

Abstract
Respiratory viral infections are associated with an increased risk of asthma, but how acute Th1 antiviral immune responses lead to chronic inflammatory Th2 disease remains undefined. We define a novel pathway that links transient viral infection to chronic lung disease with dendritic cell (DC) expression of the high-affinity IgE receptor (FcεRIα). In a mouse model of virus-induced chronic lung disease, in which Sendai virus triggered a switch to persistent mucous cell metaplasia and airway hyperreactivity after clearance of replicating virus, we found that FceRIa−/− mice no longer developed mucous cell metaplasia. Viral infection induced IgE-independent, type I IFN receptor–dependent expression of FcεRIα on mouse lung DCs. Cross-linking DC FcεRIα resulted in the production of the T cell chemoattractant CCL28. FceRIa−/− mice had decreased CCL28 and recruitment of IL-13–producing CD4+ T cells to the lung after viral infection. Transfer of wild-type DCs to FceRIa−/− mice restored these events, whereas blockade of CCL28 inhibited mucous cell metaplasia. Therefore, lung DC expression of FcεRIα is part of the antiviral response that recruits CD4+ T cells and drives mucous cell metaplasia, thus linking antiviral responses to allergic/asthmatic Th2 responses.
The risk of asthma from severe paramyxoviral infection in both human and experimental models is well documented (1–3). This virally imparted risk presents an interesting paradox; although the primary antiviral response is dominated by production of IFNα/β and IL-12, which are hallmarks of a Th1 response, rhinorrhea and mucous cell metaplasia also develop. These conditions are driven by IL-13, which is a hallmark Th2 cytokine (4, 5). The production of antiviral IgE, along with neutralizing IgG antibodies, provides a further link between these disparate responses (6–10). In fact, IgE serum concentrations have been correlated with subsequent wheezing in infants with respiratory viral infection and with the risk of otitis media with effusion in children (10, 11). How a Th1-biased response generates a Th2 phenotype is not known, although we now show that the high-affinity receptor for IgE on DCs bridges the antiviral Th1 response to the atopic/proasthmatic Th2 response.
The role of the high-affinity receptor for IgE (FcεRI) on human conventional DCs (cDCs) has been assumed to be antigen focusing, with expression being tightly regulated by serum IgE levels, much like it is on basophils (12, 13). FcεRI has not been reported on mouse DCs, and little is known of what role it might play during an antiviral response. Indeed, the role of the cDCs in an antiviral immune response is not fully understood. Initial lung cDC migration to draining lymph nodes, and subsequent antigen presentation, has been examined (14–16). However, the role of those cDCs that remain in or are attracted to the lung parenchyma during a primary response has not been evaluated. Although, in the case of secondary viral infections or challenge responses to OVA, the evidence suggests that these cells are involved in recruitment of memory effector T cells (17, 18). We have developed a mouse model of viral bronchiolitis that reproduces disease traits associated with asthma (2). In this model, FcεRI was expressed on lung cDCs only during the antiviral response, and these cells were critical for the development of postviral mucous cell metaplasia. Indeed, mice deficient in FcεRI (FceRIa−/−) failed to recruit IL-13–producing CD4+ T cells to their lungs after viral infection. Cross-linking the α-chain of FcεRI (FcεRIα) on lung cDCs produced a CD4+ T cell chemoattractant, CCL28, and blockade of this chemokine prevented development of postviral mucous cell metaplasia. Thus, these studies provide a novel insight into how the antiviral response leads to a potent Th2 response through FcεRIα on lung cDCs.
RESULTS
FceRIa−/− mice fail to develop airway mucous cell metaplasia
We infected FceRIa−/− mice or WT littermates with the mouse paramyxovirus Sendai virus (SeV). Each strain exhibited similar morbidity (as monitored by weight loss), development of an adaptive immune response (as indicated by the development of SeV-specific CD8+ T cells), and clearance of virus from the lung (based on SeV copy number) during the acute phase of viral infection (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20070360/DC1). Previous work in this model has shown that replicating virus is fully cleared by postinoculation (PI) day 12, with the subsequent development of long-lasting mucous cell metaplasia evident by PI day 21 (2). Despite a similar acute response to viral infection, we found a marked decrease in the number of Muc5ac-expressing mucous (goblet) cells in the airways of FceRIa−/− mice compared with WT mice at PI day 21 (Fig. 1). We have previously shown that Muc5ac induction by PI day 21 depends on production of IL-13 (5). Therefore, these findings suggested a link between FcεRIα expression and IL-13 production in the airway response to viral infection.
Figure 1.

Inhibition of chronic mucous cell metaplasia after viral infection inFceRIa−/−mice. (A) FceRIa −/− and WT littermate control mice were inoculated with SeV, and lung sections were immunostained for Muc5ac at PI day 0 and 21. Representative images from PI day 21 are shown. Bar, 40 μm. (B) Quantification of mucous cells based on immunostaining conditions from A. (C) Lungs from A were subjected to real-time PCR for Muc5ac mRNA. For B and C, values represent the mean ± the SEM for three to five mice. *, P < 0.05 versus WT PI day 21.
Paramyxoviral infection induces high-level expression of FcεRIα on resident lung DCs
We next identified which cell type in the lung might express FcεRIα in the setting of viral infection. In rodents, the high-affinity IgE receptor has been identified only on mast cells, basophils, and possibly eosinophils (19). However, in humans, this receptor is also found on skin and peripheral blood cDCs, as well as plasmacytoid DCs (pDCs), albeit as a trimeric form (FcεRIαγγ) that lacks the FcεRIβ chain (13, 20, 21). Therefore, we questioned whether this form of the receptor might also be found on lung parenchymal cDCs after viral infection. Using forward/side scatter characteristics and a high level of CD11c expression to identify lung cDCs, we have observed that SeV infection leads to a rapid and sustained decrease in the number of lung cDCs, but the cDC population remaining in the lung becomes more mature and differentiated (22). Therefore, we examined this population of resident cDCs in more detail.
