

Abstract
The V(D)J recombinase catalyzes DNA transposition and translocation both in vitro and in vivo. Because lymphoid malignancies contain chromosomal translocations involving antigen receptor and protooncogene loci, it is critical to understand the types of “mistakes” made by the recombinase. Using a newly devised assay, we characterized 48 unique TCRβ recombination signal sequence (RSS) end insertions in murine thymocyte and splenocyte genomic DNA samples. Nearly half of these events targeted “cryptic” RSS-like elements. In no instance did we detect target-site duplications, which is a hallmark of recombinase-mediated transposition in vitro. Rather, these insertions were most likely caused by either V(D)J recombination between a bona fide RSS and a cryptic RSS or the insertion of signal circles into chromosomal loci via a V(D)J recombination-like mechanism. Although wild-type, p53, p53 x scid, H2Ax, and ATM mutant thymocytes all showed similar levels of RSS end insertions, core-RAG2 mutant thymocytes showed a sevenfold greater frequency of such events. Thus, the noncore domain of RAG2 serves to limit the extent to which the integrity of the genome is threatened by mistargeting of V(D)J recombination.
Adaptive immunity depends on V(D)J recombination to assemble antigen receptor genes from their component gene segments during B and T cell development. The recombinase introduces double strand (ds) DNA breaks at recombination signal sequences (RSSs) that flank rearranging gene segments. An RSS consists of conserved heptamer and nonamer sequences separated by a spacer of conserved length (either 12 or 23 bp). The DNA break is made precisely at the junction between the RSS heptamer and the gene segment. These broken DNA molecules are then joined with the assistance of the nonhomologous end joining (NHEJ) family of dsDNA break repair proteins to form coding and signal joints (Fig. 1 A) (1). Mutations in genes encoding NHEJ proteins greatly diminish the frequency of productive V(D)J recombination and, when combined with mutations in p53, cause striking genomic instability and cell transformation, resulting in leukemia and lymphoma (2).
Figure 1.

The LM-TECA assay for TCRb locus RSS-fragment insertion. (A) Diagram of the germline TCRβ locus (top) and its various recombination intermediates (middle) and alternative products (bottom two lines). Triangles indicate recombination signal sequences (open triangle, RSS-12; filled triangle, RSS-23). The approximate locations of restriction sites used in LM-TECA are indicated by vertical gray arrows. (B) RSS-ended fragment excised from the TCRβ locus (thin line) shown with its reactive 39′OH groups inserting into a target DNA (thick line) elsewhere in the genome, resulting in an RSS fragment insertion event. (C) The LM-TECA method. B→ indicates a 5′ biotin-labeled primer, the white square indicates an oligonucleotide linker, and arrows indicate PCR primers (open arrow, linker primer; filled arrow, locus primer). (D) Strategy for amplification of the “other” end of an RSS fragment insertion or a balanced translocation. PCR primers are shown as arrows.
Strong evidence exists that the lymphoid V(D)J recombinase evolved from an ancient transposase (3). Consistent with this idea, genes encoding the lymphoid-specific components of the recombinase, RAG1 and RAG2, are physically linked in the genome, convergently transcribed, and lack introns in their coding regions (4). Certain aspects of the biochemistry of V(D)J recombination also resemble a transposition reaction. RAG1 contains, and its function depends on, a catalytic triad of amino acids that is common to many transposases, theD-D-E motif (5). RSSs are recognized and cleaved in a pairwise fashion, generating two blunt and two hairpin ends, as is the case in Tn10 and Hermes transposition. In vivo, the two broken RSS ends are most often ligated to one another, generating an episomal signal joint (Fig. 1 A); however, infrequently, broken RSSs can rejoin to coding ends, thereby forming what are called hybrid (Fig. 1 A, bottom) and open-and-shut joints (6). In vitro, however, RSS ends efficiently transpose, generating a variety of strand-transfer products (7–9). A truncation mutant of RAG2 (core-RAG2, consisting of amino acids 1–383 out of the 527 amino acids of the full-length protein) results in a greatly increased frequency of transposition in vitro (10–12) and hybrid joining in vivo (13). Understanding how these side reactions of V(D)J recombination are suppressed is of great significance given the frequent observation of chromosomal translocations and deletions in lymphoid malignancy (14).
V(D)J recombination can contribute to genomic instability and malignancy in several ways (15, 16). First, the recombinase can recognize so-called “cryptic” RSSs in nonantigen receptor loci (defined as a DNA sequence identical to a known functional RSS heptamer) and use them together with either bona fide RSSs or other cryptic RSSs to catalyze translocations or deletions. Previous studies revealed that the ability of cryptic RSSs associated with certain protooncogenic loci to undergo V(D)J recombination is within 10–100-fold that of a proper RSS (17). In addition, RAG proteins have the ability to recognize, cleave, and rearrange certain DNA sequences that do not resemble RSSs, but have the tendency to contain non–B-form DNA. A particularly clear example of this was reported in the major breakpoint region (Mbr) of the Bcl-2 locus, which is the most frequently rearranged oncogene in follicular lymphoma (18). The RAGs can also activate the 3′-OH group on broken RSS ends to attack duplex DNA, generating a dsDNA break in the accidental target of this reaction (9). Recently, it was reported that signal joints, previously considered inert, can reinsert into Ig or TCR loci, as well as into cryptic RSSs (19). Finally, as occurs in vitro, the RAGs can catalyze the transpositional insertion of an RSS-ended fragment into genomic DNA (Fig. 1 B). Such RAG-mediated transposition events are characterized by short, target-site sequence duplications. Only three biological examples of this have been published in the literature. In each case, an excised, signal-ended fragment from the TCRα locus was inserted into an intron within the hprt locus (20, 21). Using a retroviral reporter construct in a virally transformed pro–B cell line, Reddy et al. reported that the frequency of RSS-fragment transposition in vivo was ∼1 in 44,000 recombination events (22).
To determine which of the biochemical activities of the V(D)J recombinase contribute to genomic instability, and to understand the mechanisms that limit RSS fragment transposition and other RSS end insertion events in developing lymphocytes, we developed a methodology to detect and characterize TCRβ locus RSS end insertions, including transposition, in genomic DNA purified from mouse T lineage cells. We applied this assay to wild-type and various mutant murine DNA samples. Our results reveal that the genomes of developing thymocytes are littered with TCR signal end integration events that may contribute to genomic instability, that a high fraction of such events target cryptic RSSs, and that the noncore domain of RAG2 serves to limit these potentially deleterious events.
RESULTS
LM-TECA assay for RSS fragment insertion
We designed the ligation-mediated transferred-end capture assay (LM-TECA) to detect the insertion of a RSS-ended DNA fragment generated during V(D)J recombination into random sites around the genome (Fig. 1 C). High molecular weight genomic DNA is restriction digested, denatured, and subjected to primer extension using a biotinylated primer homologous to a region adjacent to either the Dβ1 RSS (D-side) or the Jβ1.1 RSS (J-side). Primer extension products are captured using streptavidin-coated paramagnetic beads and ligated to an oligonucleotide linker. The linker-ligated DNA is analyzed by PCR using a linker primer and a RSS fragment primer. Amplified products are purified by gel electrophoresis (Fig. 2) and characterized by DNA sequence analysis (Table I). Once the genomic location of an inserted RSS end has been determined, direct PCR can be used in an attempt to identify the sequence at the other end of the RSS fragment (Fig. 1 D, top) or the other joining product of a balanced translocation (Fig. 1 D, bottom).
Figure 2.

Gel analysis of LM-TECA PCR products. 10 0.2-μl aliquots (lanes 1–10) of captured wild-type thymus DNA were subjected to PCR analysis for J-sided RSS transposition events. The filled arrow indicates the 629-bp amplification product from the unrearranged TCRβ locus that is caused by the restriction site used to fragment the genomic DNA (Fig. 1 A, vertical gray arrow). Individual bands were excised from the gel and subjected to DNA sequence analysis. The open arrows indicate two RSS insertion events, whereas the other discrete bands correspond to random breaks along the germline TCRβ locus. Neg, a negative PCR control consisting of 63–12 cell DNA; mkr, MspI-digested pBR322.
Table I.
