolations and tissue processing. To obtain a sufficient number of CNS APCs, 12 immunocytotherapy recipients were pooled together. APCs extracted from the spleens of LCMV carrier mice (not receiving immunocytotherapy) and naive C57BL/6 mice served as controls. The extracted mononuclear cell preparations were then stained with the following antibodies: CD45.2-FITC (leukocyte marker), CD3-PE (T cells), CD19-PE (B cells), NK1.1-PE (NK cells), CD11b-PerCP-Cy5.5 (myeloid marker), and CD11c-APC (DCs). Because CD11b and CD11c can be up-regulated on cells other than APCs (e.g., T cells and NK cells), a “dump channel” was included in the flow cytometric sorts to ensure a highly enriched population of APCs that did not include contaminating leukocyte populations. CD3-PE, CD-19-PE, and NK1.1-PE were used to exclude T, B, and NK cells, respectively. Flow cytometric sorting was performed using a cell sorter (FACS Aria; BD Biosciences) to simultaneously collect DCs (CD45hiCD3−NK1.1−CD19−CD11c+), macrophages (CD45hiCD3−NK1.1−CD19−CD11c−CD11b+), and microglia (CD45low/intCD3−NK1.1−CD19−CD11b+). It should be noted that cells expressing low and intermediate levels of CD45 (i.e., microglia) were only found in the CNS preparations.
To isolate LCMV-specific memory CD8+ T cells for the stimulation cultures, C57BL/6 mice were seeded with 104 Thy1.1+DbGP33–41-specific CD8+ T cells. 1 d later, mice were infected with 105 PFU of LCMV Armstrong. Splenocytes were isolated from the mice at 45 d after infection, enriched using MACs (Miltenyi Biotec) anti-CD8 positive selection beads, and stained with anti-Thy1.1-PE. Flow cytometric sorting was then used to isolate Thy1.1+ (or DbGP33–41 specific) T cells. After the flow cytometric sorting, all cells were resuspended in RPMI 1640 (Life Technologies) containing 10% FBS, 1% penicillin/streptomycin, 1% l-glutamine, 1% Hepes, 1% nonessential amino acids, 1% sodium pyruvate, and 50 μM β-mercaptoethanol. Afterward, 50,000 Thy1.1+DbGP33–41-specific memory T cells were mixed with 100,000 of the indicated APC populations (1:2 T cell/DC ratio) in a 96-well U-bottom plate for 24 h in a humidified 37°C incubator (5% CO2). 5 μg/ml brefeldin A (Sigma-Aldrich) was added in the 19th hour of the 24-h culture period to block cytokine release and permit intracellular staining. After the incubation period, cultured cells were stained with antibodies directed against Thy1.1, CD8, IFN-γ, and TNF-α, and the cytokine-producing Thy1.1+DbGP33–41-specific CD8+ T cells were analyzed by flow cytometry.
Immunohistochemistry.
To visualize LCMV, DCs, and neurons, 6-μm frozen sections were cut, fixed with 2% formaldehyde, blocked with an avidin/biotin-blocking kit (Vector Laboratories), and stained for 1 h at room temperature with guinea pig anti-LCMV (1:1,000), 2.5 μg/ml of Armenian hamster anti-CD11c (Serotec), or 1.25 μg/ml of mouse antineuronal nuclei (anti-NeuN; Chemicon International), respectively. To block endogenous mouse antibodies, sections stained with mouse anti-NeuN were preincubated for 1 h at room temperature with 35 μg/ml of a Fab anti–mouse H and L chain antibody (Jackson ImmunoResearch Laboratories). After the primary antibody incubation, sections were washed, stained for 1 h at room temperature with a biotinylated secondary antibody (1:400; Jackson ImmunoResearch Laboratories), washed, and stained for 1 h at room temperature with streptavidin–Rhodamine Red-X (1:400; Jackson ImmunoResearch Laboratories). All sections were costained for 5 min at room temperature with 1 μg/ml DAPI (Sigma-Aldrich) to visualize cell nuclei.