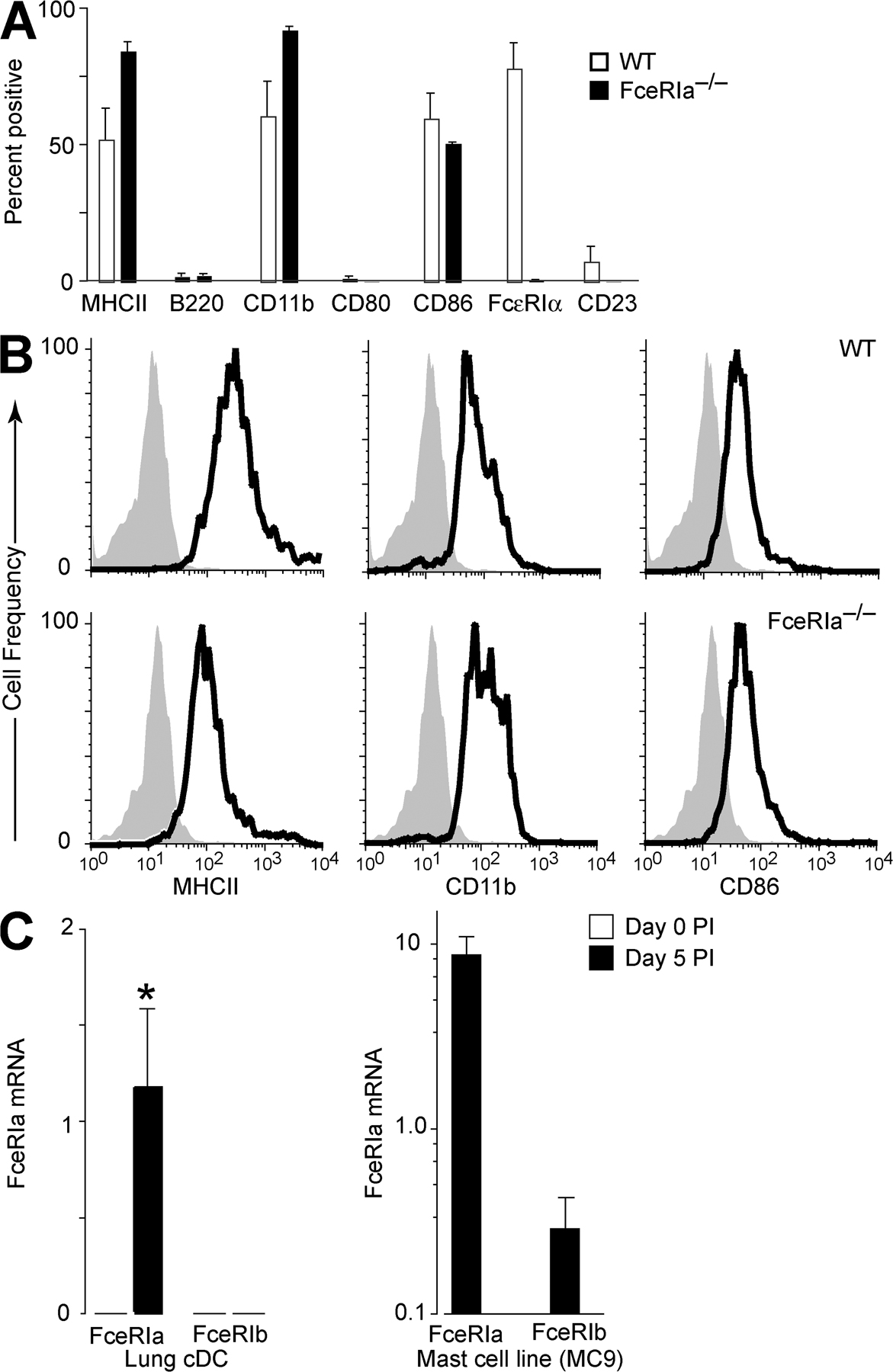
Before infection, lung cDCs failed to express FcεRI; however, by PI day 3, this population began to express the α chain of the high-affinity receptor for IgE (FcεRIα), and this expression remained detectable for 2–3 wk after SeV inoculation (Fig. 2, A and B). This response was specific to lung cDCs because there was no detectable expression of FcεRIα on cDCs isolated from draining lymph nodes or spleen during SeV infection (unpublished data). CD23, the low-affinity receptor for IgE, was not induced during the infection (Fig. 2 B). Using intranasal administration of the intravital dye CFSE, which labeled lung cells present at the time of its administration but not cells that subsequently migrated into the lung, we were able to show that FcεRIα-expressing cDCs were present in the lung at the time of the initial viral inoculation and had not migrated into the lung after the onset of infection (Fig. 2, C and D). The level of expression of FcεRIα on lung cDCs at PI day 7 was similar to levels seen on c-kit+ lung mast cells (Fig. 2, A and E). Increased expression of FcεRI after viral infection was relatively selective for cDCs and mast cells because we did not detect expression of FcεRIα on lung CD4+ or CD8+ T cells, B220+ B cells, Mac-3+ macrophages, or Gr-1+ neutrophils (unpublished data). Expression of FcεRIα was detected on 20–40% of lung pDCs at PI day 3 (unpublished data), which is consistent with reports of FcεRIαγγ on human pDCs (21). This pDC population was distinct from the lung cDC population because FcεRIα-expressing lung cDCs did not express the pDC/B cell marker B220 (Fig. S2 A, available at http://www.jem.org/cgi/content/full/jem.20070360/DC1). Expression of FcεRI did not influence cDC maturation or differentiation during viral infection. We found no difference in the levels of expression of MHC class II, CD11b, CD80, or CD86 between lung cDCs from FceRIa−/− versus WT mice (Fig. S2, A and B). Similarly, we found no influence of FcεRI on the expression of CD23.
Figure 2.

Up-regulation of FcεRIα expression on lung DCs after viral infection. (A) WT mice were inoculated with SeV, and CD11c+ lung cDCs or c-kit+ mast cells (MC) were analyzed by flow cytometry using anti-FcεRIα mAb and isotype control mAb at PI day 0 and 7. (B) Conditions in A were used for flow cytometry for FcεRIα and CD23 on the indicated PI day. Values represent the mean ± the SEM for n ≥ 6 mice. *, P < 0.05 versus PI day 0. (C) WT mice were given PBS ([–] CFSE) or CFSE ([+] CFSE) intranasally and inoculated with SeV. Lung cDCs were isolated at PI day 7, and expression of CFSE and FcεRIα was determined by flow cytometry. (D) Quantification of data from C. Values represent the mean ± the SEM for n ≥ 3 mice. (E) Lung cDCs (CD11c+) and mast cells (c-kit+) were analyzed by flow cytometry for FcεR1α, as in A. Values represent the mean ± the SEM for fold mean fluorescence intensity FcεRIα signal versus isotype control mAb for three mice per time point. (F) Cell lysates from lung cDCs (PI day 5) or MC/9 mast cells were subjected to immunoprecipitation with anti-FcεRIα mAb. Immunoprecipitated complexes (IP) were then Western blotted with anti-FcεRIβ or -FcεRIγ mAb. Representative blots are shown for two separate experiments.
Based on work with isolated cells, mice have been reported to be obligate expressers of the tetrameric form (FcεRIαβγγ) of FcεRI (23). To determine if mouse lung cDCs were, indeed, expressing the tetrameric and not trimeric form of the receptor, we analyzed mouse lung cDCs for expression of each FcεRI component chain. Immunoprecipitation of FcεRI with an anti-FcεRIα antibody, and subsequent Western blot with anti-FcεRIγ or -FcεRIβ antibodies, showed that lung cDCs isolated after SeV infection expressed FcεRIγ, but not FcεRIβ (Fig. 2 F). In contrast, MC9 mast cells expressed both FcεRIγ and FcεRIβ. In concert with these findings for FcεRI proteins, real-time PCR assays showed that lung cDCs expressed FcεRIα, but not FcεRIβ mRNA after viral infection, whereas MC9 mast cells contained high levels of both FcεRIα and FcεRIβ mRNA (Fig. S2 C). Thus, mouse lung cDCs express the trimeric (FcεRIαγγ) form of the FcεRI receptor. This is the same form of FcεRI that is expressed by human DCs, but expression on mouse cDCs requires induction by a productive viral infection. This requirement may explain the failure to detect FcεRI expression on mouse DCs in previous works.
Type I IFN receptor, but not IgE, regulates DC expression of FcεRIα
Next, we explored what component of the antiviral response is responsible for induction of FcεRIα expression on lung cDCs during a paramyxoviral infection. Serum IgE level tightly regulates expression of the high-affinity IgE receptor in humans, so we reasoned that total IgE and, more likely, SeV-specific IgE might drive FcεRI receptor expression after viral infection in mice. However, we found that serum total IgE and SeV-specific IgE do not increase until PI day 7 (Fig. 3 A). This time course lags behind the onset of expression of FcεRIα on lung cDCs, suggesting that the level of IgE is not driving the expression of FcεRI receptor under these conditions. In fact, we found that IgE-deficient (IgE −/−) mice continued to develop the same increase in FcεRIα expression on lung cDCs as their WT littermates after viral inoculation (Fig. 3 B). Therefore, IgE levels do not appear to regulate the appearance of FcεRIα on lung cDCs after viral infection in mice.