Partial DNA sequences of Dβ1 and Jβ1 signal end insertions into various chromosomal locations
| Event
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|
| Name | Geno | Chromosome breakpoint | N and P | Dβ1 Side | Gene | UCSC chr. position |
| A | 3′-RSS23 | |||||
| Germline | CACGGTG ATTCAATT | |||||
| E1-4 | WT | CCCGAGCTATcacagtgctc | AATGGCC | CACGGTG ATTCAATT | TCR Jγ1 | chr13:18678545 |
| E2-2 | WT | CCCCACTGTGctctaagcca | ACC | CACGGTG ATTCAATT | chr16:55739889 | |
| E3-4 | WT | HPAGCACCCGTG tgccaccaaa | CCG | CACGGTG ATTCAATT | chr7:69460416 | |
| EMAT2 | WT | ACACAGTGAGgtagaacgga | CACGGTG ATTCAATT | BC042698 | chr1:138847427 I. | |
| EMAT5-1 | WT | AGTGACTGTGtgggccttcc | CACGGTG ATTCAATT | C80913 (NNX3) | chr7:34900516 I. | |
| E2b-6 | cR2 | GAGCACTGTGagcgttgcca | CACGGTG ATTCAATT | lama3 | chr18:12860543 | |
| b196 | WT | TTCTGCTGTGgatcttggca | see note | CACGGTG ATTCAATT | hotspot | chr1:143968210 |
| b154 | WT | TTACACTGTGtggcagtaat | CACGGTG ATTCAATT | Scarb1 | chr5:124540600 I. | |
| EMAT5-2 | WT | GAGTCTGTGTgtcacagcag | ACGGTG ATTCAATT | DEAD box polypeptide 21 | chr10:62120725 | |
| E3-1 | WT | TCCCAACTATcacagtgctc | ATTCG | CGGTG ATTCAATT | TCR Jγ2 | chr13:18624358 |
| b220 | cR2 | TAGTGTTGTGtgcacactga | GCC | TCAATT | BC096449, unknown mRNA | chr1:152252917 |
| EL3-1 | WT | AGGACTGATGcagaaattcg | GTG | ATT | Vh J558 | chr12:112181483 I. |
| Jβ1.x Side | ||||||
| Name | Geno | 5′-RSS12 | N | Chromosome breakpoint | Gene | UCSC chr. position |
| B | ||||||
| Jβ1.1 | TCCTATGG CACTGTG | |||||
| EL2-4 | WT | TCCTATGG CACTGTG | ??????GACCGCCGTC | too short to align | ||
| EL3-1 | WT | TCCTATGG CACTGTG | GGGGTCC | aggactgatgCAGAAATTCG | Vh J558 | chr12:112181482 I. |
| S3 | WTT-cell | TCCTATGG CACTGTG | TC | tctgccactgTGCCCTCTCT | TCR Vα16 | chr14:48296888 |
| b175 | WTm/h | TCCTATGG CACTG | GG | ctgtgtcctaTCAGGGCCAA | histone deace. 7A | chr15:97821023 I. |
| S16 | WTT-cell | TCCTATGG CACTG | TGTTC | gaagaagtgtTGAGGAACAC HP | AK133119, mRNA | chr2:6346533 |
| B120 | WT | TCCTATGG C | CCTG | tgaatgtgacTGTTGCATGC | PluI in Intron | chr1:134452052 I. |
| S31 | WTT-cell | TCCTATGG | AA | gcactgtgatAGCTCGGGCT | TCRγ Jγ1 Coding | chr13:18678543 |
| b196 | WT | CCT | CCCA | ttctgctgtgGATCTTGGCA | chr1:143968211 | |
| Jβ1.2 | CTGTATTC TGATGTG | |||||
| b108 | WTm/h | CTGTATTC TGATGTG | aatccaggggCGTTAGTTCA | Aarsd1 | chr11:101526036 I. | |
| b183 | WT | CTGTATTC TGATGTG | AT | gatgtgtgtgTATGTGTGAG | C030010B13Rik | chr2:173392902 I. |
| b177 | WTm/h | CTGTATTC TGAT | aatgttgtgcTCATCCAGAC | Vγ2-Coding Seg | chr13:18703815 | |
| S1 | WTT-cell | CTGTATTC TGAT | cacagtgagtTCCACCCTGT | chr2:132423309 | ||
| b14 | WTm/h | CTGTATTC TGA | CCAA | ttttgtgtgtGTCTGTCACA | Cox4nb | chr8:119296549 I. |
| S2 | WTT-cell | CTGTATTC TGA | AG | ggccctctatGCACACACAT | Rngtt | chr4:33751106 |
| b35 | WT | CTGTATT | T | cagtgtggcaGTCAGAGGCA | AK156578, unknown mRNA | chr19:40922885 |
| Jβ1.3 | ACCCGGGA GGCTGTG | |||||
| b154 | WT | ACCCGGGA GGCTGTG | AGAG | tacactgtgtGGCAGTAAGC | Scarb1 | chr5:124540598 I. |
| E1-6 | WT | ACCCGGGA GGC | CCC | tcagtgtggtGAAGAGTCAA | Hp1bp3 | chr4:137599977 E. |
| b114 | WT | ACCCGGGA GG | GCCAC | gtctcagtggGTAAAGGCAT | dead-box helicase | chrX:11716155 I. |
| C (Events recovered from mutants) | ||||||
| Jβ1.1 | TCCTATGG CACTGTG | |||||
| b75Spl | H2AX | TCCTATGG CACTGTG | CGG | cgctgtgggaGAAGTGGAGG | hypo-LDLRA | chr2:101888293 I. |
| b235 | cR2 | TCCTATGG CACTGTG | TGG | tcacactctcAAGAATGATT | Neb1 | chr2:17416302 I. |
| E2b-6 | cR2 | TCCTATGG CAC | AGGG | cactgtgagcGTTGCCAAGC | lama3 | chr18:12860547 E. |
| Jβ1.2 | CTGTATTC TGATGTG | |||||
| b220 | cR2 | CTGTATTC TGATG | GACGAGAG | agtgttgtgtGCACACTGAG | BC096449, unknown mRNA | chr1:152252915 |
| E2a-6 | cR2 | CTGTATTC TGAT | T | tgtgtgtgagTGTGTGTGAA | chr9:120505807 | |
| Jβ1.1 | TCCTATGG CACTGTG | |||||
| b243 | p53SCID | TCCTATGG CACTGTG | GGA | tgcacgtgtgTGTTGTAGGG | Wdtc 1 | chr4:132706876 I. |
| b314 | p53SCID | TCCTATGG CACTGTG | GGTCGGTA | cagtagctgtGCTACCGACA | TCR Jδ1 | chr14:48995397 |
| b346 | p53SCID | TCCTATGG CACTGTG | TGTGTGGACC | atagctcaggTTTTCACAAG | TCR Jγ1 | chr13:18577594 |
| b340 | p53SCID | TCCTATGG CACTGTG | G | tatttacagaGACAACTTAA | repeated region | chr5:150937664 |
| b356 | p53SCID | TCCTATGG CACTG | GGTA | ggttagagttAGGGTTAGGG | AK148393, REPEAT | chr18:48387988 |
| b338 | p53SCID | TCCTATG | ATGGAG | aagtagcctaCTCACCGCCA | 114668bps 5′ of Dβ1 | chr6:41349571 |
| Jβ1.2 | CTGTATTC TGATGTG | |||||
| b256 | p53SCID | CTGTATTC TGATGTG | CC | acaaggactgCAGCCCAGTG | AF397014, unknown mRNA | chr13:17054115 |
| b292 | p53SCID | CTGTATTC TGATGTG | GGGT | ctgtgatagcTCAGGTTTTC | TCR Jγ1 | chr13:18577589 |
| b327 | p53SCID | CTGTATTC TGATG | aatgcctgttGTATTTTCCT | chr12:110724915 | ||
| b361 | p53SCID | CTGTATTC TGATG | aacttgggtgCAGTGGGGGA | 4531bps 5′ of Dβ2 | chr6:41468702 | |
| b347 | p53SCID | CTGTATTC TGAT | T | tgtgtgtgagTGTGTGTGAA | chr9:120505807 | |
| b350 | p53SCID | CTGTATTC | aaatttgagcCTTTTCAAGGHP | 432bps 5′ of Dβ1 | chr6:41463838 | |
| b328 | p53SCID | CTGT | tgctctctgtCCTTATCAGT | 467bps 3′ of Jβ1.6 | chr6:41467144 | |
Sequence of the Dβ1 (part A) or Jβ1 (parts B and C) signal end is given in upper case, with sequences aligned to reveal partial RSS deletions. N or P nucleotides are shown in the middle. RSS insertion target sequences are shown in capital letters with flanking sequence in lowercase. Underlined target sequence indicates the position of a cryptic heptamer, as tested according to Cowell et al. (44). Potential stem-loop structures are denoted with the superscript HP and shown in boldface and italic. Events in which both RSS ends of a putative RSS fragment insertion were amplified from the same DNA sample are indicated in boldface and shown diagrammatically in Fig. S1. Nucleotide positions and target gene annotation refer to the August 2005 release of the University of California Santa Cruz mouse genome browser (http://genome.ucsc.edu/cgi-bin/hgGateway?org=mouse). cR2, core-RAG2; T-cell, sorted splenic T cell; m/h, a mixture of murine thymus and HeLa cell DNA; H2Ax, H2Ax null thymus; p53scid, p53 x scid thymus.