To visualize MHC class II, frozen sections were fixed with cold (−20°C) 95% ethanol, blocked with an avidin/biotin-blocking kit, and stained for 1 h at room temperature with 1.0 μg/ml of rat anti–MHC class II (BD Biosciences). After the primary antibody incubation, sections were washed, stained for 1 h at room temperature with a biotinylated secondary antibody, washed, and stained for 1 h at room temperature with streptavidin–Rhodamine Red-X. For simultaneous analysis of GFP, MHC II, and LFA (Fig. 8), MHC II staining was combined with a primary antibody directed against 3 μg/ml LFA-1 (BD Biosciences). The LFA-1 primary was visualized with a secondary antibody conjugated to Cy5. For colabeling of LCMV and NeuN (Fig. 1, B–D), frozen sections were stained as described in the beginning of this section except that the anti-LCMV antibody was detected with an anti–guinea pig secondary antibody directly conjugated to FITC (1:750; for 1 h at room temperature). All working stocks of primary and secondary reagents were diluted in PBS containing 2% FBS.
Microscopy.
Two- and three-color organ reconstructions to visualize the distribution of LCMV, MHC class II, and GFP+DbGP33–41-specific CD8+ T cells on 6-μm frozen sections were obtained using an immunofluorescence microscope (Axiovert S100; Carl Zeiss MicroImaging, Inc.) fitted with an automated xy stage, a color digital camera (Axiocam, Carl Zeiss MicroImaging, Inc.), and a 5× objective. Registered images were captured for each field on the tissue section, and reconstructions were performed using the MosaiX function in KS300 image analysis software (Carl Zeiss MicroImaging, Inc.). Higher resolution images of LCMV-infected neurons (Fig. 1, B–D) and interactions between GFP+DbGP33–41-specific CTLs and MHC II+ APCs (Fig. 8) were captured with a confocal microscope (MRC1024; Bio-Rad Laboratories) fitted with a krypton/argon mixed gas laser (excitation at 488, 568, and 647 nm) and a 40× oil objective (Carl Zeiss MicroImaging, Inc.). All two-dimensional confocal images illustrate a single z section captured at a position approximating the midline of the cell.
Image analysis.
The percentage of CNS tissue occupied by MHC I, MHC II, and LCMV staining was calculated from sagittal brain reconstructions (see Fig.4 and previous section) using a program written for the KS300 image analysis software.
Statistical analyses.
Data handling, analysis, and graphical representations were performed using Microsoft Excel and SigmaPlot 8.0 (Systat). Statistical differences were determined by Student's t test or one-way analysis of variance (ANOVA; P < 0.05) using SigmaStat 2.0 (SigmaStat). Correlation coefficients were also calculated in SigmaStat using a Pearson Product Moment correlation test (P < 0.05).
Online supplemental material.
Fig. S1 shows the recruitment of DbGP33–41-specific CD8+ and I-AbGP61–80-specific CD4+ T cells into the CNS of immunotherapy recipients. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20060039/DC1.
Supplemental Material
Acknowledgments
This work was supported by National Institutes of Health grants NS048866-01 (to D.B. McGavern), AI09484 (to M.B.A. Oldstone), and AI045927 (to E.I. Zuniga and M.B.A. Oldstone), a grant from The Dana Foundation (to D.B. McGavern), and a grant from Novartis (to D.B. McGavern). H. Lauterbach is supported by a fellowship from Deutsche Forschungsgemeinschaf.
The authors have no conflicting financial interests.
Abbreviations used: ANOVA, analysis of variance; CNS, central nervous system; DT, diphtheria toxin; DTR, DT receptor; GP, glycoprotein; LCMV, lymphocytic choriomeningitis virus; tg, transgenic.
H. Lauterbach and E.I. Zuniga contributed equally to this paper.
References
- 1.Borrow, P., and M.B.A. Oldstone. 1997. Lymphocytic choriomeningitis virus. Lippincott-Raven Publishers, Philadelphia. 940 pp.