Figure 3.

Effect of IgE levels and IFN signaling on up-regulation of FcεRIα on lung cDCs after viral infection. (A) WT mice were inoculated with SeV, and serum was analyzed for total and SeV-specific IgE levels. Values represent the mean ± the SEM for three to six mice. (B) IgE −/− and WT control mice were inoculated with SeV, and lung cDCs were analyzed by flow cytometry using anti-FcεRIα and isotype control mAbs. (C) IFNAR −/− and WT mice were inoculated with SeV, and lung cDCs were analyzed at PI day 5, as in B. For conditions in C, quantification of fold increase in mean fluorescence intensity for FcεRIα over isotype control (D) and percentage of lung cDCs expressing FcεRIα (E). Values represent the mean ± the SEM for four to eight mice per group.
Given that initial expression of FcεRIα developed at the same time as IFN-dependent responses in this viral model, we examined whether IFN signaling was necessary for FcεRI expression. Indeed, we found that type I IFN receptor (IFNAR)–deficient (IFNAR −/−) mice no longer exhibited an increase in FcεRIα expression on lung cDCs during viral infection (Fig. 3, C–E). In contrast, mice that were deficient for CD1d, CD4, CD8, perforin, or MyD88 gene expression showed no defect in FcεRIα expression after viral infection (unpublished data). Thus, although IFNAR is critical for expression of FcεRIα on lung cDCs after viral infection, there is no substantial role for NKT cells, CD4+ T cells, or CD8+ T cells, or for the innate antiviral perforin- or MyD88-dependent Toll-like receptor pathways in this process.
We next examined the mechanism for IFN-dependent expression of FcεRI on DCs. We recognized that direct IFN treatment of human lung pDCs failed to increase FcεRIα expression (21). This finding suggested that IFN-dependent increases in FcεRI expression on lung DCs may involve other cell types besides DCs. Accordingly, we next determined whether IFNAR expression on cDCs was necessary for FcεRI expression after viral infection. For these experiments, we purified lung cDCs from IFNAR−/− and WT control mice. We verified that the cells we purified exhibited typical DC morphology (Fig. 4). Purified lung cDCs were loaded with CFSE (to discriminate from endogenous cDCs) and transferred into WT or IFNAR−/− recipients. We found that either IFNAR−/− or WT cDC transfer into WT mice allowed for expression of FcεRIα on cDCs (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20070360/DC1). In contrast, transfer of either genotype of cDCs into IFNAR−/− mice did not permit expression of FcεRIα on cDCs after viral infection. These findings imply that IFN acts through another intermediate cell to activate FcεRI expression on lung cDCs.
Figure 4.

Morphology of purified lung cDCs versus macrophages. Lung cDCs were isolated by positive immunomagnetic selection with macrophages obtained by BAL. Both cell preparations were cytocentrifuged onto slides (left two columns) and cultured in chamber slides with or without 10 μg/ml LPS for 24 h at 37°C (right two columns), and then imaged with modified Wright's stain and photomicrography. For each condition, representative photomicrographs from three separate experiments are shown. Bars, 10 μm.
Lung cDCs from virus-infected mice drive IL-13 production from CD4+ T cells
Because IL-13 is thought to drive postviral mucous cell metaplasia, and Th2 cells are thought to be a likely source of IL-13, we reasoned that generation of Th2 cells might be altered as a result of FcεRIα engagement on cDCs (5). CD8− cDCs, like those found in the lung, have been shown to drive T cell development toward a Th2 phenotype in vitro (24). Therefore, to determine if induction of IL-13–producing CD4+ T cells depended on engagement of FcεRIα, we examined IL-13 production in an in vitro cell culture system that contains CD4+ T cells and cDCs. OVA-specific CD4+ T cells (OT-II cells) were cultured with lung cDCs isolated from WT or FceRIa−/− mice with or without SeV infection. We isolated cDCs at PI day 7 and verified that these cDCs had not yet bound IgE at this time point (unpublished data). We found that OT-II CD4+ T cells produced IL-13 when cultured with antigen and cDCs, regardless of whether cDCs were isolated from WT or FceRIa−/− mice or from infected or uninfected mice (Fig. 5 A). Furthermore, cross-linking FcεRIα had no effect on T cell production of IL-13 or proliferation (Fig. 5 A and not depicted). Thus, we conclude that lung cDCs are capable of driving the development of IL-13–producing CD4+ Th2 cells, but this process does not require cDC expression or activation of FcεRIα.
Figure 5.

CCL28 production after activation of FcεRIα on lung cDCs. (A) Lung cDCs from WT or FceRIa−/− mice that were uninfected (PI day 0) or SeV-infected (PI day 7) were treated with or without anti-FcεRIα mAb or control IgG. Cells were loaded with OVA peptide and cocultured with OVA-specific CD4+ T cells (OT-II) for 4 d at 37°C, followed by ELISA of cell supernatants for IL-13. Values represent the mean ± the SEM for four to seven experiments done in triplicate. (B) Lung cDCs were purified from PI day 5 and treated with an anti-FcεRIα mAb or control IgG for 18 h at 37°C. Cell supernatants were assayed for chemotactic activity for naive CD4+ T cells with and without treatment with anti-CCL28 or -CCL27 mAb. Values represent the mean ± the SEM for three to six experiments done in triplicate. *, P < 0.05 versus IgG supernatant; **, P < 0.05 versus anti-CCL27 and no antibody treatment. (C) Lung cDCs purified at PI day 7 were loaded with anti-OVA IgE and cultured with (+) or without (–) 3 μg/ml OVA protein for 18 h at 37°C. Cellular RNA was subjected to real-time PCR for Ccl28 mRNA. Values are corrected for Gapdh mRNA level and represent the mean ± the SEM for three experiments. *, P < 0.05 versus no OVA protein. (D) FceRIa −/− and WT mice were inoculated with SeV, and lung levels of Ccl28 mRNA were determined by real-time PCR at the indicated times. Values represent the mean ± the SEM for three to five mice. (E) Lung frequencies of CD4+ T cells were quantified by flow cytometry in WT and FceRIa −/− mice at the indicated times. Values represent the mean ± the SEM for three to nine mice. (F) Lung CD4+ T cells were purified by positive immunomagnetic selection at PI day 21 and analyzed for Il-13 by real-time PCR. (G) Using conditions in F, expression of GATA-3 mRNA was analyzed by real-time PCR. (H) Percentage of total lung cells in the lymphocyte gate expressing CCR10 and CD4 by flow cytometry at PI day 0 or 10 in WT and FceRIa−/− mice. For F–H, values represent the mean ± the SEM for three mice. *, P < 0.05 versus WT mice for D–H. (I) CCR10+ CD4+ T cells and CCR10− CD4+ T cells were purified by flow cytometry at PI day 10 and subjected to real-time PCR for GATA-3 and T-bet. Values represent the mean ± the SEM for the ratio of GATA-3 to T-bet from three experiments with three mice per experiment. *, P < 0.05 versus CCR10− CD4+ T cells.