LM-TECA amplifies several classes of background events. Most of these are mechanical breaks in purified genomic DNA that are ligated and captured by the assay after initial primer extension. A second source of background events are signal end breaks, which exist in cells undergoingD-to-Jβ rearrangement. Captured signal end PCR products are of known size and can thus be identified on gels and ignored. For assays on the Dβ1-side, a third source of background events result from direct Vβ-to-Dβ1 rearrangements that occur rarely in wild-type animals and 10 to 1,000 time more frequently (unpublished data) in core-RAG2 mutant mice (23). We spent the bulk of our sequencing efforts on capturing Jβ1-sided events to avoid the confounding effects of these direct V-to-D coding joints.
As a negative control for LM-TECA, we used a RAG1 −/− mouse expressing a TCRβ chain transgene. Thymocytes from these mice contain large numbers of preT cells lacking RAG-mediated breaks or rearrangements. DNA sequence analysis of 91 uniquely sized Dβ1-side or Jβ1-side PCR products revealed random DNA breaks across this region of the TCRβ locus (unpublished data). A second negative control was designed to address the concern that PCR artifacts might generate presumed RSS insertion fragments, particularly from genomic DNA samples that contain considerable amounts of broken RSS ends. We mixed mouse thymocytes with equal numbers of human HeLa cells and then purified genomic DNA. If LM-TECA results in the generation of artifacts caused by RSS-ended DNA fragments joining with random DNA fragments, we would expect to detect “insertions” of TCRβ RSSs into human DNA. We performed numerous LM-TECA assays on this source of material and never found any mouse–human DNA recombinants; however, we did detect four insertions of TCRβ RSS fragments into mouse DNA (Table I).
Fig. 2 shows a typical set of PCR reactions in which multiple 0.2-μl aliquots from a single Jβ-side LM-TECA of wild-type thymus DNA were assayed. 9 out of the 10 reactions show a 655-nt product that corresponds to the germline TCRβ locus cleaved at the restriction site (filled arrow). This amplified fragment serves as an internal control for the assay. Individual fragments both smaller and larger than the germline fragment can be seen. From these 10 PCR reactions, 2 of the sequenced fragments proved to be signal end insertion events (open arrows). Depending on the sample, between 5 and 10% of sequenced products were RSS insertions (unpublished data), and the remainder were “background” events as noted in the previous paragraph.
Using this strategy, we were able to isolate and characterize a series of Dβ1-side or Jβ1-side RSS insertion events from primary tissues (Table I). Most of the events we detected were found in WT-thymic DNA samples. We obtained one event from total splenic DNA and five others from sorted splenic T cells. We also analyzed thymic DNA from core-RAG2 mutant mice (23) and from several DNA repair–deficient mouse strains (p53, p53 x scid, 53bp1, H2Ax, and ATM) (24–27).
Characteristics of RSS fragment insertions
The 48 RSS end insertion events we recovered mapped to 18 different chromosomes (Table I). 36 were located within or near annotated genes, including 6 into the TCRγ locus and 4 into non–RSS-containing regions of the TCRβ locus itself. 35 of the events contained N regions (nontemplated nucleotides) ranging from 1 to 10 nt in length. Interestingly, 4 of 12 Dβ1 RSS fragment insertions occurred near, but not exactly at, the RSS heptamer, deleting 1, 2, 9, or 12 nt from the RSS end. There was even more alteration of sequence among the Jβ1 RSS end insertions, with only 14 of 36 sequences containing the full heptamer, whereas in the other 22, the break deleted between 2 and 12 nt of the RSS.
Many of the chromosomal targets of RSS-end insertion were at or near RSSs or cryptic heptamers (Table I, underlined sequences). 9 of the 12 Dβ1 RSS end insertions were of this type. Two of these involved TCR Jγ RSSs, whereas the other seven breakpoints occurred within 2 nt of a consensus heptamer. The situation was somewhat different in respect to Jβ1 RSS end targets, with only 13 of 36 sequences targeted near cryptic heptamers and 4 involving bona fide RSSs (TCRβ and γ loci). Cryptic heptamers at target loci were found on either side of the RSS insertion site, suggesting that ligation events could resemble signal, hybrid, or coding joint formation.
Detecting the second end of an RSS insertion
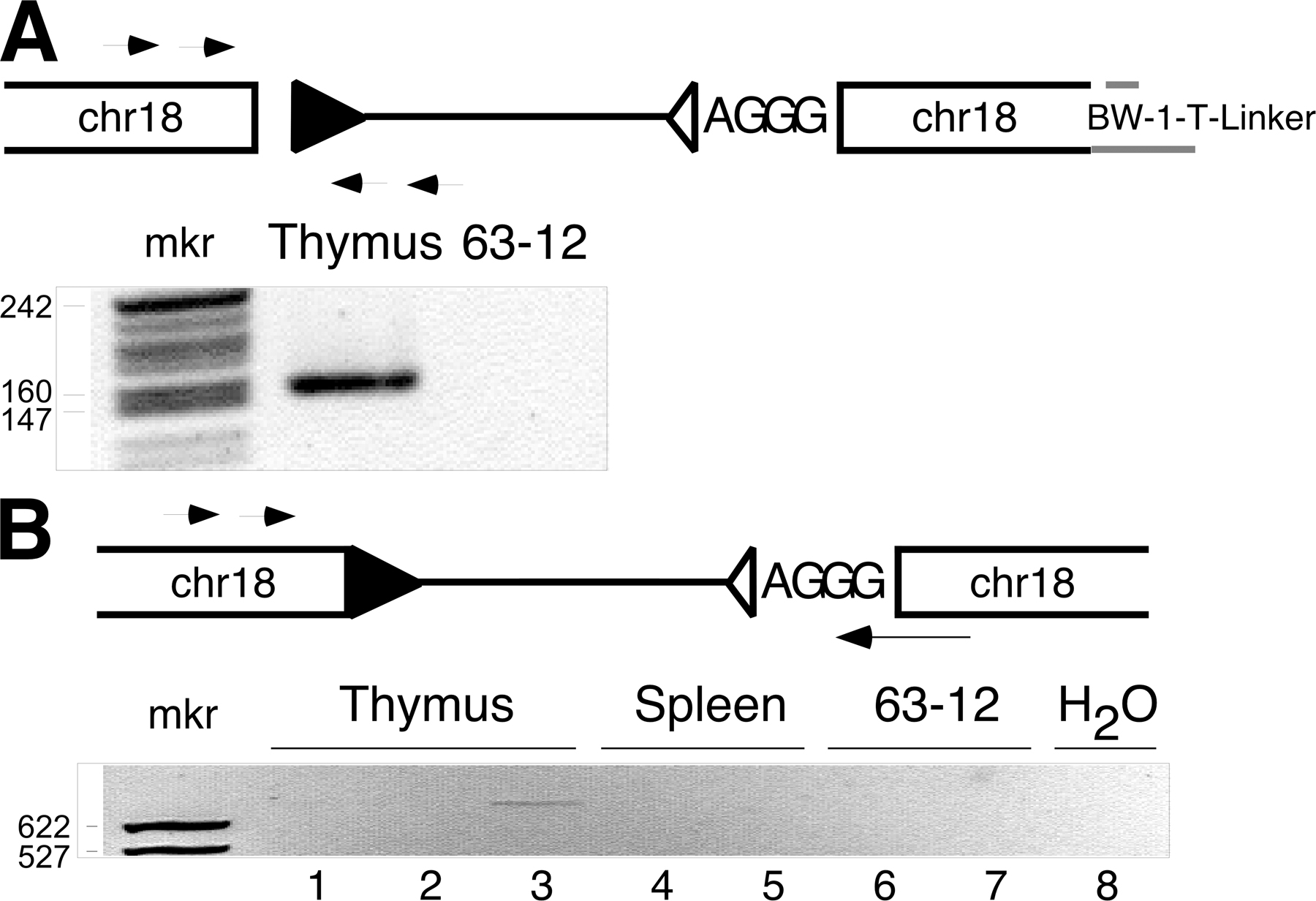
For the majority of the Dβ-side or Jβ-side RSS insertion events, we designed a specific direct PCR assay to detect possible insertion of the other end of the RSS-ended fragment into the same target locus (Fig. 1 D, Fig. 3, and Fig. 4). These assays involved nested PCRs with TCRβ-sided and target locus-sided primers. Using this strategy, we were able to identify the “other” end of a total of five events (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20070583/DC1). Event EL3-1 (Table I), for example, was an insertion of the Dβ1-to-Jβ1.1 RSS-ended fragment into a region of the IgHC locus 5′ to a V558-family gene segment (Fig. 3 and Fig. S1). This is somewhat surprising, given the inaccessibility of the IgHC V region to the V(D)J recombinase in developing T cells. In this insertion, the RSS-end identified by LM-TECA was intact (the Jβ1.1 RSS), whereas the other end, detected by direct PCR in 6 out of 6 DNA aliquots from the same thymus, had undergone a 12-nt deletion (Fig. 3 A). We were unable to detect this Dβ-side RSS insertion in splenic DNA from the same animal or in thymic DNA from any of four additional wild-type mice (Fig. 3 B, top). Dilution analysis (Fig. 3 B, bottom) led us to estimate the frequency of this RSS fragment insertion as at least 1 per ∼17,000 cells based on our ability to detect the amplicon in a sample containing as little as 100 ng of genomic DNA. Both end insertions contained N regions. Using this sequence information, we designed a PCR assay that would amplify across the entire insertion, identifying the predicted molecule in multiple separate DNA aliquots from the same thymus, but not in splenic DNA from that same animal (Fig. 3 C). The identity of the fragment was confirmed by DNA sequencing (unpublished data). The fact that we were unable to detect the EL3-1 RSS fragment insertion in splenocytes suggests the cell containing the initial transposition event had undergone multiple rounds of cell division within the thymus, but failed to be positively selected into the peripheral repertoire.