- 2.Oldstone, M.B. 1987. Immunotherapy for virus infection. Curr. Top. Microbiol. Immunol. 134:211–229. [DOI] [PubMed] [Google Scholar]
- 3.Homann, D. 2002. Immunocytotherapy. Curr. Top. Microbiol. Immunol. 263:43–65. [DOI] [PubMed] [Google Scholar]
- 4.Fazakerley, J.K., P. Southern, F. Bloom, and M.J. Buchmeier. 1991. High resolution in situ hybridization to determine the cellular distribution of lymphocytic choriomeningitis virus RNA in the tissues of persistently infected mice: relevance to arenavirus disease and mechanisms of viral persistence. J. Gen. Virol. 72:1611–1625. [DOI] [PubMed] [Google Scholar]
- 5.Pircher, H., K. Burki, R. Lang, H. Hengartner, and R.M. Zinkernagel. 1989. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 342:559–561. [DOI] [PubMed] [Google Scholar]
- 6.von Herrath, M.G., D.P. Berger, D. Homann, T. Tishon, A. Sette, and M.B. Oldstone. 2000. Vaccination to treat persistent viral infection. Virology. 268:411–419. [DOI] [PubMed] [Google Scholar]
- 7.Oldstone, M.B., P. Blount, P.J. Southern, and P.W. Lampert. 1986. Cytoimmunotherapy for persistent virus infection reveals a unique clearance pattern from the central nervous system. Nature. 321:239–243. [DOI] [PubMed] [Google Scholar]
- 8.Ransohoff, R.M., P. KivisAkk, and G. Kidd. 2003. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 3:569–581. [DOI] [PubMed] [Google Scholar]
- 9.Joly, E., L. Mucke, and M.B. Oldstone. 1991. Viral persistence in neurons explained by lack of major histocompatibility class I expression. Science. 253:1283–1285. [DOI] [PubMed] [Google Scholar]
- 10.Joly, E., and M.B. Oldstone. 1992. Neuronal cells are deficient in loading peptides onto MHC class I molecules. Neuron. 8:1185–1190. [DOI] [PubMed] [Google Scholar]
- 11.Volkert, M. 1962. Studies on immunological tolerance to LCM virus. A preliminary report on adoptive immunization of virus carrier mice. Acta. Pathol. Microbiol. Scand. 56:305–310. [PubMed] [Google Scholar]
- 12.Berger, D.P., D. Homann, and M.B. Oldstone. 2000. Defining parameters for successful immunocytotherapy of persistent viral infection. Virology. 266:257–263. [DOI] [PubMed] [Google Scholar]
- 13.Tishon, A., H. Lewicki, G. Rall, M. Von Herrath, and M.B. Oldstone. 1995. An essential role for type 1 interferon-gamma in terminating persistent viral infection. Virology. 212:244–250. [DOI] [PubMed] [Google Scholar]
- 14.Matyszak, M.K., and V.H. Perry. 1996. The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience. 74:599–608. [DOI] [PubMed] [Google Scholar]
- 15.Serafini, B., S. Columba-Cabezas, F. Di Rosa, and F. Aloisi. 2000. Intracerebral recruitment and maturation of dendritic cells in the onset and progression of experimental autoimmune encephalomyelitis. Am. J. Pathol. 157:1991–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer, H.G., U. Bonifas, and G. Reichmann. 2000. Phenotype and functions of brain dendritic cells emerging during chronic infection of mice with Toxoplasma gondii. J. Immunol. 164:4826–4834. [DOI] [PubMed] [Google Scholar]
- 17.Fischer, H.G., and G. Reichmann. 2001. Brain dendritic cells and macrophages/microglia in central nervous system inflammation. J. Immunol. 166:2717–2726. [DOI] [PubMed] [Google Scholar]
- 18.Karman, J., C. Ling, M. Sandor, and Z. Fabry. 2004. Initiation of immune responses in brain is promoted by local dendritic cells. J. Immunol. 173:2353–2361. [DOI] [PubMed] [Google Scholar]
- 19.Reichmann, G., M. Schroeter, S. Jander, and H.G. Fischer. 2002. Dendritic cells and dendritic-like microglia in focal cortical ischemia of the mouse brain. J. Neuroimmunol. 129:125–132. [DOI] [PubMed] [Google Scholar]
- 20.McMahon, E.J., S.L. Bailey, C.V. Castenada, H. Waldner, and S.D. Miller. 2005. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat. Med. 11:335–339. [DOI] [PubMed] [Google Scholar]
- 21.Greter, M., F.L. Heppner, M.P. Lemos, B.M. Odermatt, N. Goebels, T. Laufer, R.J. Noelle, and B. Becher. 2005. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat. Med. 11:328–334. [DOI] [PubMed] [Google Scholar]