Cross-linking FcεRIα on lung DCs produces the T cell chemoattractant CCL28
Because we did not detect a requirement for cross-linking FcεRIα in the development of the T cell cytokine response, we next assessed whether FcεRI might be involved in the induction of T cell chemoattractants. Lung cDCs were purified from WT mice by immunomagnetic selection at PI day 5. The purified cells were then cultured for 18 h with a cross-linking antibody against FcεRIα or a control hamster IgG. Supernatants from these cultures were then used in a modified Boyden chamber assay to assess whether a functional T cell chemoattractant had been produced. Supernatants from FcεRIα cross-linked cDCs induced significantly more CD4+ T cell migration than did IgG control supernatants, indicating that engagement of the receptor led to production of a CD4+ T cell chemoattractant (Fig. 5 B). To identify the chemokine receptor for this T cell chemoattractant, we added various chemokines (CCL5, CCL22, and CCL27) to the upper chamber and evaluated the effect on chemotaxis (Fig. S4 A, available at http://www.jem.org/cgi/content/full/jem.20070360/DC1). Only CCL27 (C-TACK) inhibited T cell migration, indicating that the CD4+ T cells were moving in response to a CCR10 agonist. Only two known CCR10 agonists, CCL27 and CCL28 (MEC), have been identified. Using blocking mAbs, we found that the chemotactic activity was entirely caused by CCL28 (Fig. 5 B). Of relevance to this work, CCL28 has been associated with both human asthma and mouse models of asthma (25–27).
To establish that CCL28 expression was inducible in cDCs with a more physiological stimulus of FcεRI activation, we loaded lung cDCs from PI day 7 with IgE against OVA. In this setting, we found that addition of OVA (by cross-linking anti-OVA IgE bound to FcεRIα on the cDC) caused a marked increase in CCL28 mRNA (Fig. 5 C). Similar results were obtained when using the anti-FcεRIα antibody to directly cross-link the receptor (unpublished data). To determine whether FcεRI activation also caused CCL28 expression in vivo, we returned to experiments with the mouse model of virus-induced lung disease. Again, we found that lung levels of CCL28 mRNA were increased after SeV infection in WT mice, and that this effect was lost in FceRIa−/− mice (Fig. 5 D). If CCL28 was being released with engagement of FcεRI, then we would expect differences in the frequency of CD4+ T cells in the lungs of FceRIa−/− and WT mice after viral infection. Indeed, we found that CD4+ T cells were significantly decreased in the lungs of FceRIa−/− mice compared with WT mice at PI day 21 (Fig. 5 E).
Because we have previously shown that mucous cell metaplasia after viral infection is dependent on IL-13 production, we next examined the amount of IL-13 and GATA-3 (a Th2-specific transcription factor) message in lung CD4+ T cells of WT and FceRIa−/− mice after SeV infection (5). We found a significant decrease in the levels of IL-13 and GATA-3 mRNA in CD4+ T cells from the lungs of FceRIa−/− compared with WT mice (Fig. 5, F and G). These findings further supported the proposal that inhibition of mucous cell metaplasia in FceRIa−/− mice was based on decreased accumulation of IL-13–producing CD4+ Th2 cells caused by a lack of production of CCL28 by the resident FcεRIα+ lung cDCs. We also examined IL-13 and GATA-3 mRNA levels in the CD4+ T cells that had or had not migrated in response to CCL28 in vitro. We found that there was much greater expression of both IL-13 and GATA-3 mRNA in T cells that migrated compared with cells that did not migrate in response to CCL28 (Fig. S4 B).
Because CCL28 binds to CCR10 to recruit immune cells, we determined the level of CCR10+ CD4+ T cells in the lungs of WT and FceRIa−/− mice before and after viral infection. We found an increase in CCR10+ CD4+ T cells in WT mice after viral infection (Fig. 5 H). Furthermore, this increase in CCR10+ CD4+ T cells did not develop in FceRIa−/− mice. We also found that CCR10+ CD4+ lung T cells from WT mice contained increased levels of GATA-3 mRNA (typically found in Th2 cells) compared with T-bet mRNA (typically found in Th1 cells; Fig. 5 I). Together, these findings provide further support for the proposal that activation of FcεRIα on cDCs leads to preferential accumulation of Th2 cells in the lung after SeV infection.
cDC reconstitution restores CD4+ T cell accumulation and mucous cell metaplasia after viral infection
To prove that FcεRIα expression on cDCs was necessary for recruitment of CD4+ Th2 cells to the lung and mucous cell metaplasia after viral infection, we performed adoptive cell transfer experiments with cDCs from WT or FceRIa−/− mice transferred into FceRIa−/− recipients, followed by SeV inoculation. We found that reconstitution with WT cDCs restored postviral mucous cell metaplasia in FceRIa−/− recipient mice (Fig. 6, A and B, and Fig. S5 A, available at http://www.jem.org/cgi/content/full/jem.20070360/DC1). In contrast, transfer of FceRIa−/− cDCs were unable to restore the development of mucous cell metaplasia after viral infection. Furthermore, we found that CD4+ T cells isolated at PI day 21 from lungs of FceRIa−/− mice that had received WT cDCs expressed IL-13 and GATA-3 at significantly higher levels than mice that received cDCs from FceRIa−/− mice (Fig. 6 C). We found no difference in the engraftment of WT versus FceRIa−/− cDCs, with both genotypes of cDCs persisting for at least 6 d after transfer to the recipient mice (Fig. S5 B). Together, these results indicate that FcεRIα on lung cDCs is critical for Th2 cell recruitment and mucous cell metaplasia after viral infection.
Figure 6.

Restoration of postviral mucous cell metaplasia in FceRIa−/− mice after lung cDC transfer. (A) Lung cDCs were isolated from WT or FceRIa −/− mice and transferred into FceRIa −/− recipients 1 d before inoculation with SeV. At PI day 21, lung sections were obtained for immunostaining for Muc5ac. Representative photomicrographs are shown. Bar, 40 μm. (B) Quantification of immunostaining in A. Values represent the mean ± the SEM for two to four recipient mice per group from two separate experiments; *, P < 0.05 versus WT DCs transferred into FceRIa−/− recipients. (C) For conditions in A, lung CD4+ T cells were purified at PI day 21 and analyzed for Il-13, GATA-3, and Gapdh mRNA by real-time Q-PCR. Values represent the mean ± the SEM from two separate experiments with two to four recipient mice per experiment. *, P < 0.05 versus FceRIα−/− cDCs transferred into FceRIa−/− recipients.
Blockade of CCL28 inhibits postviral mucous cell metaplasia
We next determined the role of CCL28 in the cascade leading to mucous cell metaplasia after viral infection. For these experiments, we treated WT mice with an anti-CCL28–blocking mAb or control IgG2b and monitored the development of mucous cell metaplasia at PI day 21. We found a significant decrease in mucous cell metaplasia after treatment with anti-CCL28 mAb compared with control IgG (Fig. 7, A and B, and Fig. S6, available at http://www.jem.org/cgi/content/full/jem.20070360/DC1). We observed no significant difference in the frequency of CD4+ T cells in the lungs of mice treated with either anti-CCL28 mAb or control IgG2b (Fig. 7 C). This finding suggests that CCL28 is a chemotactic factor for immune cells producing IL-13 and driving mucous cell metaplasia after viral infection, but additional chemoattractants also contribute to CD4+ T cell accumulation in the lung in this setting.
Figure 7.