Figure 3.

Identifying the second end of a Jβ1.1 RSS fusion to chromosome 12 (event EL3-1). (A) Diagram of the Jβ1.1 RSS-12 (open triangle) insertion into chromosome 12, which includes a 7-nt N region (sequence shown). The hypothetical structure of the other end of this transposed fragment is indicated (Dβ1 RSS-23; filled triangle) along with the nested set of PCR primers used. A negative image of the gel analysis of this PCR assay is shown below the diagram, containing the predicted 123-bp product. Lanes labeled Thymus-1 DNA are PCR reactions programmed with independent DNA samples from the same thymus. Lane 63–12 is DNA from a RAG2 −/− pro-B cell line. (B) PhosphorImages of Southern blotted PCR products from a thymus sample and a set of spleen samples from the same mouse (indicated as “1”), along with four thymus samples from different mice (top) (2–5), and a set of twofold serial dilutions of thymus DNA and a spleen sample (Spl-1) from animal 1 analyzed for the chr12-Dβ1-RSS23 insertion (bottom). (C) Diagram of the presumptive insertion event and a PCR strategy to determine if the characterized Dβ1 RSS end is contiguous with the Jβ1 RSS end. The negative image of an ethidium-stained agarose gel analysis of PCR products (740 bp) from DNA purified from four aliquots of animal 1's thymus and two aliquots of animal 1's spleen is shown. The identity of this PCR product was confirmed by DNA sequencing.
Figure 4.

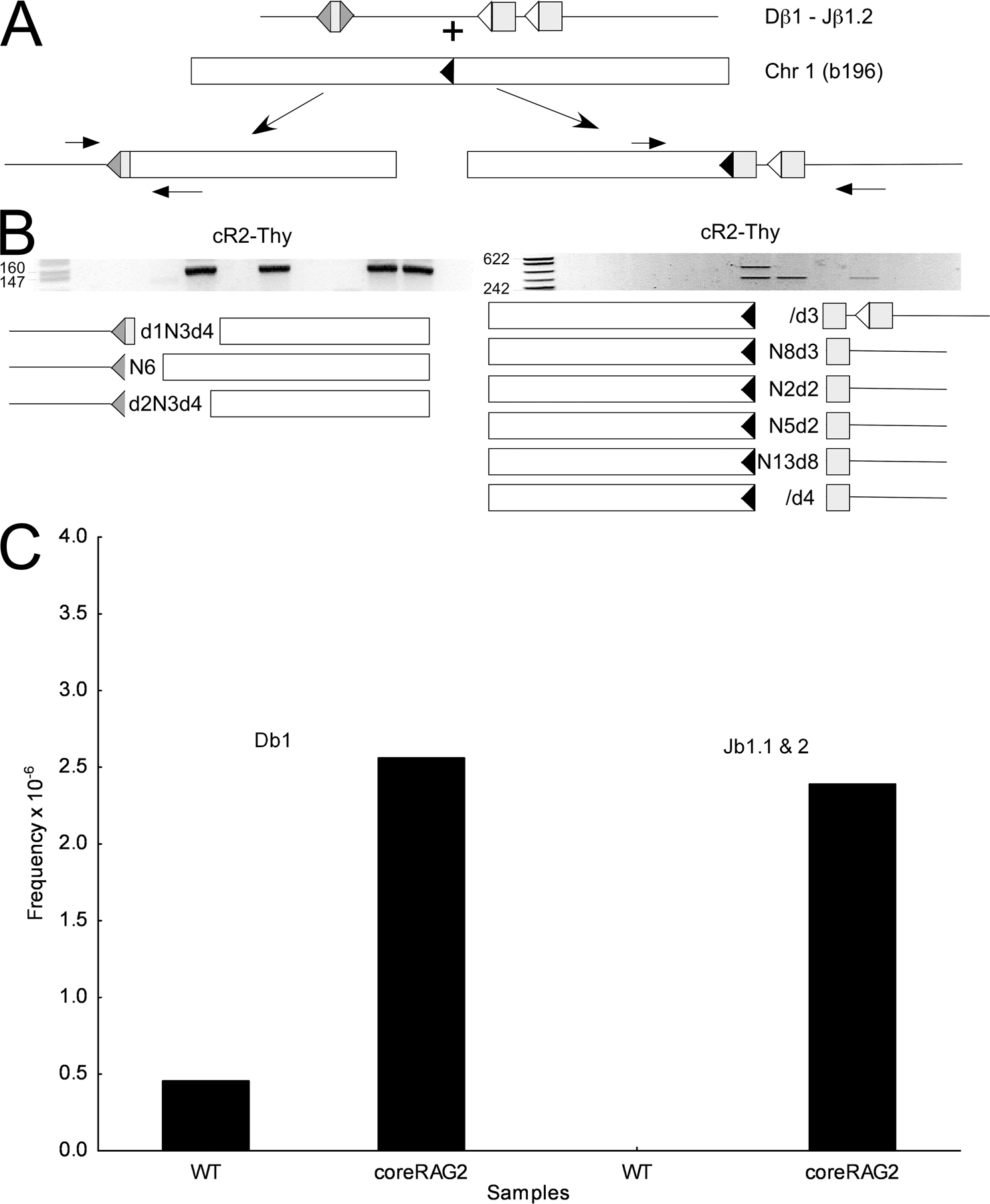
A hotspot for Dβ1 RSS transposition (event b196). (A, top) Diagram of the captured Jβ1.1-RSS12 (open triangle) integration into chromosome 1 showing the PCR strategy used to assay for the corresponding Dβ1-RSS23 (filled triangle) integration. (middle) The negative image of an ethidium-stained agarose gel analysis of PCR products (189 bp) from 6 thymus DNA samples from the same animal. (bottom) Results of DNA sequence analysis with the junction sequence shown. Lane 63–12 is a RAG-2–deficient pro-B cell line. (B) PCR analysis of the Dβ1-RSS23 insertion into chromosome 1 using two aliquots of thymus DNA from each of four different mice (1–4). The DNA sequence at the chromosome 1–Dβ1 RSS junction is indicated below each amplified product. Nucleotides in parentheses are N regions. (C) PCR analysis of Dβ1-RSS23 insertion into chromosome 1 in two separate aliquots of thymus (Thy), spleen (Spl), and bone marrow (BM) DNA from three additional mice (5–7). β-Globin control PCR products (labeled β-G) show that similar amounts of DNA were amplified in each case. The chromosome 1—Dβ1-RSS23 products were sequenced as indicated. Overlapping sequences denote multiple sequences present in a single PCR band.
A second such event, E2b-6 (Table I, Fig. S1, and Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20070583/DC1), was detected in thymus from a core-RAG2 mutant mouse. We identified the Jβ1-sided event by LM-TECA and used target locus sequence to directly amplify the corresponding Dβ1-sided event (Fig. S2 A). The Dβ1 RSS formed a precise signal joint with a cryptic heptamer in an exon of a gene called lama3 on chromosome 18, whereas the Jβ1.1 RSS had suffered a 4-nt deletion. The Jβ1.1 insertion also contained a 4-nt N region. Thymocytes with this RSS fragment insertion were rarer than those with the aforementioned EL3-1 transposition because only occasional aliquots of thymic DNA from one animal gave the expected amplification product corresponding to the complete inserted RSS-ended fragment (Fig. S2 B, lane 3; identity was confirmed by DNA sequence analysis). It, too, was undetectable in splenic DNA.