- 22.Zammit, D.J., L.S. Cauley, Q.M. Pham, and L. Lefrancois. 2005. Dendritic cells maximize the memory CD8 T cell response to infection. Immunity. 22:561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Rijt, L.S., S. Jung, A. Kleinjan, N. Vos, M. Willart, C. Duez, H.C. Hoogsteden, and B.N. Lambrecht. 2005. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J. Exp. Med. 201:981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGavern, D.B., D. Homann, and M.B. Oldstone. 2002. T cells in the central nervous system: the delicate balance between viral clearance and disease. J. Infect. Dis. 186:S145–S151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGavern, D.B., U. Christen, and M.B. Oldstone. 2002. Molecular anatomy of antigen-specific CD8(+) T cell engagement and synapse formation in vivo. Nat. Immunol. 3:918–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murali-Krishna, K., J.D. Altman, M. Suresh, D.J. Sourdive, A.J. Zajac, J.D. Miller, J. Slansky, and R. Ahmed. 1998. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 8:177–187. [DOI] [PubMed] [Google Scholar]
- 27.Cabanas, C., and F. Sanchez-Madrid. 1999. CD11c (leukocyte integrin CR4 alpha subunit). J. Biol. Regul. Homeost. Agents. 13:134–136. [PubMed] [Google Scholar]
- 28.Lin, Y., T.J. Roberts, V. Sriram, S. Cho, and R.R. Brutkiewicz. 2003. Myeloid marker expression on antiviral CD8+ T cells following an acute virus infection. Eur. J. Immunol. 33:2736–2743. [DOI] [PubMed] [Google Scholar]
- 29.Sedgwick, J.D., S. Schwender, H. Imrich, R. Dorries, G.W. Butcher, and V. ter Meulen. 1991. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc. Natl. Acad. Sci. USA. 88:7438–7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ford, A.L., A.L. Goodsall, W.F. Hickey, and J.D. Sedgwick. 1995. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J. Immunol. 154:4309–4321. [PubMed] [Google Scholar]
- 31.Santambrogio, L., S.L. Belyanskaya, F.R. Fischer, B. Cipriani, C.F. Brosnan, P. Ricciardi-Castagnoli, L.J. Stern, J.L. Strominger, and R. Riese. 2001. Developmental plasticity of CNS microglia. Proc. Natl. Acad. Sci. USA. 98:6295–6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Slifka, M.K., and J.L. Whitton. 2000. Activated and memory CD8+ T cells can be distinguished by their cytokine profiles and phenotypic markers. J. Immunol. 164:208–216. [DOI] [PubMed] [Google Scholar]
- 33.Homann, D., D.B. McGavern, and M.B. Oldstone. 2004. Visualizing the viral burden: phenotypic and functional alterations of T cells and APCs during persistent infection. J. Immunol. 172:6239–6250. [DOI] [PubMed] [Google Scholar]
- 34.Jung, S., D. Unutmaz, P. Wong, G. Sano, K. De los Santos, T. Sparwasser, S. Wu, S. Vuthoori, K. Ko, F. Zavala, et al. 2002. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 17:211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klavinskis, L.S., R. Geckeler, and M.B. Oldstone. 1989. Cytotoxic T lymphocyte control of acute lymphocytic choriomeningitis virus infection: interferon gamma, but not tumour necrosis factor alpha, displays antiviral activity in vivo. J. Gen. Virol. 70:3317–3325. [DOI] [PubMed] [Google Scholar]
- 36.de la Torre, J.C., G. Rall, C. Oldstone, P.P. Sanna, P. Borrow, and M.B. Oldstone. 1993. Replication of lymphocytic choriomeningitis virus is restricted in terminally differentiated neurons. J. Virol. 67:7350–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shortman, K., and Y.J. Liu. 2002. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2:151–161. [DOI] [PubMed] [Google Scholar]
- 38.Guidotti, L.G., and F.V. Chisari. 2001. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu. Rev. Immunol. 19:65–91. [DOI] [PubMed] [Google Scholar]
- 39.Probst, H.C., K. Tschannen, B. Odermatt, R. Schwendener, R.M. Zinkernagel, and M. Van Den Broek. 2005. Histological analysis of CD11c-DTR/GFP mice after in vivo depletion of dendritic cells. Clin. Exp. Immunol. 141:398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dutko, F.J., and M.B. Oldstone. 1983. Genomic and biological variation among commonly used lymphocytic choriomeningitis virus strains. J. Gen. Virol. 64:1689–1698. [DOI] [PubMed] [Google Scholar]
- 41.Brooks, D.G., L. Teyton, M.B. Oldstone, and D.B. McGavern. 2005. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. J. Virol. 79:10514–10527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGavern, D.B., and P. Truong. 2004. Rebuilding an immune-mediated central nervous system disease: weighing the pathogenicity of antigen-specific versus bystander T cells. J. Immunol. 173:4779–4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaech, S.M., S. Hemby, E. Kersh, and R. Ahmed. 2002. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 111:837–851. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}