Effect of CCL28 blockade on mucous cell metaplasia after viral infection. (A) WT mice were inoculated with SeV and treated with anti-CCL28 mAb or control IgG2b mAb every other day starting on PI day 3. At PI day 21, lung sections were subjected to immunostaining for Muc5ac, and representative photomicrographs are shown. Bar, 40 μm. (B) Quantification of immunostaining in A. (C) For conditions in A, the frequency of lung CD4+ T cells was monitored by flow cytometry. Values represent the mean ± the SEM for four mice per group. *, P < 0.05 versus control IgG2b treatment.
DISCUSSION
In this study, we show that respiratory viral infection can lead to expression and activation of FcεRI on lung cDCs and thereby drive the development of mucous cell metaplasia even after the inciting infection is cleared. We examined lung cDCs because we previously found that the migration and maturation of this cell population was tightly regulated during respiratory viral infection (22). Indeed, the potent effects of viral infection on DCs might explain why we have detected FcεRI on DCs in the mouse, whereas others have not previously found this receptor on DCs (28).
We have also shown that expression of FcεRI in the setting of a productive viral infection requires an active type I IFN response. In fact, expression of FcεRI on cDCs quickly returns to low levels with resolution of the antiviral response, such that expression is nearly undetectable by 21 d after viral inoculation. Previous work has shown that IgE levels regulate FcεRI expression on mast cells. In addition, the low levels of FcεRI expression found on mast cells from IgE−/− mice were attributed to IL-4 production (29). These findings now indicate that FcεRI expression on cDCs is also regulated by type I IFN. Moreover, expression on cDCs can be regulated independent of IgE levels because FcεRI levels return to baseline despite a persistent increase in IgE levels after viral infection. Thus, FcεRI on cDCs may be selectively regulated by type I IFN, at least in the mouse. Human cDCs express FcεRI at baseline, but whether these levels are increased by viral infection still needs to be determined (12, 13).
In that regard, the consequences of IgE production and IgE–FcεRI interaction in the setting of viral infection were also uncertain. Any possible pathogenic role for IgE-dependent signals remained undefined, despite recognition that viral infections stimulate potent IgE responses along with neutralizing IgG antibodies (6–10). Furthermore, increased antiviral IgE production is closely associated with subsequent wheezing in infants after respiratory viral infection (10). Moreover, the two major pathogens linked to asthma pathogenesis, i.e., respiratory syncytial virus (a paramyxovirus-like SeV) and rhinovirus, are both known to potently induce the production of IgE in humans (10, 30–33). Our findings provide for a novel IgE-dependent mechanism in which an antiviral response characterized by IFN production drives expression of FcεRI on lung cDCs. IgE engagement of the receptor leads to release of soluble mediators that generate an atopic response characterized by Th2 cell activation and IL-13 production. Consistent with this mechanism, others showed that passive transfer of antiviral IgE can increase the airway hyperreactivity caused by respiratory syncytial virus infection in mice, and that FcεRI was necessary for this IgE-dependent response (34). This effect was assumed to be caused by mast cell activation during the viral infection. However, given the very low dissociation kinetics of FcεRI-bound IgE (Kd of ∼10−9–10−10 M), it is not clear that IgE during a primary viral infection would bind to mast cell FcεRI in any significant quantity during the acute infection (35). It is possible that the rapid expression of FcεRI on lung cDCs would make these cells a better target cell for binding antiviral IgE because this cell population would have a significant number of unoccupied receptors. Although we did not study this issue directly, it is quite possible that virus-induced airway hyperreactivity, like mucous cell metaplasia, is a result of engagement of FcεRI on lung cDCs and not mast cells or basophils.
The actions of FcεRI found on mouse lung cDCs are distinct from the previously described role of FcεRIα signaling on human DCs. In the human cells, FcεRI is thought to help target antigens to DCs (12). Because IgE production lags behind expression of FcεRI, it appears that IgE may act as a signaling molecule for lung cDCs. Once an effective adaptive immune response has been generated, virus-specific IgE will be produced, bind to the cDCs, and be cross-linked by viral antigen. This signals cDCs to produce CCL28 and recruit effector Th2 cells. Because Th2 recruitment to the lung is antigen nonspecific, it is possible that this mechanism contributes in other ways to allow the innate immune system to influence the antiviral response (36). For example, regulatory T cells express CCR10 and may be another important target of CCL28 (37). Thus, FcεRI engagement on lung cDCs may provide mechanisms to down-regulate an ongoing Th1 response in the lung after viral infection.
These results may also bear on the increased incidence of asthma and allergic diseases that are being observed in more industrialized countries. This observation has led to the “hygiene hypothesis,” in which cleaner living conditions and decreased childhood bacterial infections are thought to drive an increase in allergic disease (38). Our results suggest an additional mechanism that may explain the increased incidence of asthma in the urban environment. In this setting, the increased population density, as well as other factors, may lead to increased transmission of respiratory viral infections. If viral respiratory illnesses drive FcεRI expression on lung cDCs in humans, this process could lead to an increase in lung Th2 responses in individuals with genetic susceptibility to this type of response. This mechanism is consistent with the increased incidence of asthma now being found for children living in the inner city (39).
In summary, we have defined a new scheme for mucous cell metaplasia after viral infection that depends on an IFNAR–FcεRI–CCL28–IL-13 immune axis (Fig. S7, available at http://www.jem.org/cgi/content/full/jem.20070360/DC1). This mechanism involves cDC expression of FcεRI and production of CCL28 in concert with T cell production of IL-13 and consequent mucous cell metaplasia. This immune axis may be more active after severe infection to help explain why more severe respiratory viral infections are associated with an increased risk of asthma. For example, the level of IgE production after SeV infection in mice appears to depend on the severity of the infection (unpublished data). The scheme also implies that exposure to even nonviral IgE and antigen during the resolution of the viral response might also lead to increased Th2 recruitment to the lung driven by additional FcεRI cross-linking and CCL28 production. This mechanism would thereby provide for an allergic-type response to nonviral antigens because Th2 cells that had migrated to the lung could be directed against nonviral antigens. In the atopic individual, these Th2 cells could lead to the development of asthma, or an exacerbation of the disease in patients with established asthma. Each of these mechanisms is distinct from previous proposals for IgE–FcεRI function in the traditional response to parasitic infection or allergen exposure. Thus, viral antigens and the consequent IFN-dependent antiviral response may also trigger an “allergic” cascade, leading to disease traits that are characteristic of asthma and other chronic airway diseases.
MATERIALS AND METHODS
Reagents and antibodies.
Anti-CD16/-CD32 antibodies (clone 2.4G2; obtained from C. Pham, Washington University, St. Louis, MO) were used for flow cytometry, as previously described (40). Anti–mouse CCL28 mAb or control rat IgG2b was obtained from R&D Systems. PE- or allophycocyanin-labeled antibodies against mouse CD4, CD8, CD11c, CD23, c-Kit, FcεRIα (clone MAR-1), and IgE, as well as isotype control IgGs (rat and Armenian hamster), were obtained from eBioscience or BD PharMingen. SeV-specific CD8+ T cells were monitored using tetrameric MHC/peptide reagents for the immunodominant epitope of SeV nucleoprotein (NP324-332) or control OVA peptide (SIINFEKL) complexed with K(b) provided by the National Institute of Allergy and Infectious Diseases (NIAID) Tetramer Core Facility (41). Purified anti-FcεRIα mAb (clone MAR-1; eBioscience) was used for immunoprecipitation, and anti-FcεRIβ mAb (clone JRK) obtained from J. Rivera (National Institutes of Health, Bethesda, MD) or rabbit anti-FcεRIγ Ab (Millipore) was used for detection of the immune complex in Western blotting. For cross-linking experiments, functional grade Armenian hamster anti–murine FcεRIα or functional grade Armenian hamster IgG isotype control antibodies were obtained from eBioscience. A secondary goat anti–Armenian hamster IgG antibody purchased from Jackson ImmunoResearch Laboratories was also used.