A third event led us to define a “hotspot” for RSS end insertion in developing T cells. Event b196, detected by LM-TECA, consisted of a partial deletion of the Jβ1.1 RSS ligated to a region of chromosome 1 that lacks nearby annotated genes (Fig. 4 and Fig. S1). Direct PCR assays allowed us to identify the other end of this putative RSS fragment insertion, which consists of the precise fusion of an intact Dβ1 RSS to a cryptic heptamer at the chromosome 1 site. Remarkably, we recovered multiple different sequences for this insertion junction from the same and from different thymuses, thus identifying this region of chromosome 1 as a hotspot for RSS end insertion (Fig. 4, B and C). The sequences all contained full-length Dβ1 heptamers, but differed in the nature of N regions inserted within the joined fragment. We did not detect the inserted Dβ1 RSS in a limited number of 1 μg DNA aliquots from spleen or bone marrow, suggesting that this event might have a negative effect on T cell development or viability (Fig. 4 C). We were unable to detect by PCR a single amplicon containing both RSS ends inserted into the b196 hotspot (unpublished data). Given the apparent frequency of Dβ1 RSS insertion at this cryptic heptamer, we went on to examine this site for reciprocal translocation events between the Dβ1 coding end and the coding end generated by cryptic heptamer cleavage. We were able to detect this recombination event, as well as the formation of a hybrid joint between the cryptic RSS and the Jβ1.1 and Jβ1.2 coding elements, by direct PCR in multiple independent aliquots of thymus DNA (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20070583/DC1). The presence of RSS insertions (either RSS fragment insertions or chromosomal translocations) to this same location in multiple mice allowed us to use this event as a measure of RSS end insertion frequency (see the following section).
The frequency of RSS insertion events in thymocyte DNA from wild-type and mutant mice
Scid mice contain a mutation in the gene encoding DNA-PK, which is a kinase involved in dsDNA break repair and V(D)J recombination. They are deficient in coding and, to a lesser extent, signal joint formation and produce few mature lymphocytes (28, 29). Coding joints that are formed often contain large deletions. Breeding the scid mutation onto a p53 −/− genetic background results in the partial rescue of T cell development to the double-positive stage and allows accumulation of TCRβ locus coding joints (25). We reasoned that such mice might exhibit more readily detectable RSS insertion events because of the inefficiency of proper joint formation. Using LM-TECA, we found multiple J-sided events in p53 x scid mice (Table I). In 7 of 13 instances, the Jβ1 RSS underwent modest (2–11 nt) deletion before insertion, as compared with 12 of 18 instances of deletion in wild-type mice (2–12 nt). Interestingly, only 3 of the 13 target loci in scid x p53 mice, compared with 7 of 18 in wild-type mice, contained authentic or cryptic RSS sequences. Also, 7 of the 13 targets in p53 x scid thymus were loci that undergo V(D)J recombination in that tissue, compared with only 3 out of 18 such targets in wild-type.
To more precisely compare the frequency of RSS fragment insertion among thymocytes from various mutant and wild-type mice, we took advantage of the b196 RSS end insertion hotspot on chromosome 1 described in the previous section. We performed sets of 85 identical direct PCR assays for b196 Dβ-sided RSS insertion on 1-μg aliquots of DNA and used the number of negative samples to calculate the frequency of this insertion based on the Poisson distribution (see Materials and methods). The frequencies of b196 RSS insertion in thymus samples from the DNA damage detection mutants p53, ATM, and H2Ax were within twofold of the wild-type frequency (Fig. 5). Remarkably, the b196 RSS insertion was undetectable in 53bp1 and p53 x scid mutant mice (Fig. 5; frequency was at least 15-fold lower than wild type), although the region of DNA surrounding the hotspot had the identical sequence in all strains of mice in this study (not depicted).
Figure 5.

The frequency of d- or J-side RSS insertion into the b196 hotspot in wild-type and mutant mice. 85 1-μg (∼166,000 genome) samples of thymus or spleen DNA from the indicated mice were analyzed by PCR for either D or J RSS hotspot insertion events. Frequency was calculated according to the Poisson distribution. Error bars on the cR2 Thy and WT Thy samples indicate analysis of independent sets of 85 PCR assays from each of 3 different animals.
As noted in the Introduction, there is biochemical evidence that suggests that the noncore domain of RAG2 suppresses RAG-mediated RSS transposition (10–12). This led us to examine core-RAG2 mutant mouse thymus for examples of TCRβ fragment RSS insertion (23). Using the b196 hotspot assay, we found that TCRβ RSS insertion was sevenfold greater in core-RAG2 compared with wild-type thymus (Fig. 5). Of the four novel Jβ1 RSS end insertions we identified by LM-TECA in core-RAG2 thymus DNA, we were able to detect the corresponding Dβ1 RSS end insertion twice (Table I, events E2b-6 and b220). In both of these instances, the Dβ1 and Jβ1 RSS ends were inserted at or very near cryptic heptamers in the target locus. There was no evidence of target site duplication in either case.
Signal circles as a potential source of DNA ends for RSS insertion
More than half (26 of 48) of the RSS insertion events detected by LM-TECA displayed partial deletion of the TCRβ RSS, and 15 others contained N regions adjacent to full-length RSS ends (Table I). These events cannot be accounted for by a simple transposition mechanism in which the RAGs remain bound to a broken RSS end and catalyze attack by the 3′ hydroxyl group on a DNA duplex (Fig. 1 B) because such a mechanism would not result in deletion of RSS sequences or N region addition. Therefore, we went on to test the hypothesis that a signal circle might serve as donor for RAG-catalyzed RSS fragment insertion.
To test this potential mechanism, we inserted a PGK-neo cassette into plasmid clones containing (a) a precise Dβ1-Jβ1 signal joint; (b) a signal joint with a partially deleted RSS-23 heptamer; and (c) a signal joint with intact RSS ends separated by a 6-nucleotide N region (Fig. 6 A). Each intact circular plasmid was then transfected into a core-RAG2 mutant AMuLV-transformed preB cell line (23) that had been treated with the Abl-kinase inhibitor STI-571 to induce high levels of RAG expression and endogenous Vκ-to-Jκ gene rearrangement (30). Stable transfectants were selected with G418, and purified genomic DNA from each culture was assayed by PCR for plasmid RSS insertion at Vκ or Jκ RSSs associated with gene segments undergoing recombination in these cells. We detected insertion of the wild-type (a) and the N region–containing (c) signal joint plasmids at Vκ RSS-12 sequences (Fig. 6, B and C). These insertions most often had a precise signal joint (Fig. 6 C) and diverse “coding” joints (Fig. 6 B and not depicted). Furthermore, each of 10 sequenced insertions of construct c contained portions of the donor plasmid's N region in a joint with the Vκ coding segment, indicating that RAG cleavage occurred at the RSS-23 heptamer within the signal joint construct. Thus, a subset of imprecise signal circles can serve as a source genomic instability. Interestingly, we failed to detect any insertions into Jκ RSS-23 sequences.
Figure 6.

Signal joints can undergo recombination with chromosomal RSSs. (A) Diagram of a strategy to detect insertion of a transfected recombination signal circle at a Vκ RSS. The arrows indicate PCR primers used to detect RSS insertion events. Triangles represent RSSs (hatched, TCRβ D or J; unfilled, Vκ RSS-12). (B) Agarose gel analysis of PCR assays (primers a and b) for RSS insertion in genomic DNA purified from cells transfected with wild-type (lane 1), RSS-23 mutant (lane 2), or N region–containing (lane 3) TCRβ signal circle plasmids. Arrow denotes the predicted ∼119-bp PCR products that were confirmed by DNA sequence analysis. (C) Agarose gel analysis of PCR assays (primers c and d) using DNA purified from cells transfected with wild-type (lanes 1 and 2) and N region–containing (lanes 3 and 4) signal joint plasmids. PCR products were either digested (+) or not (-) with restriction endonuclease ApaL1, which cleaves precise signal joints. Filled arrow, predicted 142-bp PCR product; open arrow, 115-bp ApaLI-cleaved product.
DISCUSSION
The V(D)J recombinase generates dsDNA breaks at the junctions between RSSs and Ig or TCR gene segments. It then relies on the NHEJ pathway to assemble these broken molecules to form the variable exons of antigen receptor genes and to safely discard RSS-ended DNA fragments. Most often, RSS-ended fragments are joined to form extrachromosomal signal circles. Previous biochemical and transfection experiments have shown that the V(D)J recombinase catalyzes various other reactions that might result in chromosomal translocations and RSS fragment transpositions or insertions (1). We designed the LM-TECA assay to allow us to detect and characterize in an unbiased fashion such events involving the D-J region of the TCRβ locus in developing thymocytes. Using this approach, we found that rearranged genomes of the lymphoid compartment are littered with remnants of V(D)J recombination reaction intermediates.