Mouse generation and handling.
C57BL/6; BALB/c; B6.129S2-CD4+tm1Mak/J (CD4-null); B6.129S2-Cd8atm1Mak/J (CD8-null); and C57BL/6-Prf1tm1Sdz/J (perforin-null) mice were obtained from The Jackson Laboratory. TgN(OT-II.2a)Rag1tm1 (OT-II) mice on a C57BL6 background were obtained from the Taconic NIAID Emerging Models Program (Germantown, NY). C57BL6 mice deficient in CD1d were obtained from A. Bendelac (University of Chicago, Chicago, IL); IFNAR-null mice on a C57BL6 background were obtained from J. Sprent (The Scripps Research Institute, La Jolla, CA); MyD88-deficient mice and WT littermates on a mixed B6/129 background were a gift from M. Colonna (Washington University, St. Louis, MO); FcεRIα-deficient mice on a C57BL6 background were obtained from J.-P. Kinet (Harvard Medical School, Boston, MA); and IgE-deficient mice on a BALB/c background were obtained from H. Oettgen (Harvard Medical School, Boston, MA). Mice 6–20 wk of age were used for all experiments under protocols approved by the University Animal Studies Committee.
Mice were inoculated with SeV (Fushimi strain; American Type Culture Collection [ATCC]) or UV-inactivated SeV, as previously described, using an inoculum of 2 × 105 pfu, and they were monitored daily for weight and activity. To track lung cDCs, mice were anesthetized and given 30 μl of 5 mM CFSE intranasally, as previously described (2, 3, 5, 14). For blockade of CCL28, mice were given 100 μg of anti-CCL28 mAb or control IgG2b subcutaneously every other day from PI day 3 to 19. Mucous cell metaplasia and Muc5ac mRNA in lungs were assessed at PI day 21 as previously described (2, 3, 5).
Lung cDC isolation.
For cDC isolation, mice were killed, and intracardiac injection of PBS was used to flush out the pulmonary and systemic circulations. BAL was performed to remove any cells in the bronchiolar or alveolar space. Next, 1 ml of digest media was injected intratracheally. Digest medium consisted of DME supplemented with 5% fetal calf serum, penicillin/streptokinase, 10 mM Hepes, 250 U/ml collagenase I (Worthington Biochemical), 50 U/ml DNase I (Worthington Biochemical), and 0.01% hyaluronidase (Sigma-Aldrich). The lungs were carefully dissected from the trachea, main-stem bronchi, draining lymph nodes, and surrounding tissue. The lungs were minced and incubated in digest media for 1 h at 37°C. During the final 15 min of incubation time, EDTA was added to a final concentration of 2 mM. After digestion, the cell mixture was passed through a 40-μm pore cell strainer to generate single-cell suspensions, and erythrocytes were removed by hypotonic lysis. Cell viability was assessed by trypan blue exclusion. An analogous procedure was performed for removal of DCs from spleen or draining lymph nodes, as well as for isolation of T cells from C57BL6 or OT-II splenic tissue. Lung cDCs were identified (and other cell populations, such as macrophages, were excluded) by flow cytometry, as previously described (22).
Cell purification and culture.
DCs were purified from lung cell suspensions using the MACS system (Miltenyi Biotec) for positive immunomagnetic selection with anti-CD11c microbeads. Using two serial purifications, cDC purity was ≥95%. Cells obtained in this manner had typical cDC morphology (Fig. 4). Purified cDCs were cultured with or without cross-linking antibodies in complete RPMI (Sigma-Aldrich) supplemented with 10% fetal calf serum and penicillin/streptokinase (Invitrogen) for 18 h at 37°C. For T cell proliferation experiments, cDCs were subjected to 200 cGyn before being loaded with 0.3 μM of OVA peptide (OVA323-339). CD4+ T cells were purified from OT-II spleens using the Miltenyi-Biotec MACS system for positive immunomagnetic selection. T cell purities were ≥95%. Irradiated cDCs (104 per condition) were cultured for 4 d with 105 OVA-specific CD4+ T cells with or without cross-linking antibodies. For T cell proliferation, CD4+ T cells were labeled with 5 μM CFSE (Invitrogen) before culture, and cell proliferation was monitored by CFSE dilution, as previously described (42). For some experiments, lung cDCs were loaded with 10 μg/ml mouse anti-OVA IgE (Serotec) for 48 h at 37°C, washed, and incubated with or without 3 μg/ml OVA protein (Sigma-Aldrich) for 18 h at 37°C, followed by mRNA isolation from cell pellets. Mouse MC/9 mast cells were obtained from the ATCC and were maintained in ATCC complete growth medium (DME with 2 mM l-glutamine, 0.05 mM 2-ME, 4.5 g/liter glucose, and 1.5 g/liter sodium bicarbonate) supplemented with 10% fetal calf serum and 10% Rat T-Stim (BD Biosciences) (43).
Immunoprecipitation and Western blot analysis.
Immunoprecipitation of FcεRIα from lung cDCs and MC/9 mast cells was performed as previously described (44, 45). In brief, 3 × 106 cells were solubilized in 100 mM Tris containing 0.5% Nonidet P-40, 10% glycerol, 5 M NaCl, 100 mM MgCl2, and enhanced inhibitor supplement consisting of a 20× concentrate of complete edium (80 mM benzamidine HCL, 50 mM ε-caproic acid, 16 mM iodoacetamide, 10 μg/ml leupeptin, 10 μg/ml pepstatin, and 100 μg/ml soybean trypsin inhibitor). Aliquots of samples containing 500 μg protein were immunoprecipitated using 20 μl of protein A–Sepharose beads conjugated to 25 μg of rabbit secondary antibody. Immune complexes were separated by nonreducing PAGE, and then detected by Western blotting onto an Immobilon-P PVDF membrane (Millipore), probed with anti-FcεRIβ or -FcεRIγ antibodies, and imaged with chemiluminescence (SuperSignal West Pico kit; Pierce Chemical Co.).
Adoptive transfer protocol.
Lung cDCs were purified from C57BL6, IFNAR−/−, or FceRIa−/− mice and delivered intranasally to anesthetized recipient C57BL6 or FceRIa−/− mice (2.5 × 105 cells in 30 μl of PBS given 1 d before SeV inoculation). In some experiments, cDCs were loaded with either CFSE or TAMRA-SE (Invitrogen), as previously described (40). No difference in transfer efficiency (engraftment) was seen with either dye (unpublished data).
Chemotaxis assay.
Chemotaxis was monitored with a modified Boyden chamber, as previously described (46). For these experiments, CD4+ T cells were purified from naive C57BL6 spleen, and 2.5 × 104 cells in 50 μl were loaded in the upper chamber, which was separated from the lower chamber by a 5-μm membrane (Neuro Probe, Inc.). A 25-μl aliquot of a 1:3 dilution of supernatant from lung cDC cultures was placed in the lower chamber. In some experiments, control rat IgG2b, rat anti–mouse CCL27 antibody, or rat anti–mouse CCL28 antibody (100 μg/ml) was added to the lower chamber. After a 3-h incubation at 37°C, the number of cells that had transmigrated into the lower chamber was determined by flow cytometry, as previously described (46). Results are calculated as a migration index that represents the ratio of cells migrating in each condition compared with the number migrating to CCL21 control (1 μg/ml; R & D Systems).
Real-time PCR assay.
mRNA was isolated with the RNeasy kit (Qiagen) or Trizol (Sigma-Aldrich) and used to generate cDNA with Applied Biosystems cDNA archive kit as per the manufacturer's instructions. Quantitative real-time PCR assays were performed using an Applied Biosystems 7500 Fast real-time PCR system and TaqMan fast master mix. In addition to the TaqMan rodent GAPDH control, the following TaqMan gene expression arrays (mouse primer and probe sets) for PCR were obtained from Applied Biosystems: IL-13 (assay ID Mm00434204_m1), Muc5ac (assay ID Mm01276725_g1), CCL28 (assay ID Mm00445039_m1), GATA-3 (assay ID Mm00484683_m1), FcεRIα (assay ID Mm00438867_m1), and FcεRIβ (assay ID Mm00442780_m1). For FcεRIα and FcεRIβ mRNA levels, results represent arbitrary units (calculated as 2[CTMaximum−CTgene]) per ng of total RNA. For IL-13, Muc5ac, GATA-3, and CCL28 mRNA levels, results represent copy number per Gapdh copy number based on a standard curve with the known copy number of the target and Gapdh mRNA.
Measurement of IgE and IL-13.
Total IgE from mouse serum was determined by ELISA using capture and detection anti-IgE antibody pairs (BD PharMingen) in 96-well MaxiSorp plates (Nalge Nunc). Purified mouse IgE (BD PharMingen) was used as a control. For detection of SeV-specific IgE, SeV-coated plates from Charles River Laboratories were substituted for capture IgE antibody-coated plates. Because no control anti-SeV IgE antibodies are available, values represent absolute absorbance units. IL-13 levels were determined using a commercially available ELISA kit (Invitrogen).
Statistical analyses.
Unless otherwise stated, all data are presented as the mean ± the SEM. Student's t test was used to assess statistical significance between means. For nonnormally distributed data, comparison of medians was made using a Mann-Whitney U test. For comparison of ratios, Wilcoxin Signed Rank was used. In all cases, significance was set at P < 0.05.
Online supplemental material.
FceRIa−/− and WT mice respond similarly to SeV infection, as shown in Fig. S1. Surface marker expression on lung cDCs from FceRIa−/− and WT mice are similar, as shown in Fig. S2. Fig. S3 shows that IFNAR expression on lung cDCs is not necessary for FcεRIα expression on lung cDCs. Fig. S4 shows the effect of cDC FcεRI activation on chemotaxis of CD4+ T cells. The ability of WT, but not FceRIa−/−, cDC to restore postviral-induced mucous cell metaplasia in FceRIa−/− mice is demonstrated in Fig. S5. Fig. S6 shows that postviral Muc5ac mRNA induction is inhibited with CCL28 blockade. As shown in Fig. S7, our data support a scheme whereby an IFN–FcεRI–CCL28–IL-13 signaling pathway regulates mucous cell metaplasia after viral infection. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20070360/DC1.
Supplemental Material
Acknowledgments
The authors wish to thank Drs. Charles Parker and Richard Hotchkiss for helpful discussions, and Drs. Albert Bendelac, Marco Colonna, Jean-Pierre Kinet, Hans Oettgen, Christine Pham, Juan Rivera, and Jonathan Sprent for generous gifts of mice and/or reagents.
This work was supported by grants from the National Institutes of Health (NIAID and National Heart, Lung, and Blood Institute) and Genentech/Novartis, as well as the Martin Schaeffer Fund and Alan A. and Edith L. Wolff Charitable Trust.
The authors declare that they have no competing financial interests.
Abbreviations used: ATCC, American Type Culture Collection; IFNAR, type I IFN receptor; PI, postinnoculation; SeV, Sendai virus.
References
- 1.Sigurs, N. 2002. A cohort of children hospitalised with acute RSV bronchiolitis: impact on later respiratory disease. Paediatr. Respir. Rev. 3:177–183. [DOI] [PubMed] [Google Scholar]
- 2.Walter, M.J., J.D. Morton, N. Kajiwara, E. Agapov, and M.J. Holtzman. 2002. Viral induction of a chronic asthma phenotype and genetic segregation from the acute response. J. Clin. Invest. 110:165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel, A.C., J.D. Morton, E.Y. Kim, Y. Alevy, S. Swanson, J. Tucker, G. Huang, E. Agapov, T.E. Phillips, M.E. Fuentes, et al. 2006. Genetic segregation of airway disease traits despite redundancy of calcium-activated chloride channel family members. Physiol. Genomics. 25:502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen, Y., P. Thai, Y.H. Zhao, Y.S. Ho, M.M. DeSouza, and R. Wu. 2003. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J. Biol. Chem. 278:17036–17043. [DOI] [PubMed] [Google Scholar]
- 5.Tyner, J.W., E.Y. Kim, K. Ide, M.R. Pelletier, W.T. Roswit, J.D. Morton, J.T. Battaile, A.C. Patel, G.A. Patterson, M. Castro, et al. 2006. Blocking airway mucous cell metaplasia by inhibiting EGFR antiapoptosis and IL-13 transdifferentiation signals. J. Clin. Invest. 116:309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alexeyev, O.A., C. Ahlm, J. Billheden, B. Settergren, G. Wadell, and P. Juto. 1994. Elevated levels of total and Puumala virus-specific immunoglobulin E in the Scandinavian type of hemorrhagic fever with renal syndrome. Clin. Diagn. Lab. Immunol. 1:269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calenoff, E., J.C. Zhao, E.L. Derlacki, W.H. Harrison, K. Selmeczi, J.C. Dutra, I.R. Olson, and D.G. Hanson. 1995. Patients with Meniere's disease possess IgE reacting with herpes family viruses. Arch. Otolaryngol. Head Neck Surg. 121:861–864. [DOI] [PubMed] [Google Scholar]
- 8.Koraka, P., B. Murgue, X. Deparis, T.E. Setiati, C. Suharti, E.C. van Gorp, C.E. Hack, A.D. Osterhaus, and J. Groen. 2003. Elevated levels of total and dengue virus-specific immunoglobulin E in patients with varying disease severity. J. Med. Virol. 70:91–98. [DOI] [PubMed] [Google Scholar]
- 9.Votava, M., D. Bartosova, A. Krchnakova, K. Crhova, and L. Kubinova. 1996. Diagnostic importance of heterophile antibodies and immunoglobulins IgA, IgE, IgM and low-avidity IgG against Epstein-Barr virus capsid antigen in children. Acta Virol. 40:99–101. [PubMed] [Google Scholar]
- 10.Welliver, R.C., M. Sun, D. Rinaldo, and P.L. Ogra. 1986. Predictive value of respiratory syncytial virus-specific IgE responses for recurrent wheezing following bronchiolitis. J. Pediatr. 109:776–780. [DOI] [PubMed] [Google Scholar]
- 11.Chantzi, F.M., D.A. Kafetzis, T. Bairamis, C. Avramidou, N. Paleologou, I. Grimani, N. Apostolopoulos, and N.G. Papadopoulos. 2006. IgE sensitization, respiratory allergy symptoms, and heritability independently increase the risk of otitis media with effusion. Allergy. 61:332–336. [DOI] [PubMed] [Google Scholar]