LM-TECA permits the recovery and sequence analysis of single chromosomally integrated RSS ends. We then used public database genome sequence information to design direct PCR strategies in efforts to recover the “other end” of a potential RSS fragment insertion or balanced chromosomal translocation. Using this strategy, we were able to demonstrate that two of the 36 Jβ1 RSS end integrations were actually part of contiguous Dβ1-Jβ1 RSS fragment insertions. In three other cases, we could detect Dβ1 RSS integration events involving the other side of the target locus break, but were unable to amplify the full contiguous event. Many other RSS end integration events could not be paired with corresponding partner ends. This is probably because of the inefficiency of our assays and the likelihood that not all of the original events occurred in cells that underwent subsequent clonal expansion. Small clonal populations bearing paired Dβ1-Jβ1 RSS insertion events might still be missed simply because of their infrequency in the entire thymic population. In only one case, the b196 hotspot, did we detect a second fragment corresponding to a balanced translocation.
Mechanisms of RSS fragment insertion
Perhaps the most surprising aspect of this study is that we did not observe RAG-mediated RSS fragment transposition. None of the events in which we recovered both RSS ends contained the short target site duplications characteristic of RAG-mediated RSS transposition. The RSS fragment insertions we did detect can be placed into two broad categories—one in which the recombination break point in the target locus is at or near a “cryptic” heptamer and one in which there is no such target sequence present (Table I) (15). Of the Dβ1 RSS-23 events, nearly all the targeted chromosomes (9 of 12) contain complete RSS heptamers. In contrast, only 13 of 36 Jβ1 RSS-12 insertions involve chromosomal targets with potential cryptic heptamers. This observation raises the possibility that RSS-12 (Jβ) sequences may differ from RSS-23 sequences in their potential for chromosomal insertion or the nature of the events in which they participate. Perhaps, as suggested previously, RSS-12 RAG complexes can attack phosphoester bonds in duplex DNA regardless of DNA sequence (9). Subsequent transesterification can result in dsDNA breakage with formation of a hairpin end that can be joined to the TCR gene-segment hairpin. However, almost every Jβ1 RSS insertion was associated with the addition of nontemplated nucleotides, an observation that may not be consistent with this mechanism. Alternatively, it is possible that RSS-12 ends can pair with non-RSS sequences that can serve as recombinase targets because of their unique structural features. Precedent for such a situation involving the breakage cluster region within the Bcl-2 protooncogene was noted in the Introduction (18).
The observation that 4 of the Dβ1 and 20 of the Jβ1 RSS end insertions had lost between 1 and 12 nt of RSS sequence raises the possibility that the reaction that forms these joints may not proceed directly from a free signal end intermediate. One interesting possibility is suggested by the work of Vanura et al., who recently showed that plasmids containing signal joints could undergo V(D)J recombination with plasmids containing proper RSSs in cotransfected cell lines and excised signal circles could integrate back into chromosomal RSSs in vivo (19). Neiditch et al. reported that signal joints undergo continuous RAG-mediated cleavage and rejoining until the recombinase is inactivated during lymphoid development (31). Signal joint cleavage would cease, however, if rejoining were accompanied by nucleotide deletion on one or the other RSS. In this case, an “imprecise” signal joint might undergo synapsis, followed by V(D)J recombination in trans with a cryptic RSS, resulting in the insertion of the signal circle into the chromosome. In this case, the mutant RSS would function as a coding end. We showed that just this type of RAG-mediated insertion reaction could occur in cells performing V(D)J recombination (Fig. 6). Our observation that a majority of the Jβ1 RSS-end insertions contain multiple-nucleotide RSS deletions is also consistent with this possibility.
Our failure to observe the target site duplications characteristic of RAG-mediated RSS transposition (7, 8) in any of the five events in which we recovered both RSS ends, as well as the high frequency of events targeting cryptic heptamers, are in potential conflict with a recent study from Reddy et al. These investigators used a chromosomally integrated transposition reporter construct to estimate the frequency of RSS fragment transposition as 1 per ∼44,000 V(D)J recombination events (32). In that study, however, only ∼1/3 of the recovered events were of this type. Most other events consisted of integrations at or near RSSs in the Ig κ locus that was actively rearranging in the transfected cells. These events were similar to what we observed (Fig. 6). It is possible that constraints imposed by native Ig or TCR locus chromatin structure effectively suppress true transposition events. Alternatively, such events might fall below the limits of detection of the LM-TECA assay.
The frequency of RSS fragment insertion
Although the design of LM-TECA is qualitative, one can nonetheless make inferences regarding the frequency of RSS end insertion based on how the assay is performed. Each assay starts with 40 μg of genomic DNA containing a total of ∼13.3 × 106 TCRβ alleles. If each step was fully efficient (which is unlikely), then each microliter of the final recovered fraction would contain events from 133,000 TCRβ alleles. From 4 μl of this fraction, we were able to recover, on average, two RSS end insertion events (unpublished data). Thus, the frequency of TCRβ D–J interval RSS fragment insertion in vivo is at least 1 in 266,000 genomes. We have measured the recovery of TCRβ locus sequences after biotinylated primer extension to be ∼6% (unpublished data), and previous studies allow us to estimate the efficiency of linker ligation as <5% (33), thus allowing us to adjust our estimated transposition frequency to at least 1 event per 800 genomes or ∼125,000 events per young mouse thymus. This is likely an underestimate because some RSS end insertion events may be deleterious or may diminish the chance of functional TCRβ variable exon assembly.
RSS end insertion in mutants
We used direct PCR assays of Dβ or Jβ RSS end insertion into the b196 hotspot (Fig. 4) to compare insertion frequencies among a panel of mutant mice (Fig. 5). To our initial surprise, ATM mutant mice, which, based on a recent reporter construct study, might be expected to show an increased frequency of transposition (34), showed a frequency of RSS end insertion, in this case translocation, similar to that of wild-type. The recent ATM study, however, showed defects in the processing of coding, but not signal ends. This study focuses on signal end behavior that appeared unaffected in that study (34). We also failed to detect increased hotspot RSS translocation in p53, H2Ax, and scid mutant thymocytes (Fig. 5). This may be caused by the fact that the RSS end insertion events we observed resemble mistargeted V(D)J recombination more than they do aberrant NHEJ.
In contrast, thymocytes from core-RAG2 mutant mice showed an approximately sevenfold increase in RSS end translocation frequency. This is interesting in light of previous reports that recombinase containing core-RAG2 catalyzes transposition in vitro with much greater frequency than recombinase containing wild-type RAG2 (10–12). In contrast, TCRβ RSS end insertions lacked the target site duplications characteristic of the in vitro RAG-mediated transposition reaction (Table I). The C-terminal region of RAG2, which is missing in core-RAG2 mice, interacts with nucleosomal histones (35). Perhaps this interaction contributes to the stability of the postcleavage RSS complex and serves to limit RSS fragment insertions. This function of RAG2 might be considered analogous to the newly appreciated role of ATM in enhancing the stability of the postcleavage coding end complex (34). Alternatively, the core-RAG2 mutant recombinase may be more permissive for RSS synapsis with cryptic sites in trans.
Our data regarding RSS end insertion frequency must be interpreted with caution because the hotspot assay measures rearrangement of a small number of TCRβ RSS ends with a single cryptic RSS on a different chromosome. This event may or may not be representative of the entire class of RSS end translocations involving native and cryptic RSSs. Of the 48 RSS insertion events characterized in this study, b196 was the only site targeted by more than one independent event, and the only one that displayed evidence of a translocation. Also, although we were unable to detect hotspot RSS end insertion in p53 x scid thymus, LM-TECA identified an array of other RSS insertion events in this same material (Table I).
Protecting the genome
Lymphoid leukemias and lymphomas are invariably associated with chromosomal translocations or deletions that affect protooncogenes, and many of these have the characteristics of aberrant V(D)J recombination (14, 16, 36). The mouse provides an excellent model system with which to study genes that limit and mutations that predispose to this type of developmentally regulated genomic instability. Young mice generate between 10 and 30 million thymocytes each day. During their ontogeny, each of these cells must rearrange at least one allele at two different antigen receptor loci (αβ or γδ). Thus, the lifetime total number of dsDNA breaks introduced by the V(D)J recombinase in mouse thymus is truly enormous. Given the ability of the RAG proteins to catalyze various strand-invasion reactions in vitro, including RSS fragment transposition, it is perhaps surprising how infrequent such events appear to be in vivo. This may be caused by several constraints on the recombinase in developing lymphocytes. First, RAG-mediated dsDNA cleavage in vivo requires synapsis between an RSS-12 and -23 sequence (37). Recent data from several groups has led to the suggestion that synapsis might be regulated by RAG-independent chromosome contraction (38). In addition, the frequency of interallelic V(D)J recombination is far lower than recombination in cis. Thus, chromatin structure and the compartmentalization of various chromosomal regions within the nucleus might serve to limit opportunities for transposition and translocation (39). Second, the same DNA–protein and protein–protein interactions that allow the RAGs to bind to RSSs and contribute to their synapsis are likely involved in holding RSS ends together after pairwise cleavage. The involvement of the RAG proteins themselves in protection against transposition may be of clinical significance in light of a recent epidemiological study that associated human lymphoma with a genetic polymorphism in RAG1 (40). Third, unbalanced translocations and some RSS fragment insertions are likely to disrupt essential cellular functions and result in lethality. In fact, the vast majority of developing thymocytes fail to assemble a self-tolerant, positively selectable TCR and undergo apoptosis, reducing the frequency of surviving cells with aberrant recombination events. To our surprise, null mutations in the genes encoding p53, ATM, H2Ax, and DNA-PK did not seem to result in an increase in cells carrying RSS transposition events. We are continuing to survey mutations in NHEJ pathway proteins, as well as genes encoding “guardians of the genome” for effects on aberrant V(D)J recombination.
MATERIALS AND METHODS
Animals and cell lines.
63-12 and cR2-25, A-MuLV–transformed pro-B cell lines from RAG2−/− (41), and core-RAG2 mutant (23) bone marrow, respectively, were cultured in RPMI supplemented with 10% fetal calf serum and antibiotics. Human embryonic kidney 293T cells were cultured in DME supplemented with 10% fetal calf serum and antibiotics. RAG2−/− x TCRβ, core-RAG2, H2Ax−/−, ATM−/−, p53−/−, 53bp1−/−, and scid −/− mice have each been previously described (23–27, 42). Animal use was approved by the Animal Care and Use Committee at University of California Berkeley.
LM-TECA assay.
Approximately 40 μg of DNA (8 × 106 cells embedded within 8 gel plugs) were digested overnight with the restriction enzymes MaeIII or RsaI (NEB) at 37°C. Plugs were rinsed with TE (10 mM Tris-HCl, pH 8.0, and 0.1mM EDTA) and digested genomic DNA was mixed with 1× PCR buffer (10 mM Tris-HCl, pH 8.3, 50 mM KCl, 1.5 mM MgCl2, and 0.001% gelatin), 0.2 mM dNTPs, 175 pmol of a 5′-biotinylated oligonucleotide (Table S1, available at http://www.jem.org/cgi/content/full/jem.20070583/DC1), and 20 U of a hot-start Taq polymerase (JumpStart; Sigma-Aldrich) in a final volume of 800 μl. The mixture was denatured at 95°C and the primer extension reaction was incubated at 68°C for 1 h, and then terminated by the addition of 2.5 μl of 500 mM EDTA, pH 8.0, and chilled on ice for 10 min. The DNA gel plug was soaked overnight in 50 ml TE at 4°C and, finally, washed twice with 50 ml TE. Agarose was digested with β-agarase following manufacturers directions (NEB), and undigested agarose was removed by high-speed centrifugation and discarded.
The purified genomic DNA was magnetically fractionated, and then ligated to 20 pmol of linker (annealed BW-1-T and BW-2) with a 3′ T overhang in a 150-μl reaction containing 1× ligation buffer (Invitrogen), and 15 U of T4 DNA ligase (Invitrogen) at 14°C for 48 h. Ligations were terminated by incubation at 68°C and brought to 250 μl with the addition of 100 μl of 0.1 M NaCl TE. DNA was once again fractionated on a streptavidin column (no further beads were added). The final ligated DNA fractions were then subjected to a heminested PCR (see Supplemental materials and methods available at http://www.jem.org/cgi/content/full/jem.20070583/DC1) and analyzed on an ethidium-stained agarose gel. Individual gel bands were excised (Zymoclean; Zymo Research) and subjected to automated cycle sequencing. Those samples that yielded novel genomic DNA sequences adjacent to the Jβ1 or Dβ1 RSS sequences were further analyzed by direct PCR. PCR products were excised from agarose gels, directly ligated to a cloning vector, and subjected to DNA sequence determination. A detailed protocol is available in the Supplemental materials and methods.
Determination of hotspot transposition frequency.
The frequency of RSS end integration into the b196 hotspot was determined by performing nested PCR on a series of 85 identical 1-μg aliquots of genomic DNA from single thymuses. This secondary reaction was analyzed on an ethidium-stained 3% agarose gel. Positive reactions were counted and some were sequenced to verify their authenticity. The Poisson distribution was used to determine the frequency of the event (frequency = −lnP°/number of genomes per reaction, where P° = negative reactions/total reactions) (43).
Transfection assay.
cR2-25 cells were diluted to 200,000 cells/ml and treated with 2.5 μM STI-571 for 4 h at 37°C. Cells were then washed twice with PBS, returned to media without STI-571, and 12 h later electroporated with 4 μg of signal-joint plasmid using an Amaxa Nucleofector device (Amaxa, Inc.). 48 h later, 1 mg/ml G418 was added to each culture. After 2 wk of selection, genomic DNA was purified and assayed by PCR for insertion of the signal joint plasmid RSSs into chromosomal Vκ or Jκ gene segments. Amplified products were cloned using a Topo PCR cloning kit (Invitrogen) and sequenced.
Online supplemental material.
Fig. S1 is a diagram of the RSS insertion events in which both sides of a Dβ1 RSS–Jβ1 RSS insertion were identified in DNA from a single thymus. Fig. S2 shows an additional example of an RSS fragment insertion identified in thymus DNA from a core-RAG2 mouse. Fig. S3 shows examples of how the b196 chromosomal hotspot can be involved in the formation of chromosomal translocations that resemble coding and hybrid joints. Table S1 provides sequences of DNA primers used in these studies. We also provide a Supplemental materials and methods, which includes a more detailed protocol for the LM-TECA assay. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20070583/DC1.
Supplemental Material
Acknowledgments
The authors thank Patty Garcia for cloning the signal circle PGK-neo constructs and Dan Huang for expert technical assistance. The manuscript was improved by the thoughtful criticisms of Astar Winoto and various members of the Schlissel laboratory. We also thank Matt Schrettner for sample preparation and careful proofreading of Table I.
M.S. Schlissel and J.D. Curry acknowledge National Institutes of Health support (AI40227), and C.J. Guidos and J.S. Danska acknowledge grant support from a Leukemia and Lymphoma Society Specialized Center of Research grant and Genome Canada, with funds through the Ontario Genomics Institute.
The authors have no conflicting financial interests.
Abbreviations used: ds, double strand; LM-TECA, ligation-mediated transferred-end capture assay; NHEJ, nonhomologous end joining; RSS, recombination signal sequence.
References
- 1.Gellert, M. 2002. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu. Rev. Biochem. 71:101–132. [DOI] [PubMed] [Google Scholar]
- 2.Mills, K.D., D.O. Ferguson, and F.W. Alt. 2003. The role of DNA breaks in genomic instability and tumorigenesis. Immunol. Rev. 194:77–95. [DOI] [PubMed] [Google Scholar]
- 3.Lewis, S.M., and G.E. Wu. 1997. The origins of V(D)J recombination. Cell. 88:159–162. [DOI] [PubMed] [Google Scholar]
- 4.Oettinger, M.A., D.G. Schatz, C. Gorka, and D. Baltimore. 1990. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science. 248:1517–1523. [DOI] [PubMed] [Google Scholar]
- 5.Jones, J.M., and M. Gellert. 2004. The taming of a transposon: V(D)J recombination and the immune system. Immunol. Rev. 200:233–248. [DOI] [PubMed] [Google Scholar]
- 6.Lewis, S.M., J.E. Hesse, K. Mizuuchi, and M. Gellert. 1988. Novel strand exchanges in V(D)J recombination. Cell. 55:1099–1107. [DOI] [PubMed] [Google Scholar]
- 7.Agrawal, A., Q.M. Eastman, and D.G. Schatz. 1998. Transposition mediated by RAG1 and RAG2 and its implications for the evolution of the immune system. Nature. 394:744–751. [DOI] [PubMed] [Google Scholar]
- 8.Hiom, K., M. Melek, and M. Gellert. 1998. DNA transposition by the RAG1 and RAG2 proteins: a possible source of oncogenic translocations. Cell. 94:463–470. [DOI] [PubMed] [Google Scholar]
- 9.Melek, M., and M. Gellert. 2000. RAG1/2-mediated resolution of transposition intermediates: two pathways and possible consequences. Cell. 101:625–633. [DOI] [PubMed] [Google Scholar]
- 10.Tsai, C.L., and D.G. Schatz. 2003. Regulation of RAG1/RAG2-mediated transposition by GTP and the C-terminal region of RAG2. EMBO J. 22:1922–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swanson, P.C., D. Volkmer, and L. Wang. 2004. Full-length RAG-2, and not full-length RAG-1, specifically suppresses RAG-mediated transposition but not hybrid joint formation or disintegration. J. Biol. Chem. 279:4034–4044. [DOI] [PubMed] [Google Scholar]
- 12.Elkin, S.K., A.G. Matthews, and M.A. Oettinger. 2003. The C-terminal portion of RAG2 protects against transposition in vitro. EMBO J. 22:1931–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sekiguchi, J.A., S. Whitlow, and F.W. Alt. 2001. Increased accumulation of hybrid V(D)J joins in cells expressing truncated versus full-length RAGs. Mol. Cell. 8:1383–1390. [DOI] [PubMed] [Google Scholar]
- 14.Kuppers, R., and R. Dalla-Favera. 2001. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene. 20:5580–5594. [DOI] [PubMed] [Google Scholar]
- 15.Roth, D.B. 2003. Restraining the V(D)J recombinase. Nat. Rev. Immunol. 3:656–666. [DOI] [PubMed] [Google Scholar]
- 16.Schlissel, M.S., C.R. Kaffer, and J.D. Curry. 2006. Leukemia and lymphoma a cost of ng business for adaptive immunity. Genes Dev. 20:1539–1544. [DOI] [PubMed] [Google Scholar]
- 17.Raghavan, S.C., I.R. Kirsch, and M.R. Lieber. 2001. Analysis of the V(D)J recombination efficiency at lymphoid chromosomal translocation breakpoints. J. Biol. Chem. 276:29126–29133. [DOI] [PubMed] [Google Scholar]
- 18.Raghavan, S.C., P.C. Swanson, X. Wu, C.L. Hsieh, and M.R. Lieber. 2004. A non-B-DNA structure at the Bcl-2 major breakpoint region is cleaved by the RAG complex. Nature. 428:88–93. [DOI] [PubMed] [Google Scholar]
- 19.Vanura, K., B. Montpellier, T. Le, S. Spicuglia, J.M. Navarro, O. Cabaud, S. Roulland, E. Vachez, I. Prinz, P. Ferrier, et al. 2007. In Vivo Reinsertion of Excised episomes by the V(D)J recombinase: a potential threat to genomic stability. PLoS Biol. 5:e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Messier, T.L., J.P. O'Neill, and B.A. Finette. 2006. V(D)J recombinase mediated inter-chromosomal HPRT alterations at cryptic recombination signal sequences in peripheral human T cells. Hum. Mutat. 27:829. [DOI] [PubMed] [Google Scholar]
- 21.Messier, T.L., J.P. O'Neill, S.M. Hou, J.A. Nicklas, and B.A. Finette. 2003. In vivo transposition mediated by V(D)J recombinase in human T lymphocytes. EMBO J. 22:1381–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reddy, Y.V., E.J. Perkins, and D.A. Ramsden. 2006. Genomic instability due to V(D)J recombination-associated transposition. Genes Dev. 20:1575–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang, H.E., L.Y. Hsu, D. Cado, L.G. Cowell, G. Kelsoe, and M.S. Schlissel. 2002. The “dispensable” portion of RAG2 is necessary for efficient V-to-DJ rearrangement during B and T cell development. Immunity. 17:639–651. [DOI] [PubMed] [Google Scholar]
- 24.Celeste, A., S. Petersen, P.J. Romanienko, O. Fernandez-Capetillo, H.T. Chen, O.A. Sedelnikova, B. Reina-San-Martin, V. Coppola, E. Meffre, M.J. Difilippantonio, et al. 2002. Genomic instability in mice lacking histone H2AX. Science. 296:922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guidos, C.J., C.J. Williams, I. Grandal, G. Knowles, M.T. Huang, and J.S. Danska. 1996. V(D)J recombination activates a p53-dependent DNA damage checkpoint in scid lymphocyte precursors. Genes Dev. 10:2038–2054. [DOI] [PubMed] [Google Scholar]
- 26.Ward, I.M., K. Minn, J. van Deursen, and J. Chen. 2003. p53 binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 23:2556–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu, Y., T. Ashley, E.E. Brainerd, R.T. Bronson, M.S. Meyn, and D. Baltimore. 1996. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 10:2411–2422. [DOI] [PubMed] [Google Scholar]
- 28.Schuler, W., I. Weiler, A. Schuler, R. Phillips, N. Rosenberg, T.W. Mak, and M.J. Bosma. 1986. Rearrangement of antigen receptor genes is defective in mice with severe combined immune deficiency. Cell. 46:963–974. [DOI] [PubMed] [Google Scholar]
- 29.Fukumura, R., R. Araki, A. Fujimori, Y. Tsutsumi, A. Kurimasa, G.C. Li, D.J. Chen, K. Tatsumi, and M. Abe. 2000. Signal joint formation is also impaired in DNA-dependent protein kinase catalytic subunit knockout cells. J. Immunol. 165:3883–3889. [DOI] [PubMed] [Google Scholar]
- 30.Muljo, S.A., and M.S. Schlissel. 2003. A small molecule Abl kinase inhibitor induces differentiation of Abelson virus-transformed pre-B cell lines. Nat. Immunol. 4:31–37. [DOI] [PubMed] [Google Scholar]
- 31.Neiditch, M.B., G.S. Lee, L.E. Huye, V.L. Brandt, and D.B. Roth. 2002. The V(D)J recombinase efficiently cleaves and transposes signal joints. Mol. Cell. 9:871–878. [DOI] [PubMed] [Google Scholar]
- 32.Reddy, Y.V., E.J. Perkins, and D.A. Ramsden. 2006. Genomic instability due to V(D)J recombination-associated transposition. Genes Dev. 20:1575–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Curry, J.D., L. Li, and M.S. Schlissel. 2005. Quantification of Jkappa signal end breaks in developing B cells by blunt-end linker ligation and qPCR. J. Immunol. Methods. 296:19–30. [DOI] [PubMed] [Google Scholar]
- 34.Bredemeyer, A.L., G.G. Sharma, C.Y. Huang, B.A. Helmink, L.M. Walker, K.C. Khor, B. Nuskey, K.E. Sullivan, T.K. Pandita, C.H. Bassing, and B.P. Sleckman. 2006. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination.Nature. 442:466–470. [DOI] [PubMed] [Google Scholar]
- 35.West, K.L., N.C. Singha, P. De Ioannes, L. Lacomis, H. Erdjument-Bromage, P. Tempst, and P. Cortes. 2005. A direct interaction between the RAG2 C terminus and the core histones is required for efficient V(D)J recombination. Immunity. 23:203–212. [DOI] [PubMed] [Google Scholar]
- 36.Marculescu, R., K. Vanura, B. Montpellier, S. Roulland, T. Le, J.M. Navarro, U. Jager, F. McBlane, and B. Nadel. 2006. Recombinase, chromosomal translocations and lymphoid neoplasia: targeting mistakes and repair failures. DNA Repair (Amst.). 5:1246–1258. [DOI] [PubMed] [Google Scholar]
- 37.Tillman, R.E., A.L. Wooley, M.M. Hughes, T.D. Wehrly, W. Swat, and B.P. Sleckman. 2002. Restrictions limiting the generation of DNA double strand breaks during chromosomal V(D)J recombination. J. Exp. Med. 195:309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sen, R., and E. Oltz. 2006. Genetic and epigenetic regulation of IgH gene assembly. Curr. Opin. Immunol. 18:237–242. [DOI] [PubMed] [Google Scholar]
- 39.Roix, J.J., P.G. McQueen, P.J. Munson, L.A. Parada, and T. Misteli. 2003. Spatial proximity of translocation-prone gene loci in human lymphomas. Nat. Genet. 34:287–291. [DOI] [PubMed] [Google Scholar]
- 40.Hill, D.A., S.S. Wang, J.R. Cerhan, S. Davis, W. Cozen, R.K. Severson, P. Hartge, S. Wacholder, M. Yeager, S.J. Chanock, and N. Rothman. 2006. Risk of non-Hodgkin lymphoma (NHL) in relation to germline variation in DNA repair and related genes. Blood. 108:3161–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shinkai, Y., G. Rathbun, K. Lam, E. Oltz, V. Stewart, M. Mendelsohn, J. Charron, M. Datta, F. Young, A.M. Stall, and F.W. Alt. 1992. RAG-2 deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 68:855–867. [DOI] [PubMed] [Google Scholar]
- 42.Shinkai, Y., S. Koyasu, K. Nakayama, K.M. Murphy, D.Y. Loh, E.L. Reinherz, and F.W. Alt. 1993. Restoration of T cell development in RAG-2-deficient mice by functional TCR transgenes. Science. 259:822–825. [DOI] [PubMed] [Google Scholar]
- 43.Luria, S.E., and M. Delbruck. 1943. Mutations of bacteria from virus sensitivity to virus resistance. Genetics. 28:491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cowell, L.G., M. Davila, K. Yang, T.B. Kepler, and G. Kelsoe. 2003. Prospective estimation of recombination signal efficiency and identification of functional cryptic signals in the genome by statistical modeling. J. Exp. Med. 197:207–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}