- 12.Maurer, D., C. Ebner, B. Reininger, E. Fiebiger, D. Kraft, J.P. Kinet, and G. Stingl. 1995. The high affinity IgE receptor (Fc epsilon RI) mediates IgE-dependent allergen presentation. J. Immunol. 154:6285–6290. [PubMed] [Google Scholar]
- 13.Foster, B., D.D. Metcalfe, and C. Prussin. 2003. Human dendritic cell 1 and dendritic cell 2 subsets express FcepsilonRI: correlation with serum IgE and allergic asthma. J. Allergy Clin. Immunol. 112:1132–1138. [DOI] [PubMed] [Google Scholar]
- 14.Legge, K.L., and T.J. Braciale. 2003. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity. 18:265–277. [DOI] [PubMed] [Google Scholar]
- 15.Usherwood, E.J., T.L. Hogg, and D.L. Woodland. 1999. Enumeration of antigen-presenting cells in mice infected with Sendai virus. J. Immunol. 162:3350–3355. [PubMed] [Google Scholar]
- 16.Crowe, S.R., S.J. Turner, S.C. Miller, A.D. Roberts, R.A. Rappolo, P.C. Doherty, K.H. Ely, and D.L. Woodland. 2003. Differential antigen presentation regulates the changing patterns of CD8+ T cell immunodominance in primary and secondary influenza virus infections. J. Exp. Med. 198:399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zammit, D.J., L.S. Cauley, Q.M. Pham, and L. Lefrancois. 2005. Dendritic cells maximize the memory CD8 T cell response to infection. Immunity. 22:561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Rijt, L.S., S. Jung, A. Kleinjan, N. Vos, M. Willart, C. Duez, H.C. Hoogsteden, and B.N. Lambrecht. 2005. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J. Exp. Med. 201:981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dombrowicz, D., B. Quatannens, J.-P. Papin, A. Capron, and M. Capron. 2000. Expression of a functional Fc{epsilon }RI on rat eosinophils and macrophages. J. Immunol. 165:1266–1271. [DOI] [PubMed] [Google Scholar]
- 20.Bieber, T., H. de la Salle, A. Wollenberg, J. Hakimi, R. Chizzonite, J. Ring, D. Hanau, and C. de la Salle. 1992. Human epidermal langerhans cells express the high affinity receptor for immunoglobulin E (FcεRI). J. Exp. Med. 175:1285–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schroeder, J.T., A.P. Bieneman, H. Xiao, K.L. Chichester, K. Vasagar, S. Saini, and M.C. Liu. 2005. TLR9- and FcepsilonRI-mediated responses oppose one another in plasmacytoid dendritic cells by down-regulating receptor expression. J. Immunol. 175:5724–5731. [DOI] [PubMed] [Google Scholar]
- 22.Grayson, M.H., M.S. Ramos, M.M. Rohlfing, R. Kitchens, H.D. Wang, A. Gould, E. Agapov, and M.J. Holtzman. 2007. Controls for lung dendritic cell maturation and migration during respiratory viral infection. J. Immunol. 179:1438–1448. [DOI] [PubMed] [Google Scholar]
- 23.Blank, U., C. Ra, L. Miller, K. White, H. Metzger, and J.P. Kinet. 1989. Complete structure and expression in transfected cells of high affinity IgE receptor. Nature. 337:187–189. [DOI] [PubMed] [Google Scholar]
- 24.Maldonado-Lopez, R., T. De Smedt, P. Michel, J. Godfroid, B. Pajak, C. Heirman, K. Thielemans, O. Leo, J. Urbain, and M. Moser. 1999. CD8α+ and CD8α− subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J. Exp. Med. 189:587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.English, K., C. Brady, P. Corcoran, J.P. Cassidy, and B.P. Mahon. 2006. Inflammation of the respiratory tract is associated with CCL28 and CCR10 expression in a murine model of allergic asthma. Immunol. Lett. 103:92–100. [DOI] [PubMed] [Google Scholar]
- 26.Wang, W., H. Soto, E.R. Oldham, M.E. Buchanan, B. Homey, D. Catron, N. Jenkins, N.G. Copeland, D.J. Gilbert, N. Nguyen, et al. 2000. Identification of a novel chemokine (CCL28), which binds CCR10 (GPR2). J. Biol. Chem. 275:22313–22323. [DOI] [PubMed] [Google Scholar]
- 27.John, A.E., M.S. Thomas, A.A. Berlin, and N.W. Lukacs. 2005. Temporal production of CCL28 corresponds to eosinophil accumulation and airway hyperreactivity in allergic airway inflammation. Am. J. Pathol. 166:345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finkelman, F.D., M.E. Rothenberg, E.B. Brandt, S.C. Morris, and R.T. Strait. 2005. Molecular mechanisms of anaphylaxis: lessons from studies with murine models. J. Allergy Clin. Immunol. 115:449–457. [DOI] [PubMed] [Google Scholar]
- 29.Yamaguchi, M., C.S. Lantz, H.C. Oettgen, I.M. Katona, T. Fleming, I. Miyajima, J.-P. Kinet, and S.J. Galli. 1997. IgE enhances mouse mast cell FcεRI expression in vitro and in vivo: evidence for a novel amplification mechanism in IgE-dependent reactions. J. Exp. Med. 185:663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skoner, D.P., W.J. Doyle, E.P. Tanner, J. Kiss, and P. Fireman. 1995. Effect of rhinovirus 39 (RV-39) infection on immune and inflammatory parameters in allergic and non-allergic subjects. Clin. Exp. Allergy. 25:561–567. [DOI] [PubMed] [Google Scholar]
- 31.Las Heras, J., and V.L. Swanson. 1983. Sudden death of an infant with rhinovirus infection complicating bronchial asthma: case report. Pediatr. Pathol. 1:319–323. [DOI] [PubMed] [Google Scholar]
- 32.Rager, K.J., J.O. Langland, B.L. Jacobs, D. Proud, D.G. Marsh, and F. Imani. 1998. Activation of antiviral protein kinase leads to immunoglobulin E class switching in human B cells. J. Virol. 72:1171–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kusel, M.M., N.H. de Klerk, T. Kebadze, V. Vohma, P.G. Holt, S.L. Johnston, and P.D. Sly. 2007. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J. Allergy Clin. Immunol. 119:1105–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dakhama, A., J.-W. Park, C. Taube, K. Chayama, A. Balhorn, A. Joetham, X.-D. Wei, R.-H. Fan, C. Swasey, N. Miyahara, et al. 2004. The role of virus-specific immunoglobulin E in airway hyperresponsiveness. Am. J. Respir. Crit. Care Med. 170:952–959. [DOI] [PubMed] [Google Scholar]
- 35.Garman, S.C., B.A. Wurzburg, S.S. Tarchevskaya, J.P. Kinet, and T.S. Jardetzky. 2000. Structure of the Fc fragment of human IgE bound to its high-affinity receptor Fc epsilonRI alpha. Nature. 406:259–266. [DOI] [PubMed] [Google Scholar]
- 36.Stephens, R., D.A. Randolph, G. Huang, M.J. Holtzman, and D.D. Chaplin. 2002. Antigen-nonspecific recruitment of Th2 cells to the lung as a mechanism for viral infection-induced allergic asthma. J. Immunol. 169:5458–5467. [DOI] [PubMed] [Google Scholar]
- 37.Eksteen, B., A. Miles, S.M. Curbishley, C. Tselepis, A.J. Grant, L.S. Walker, and D.H. Adams. 2006. Epithelial inflammation is associated with CCL28 production and the recruitment of regulatory T cells expressing CCR10. J. Immunol. 177:593–603. [DOI] [PubMed] [Google Scholar]
- 38.Bufford, J.D., and J.E. Gern. 2005. The hygiene hypothesis revisited. Immunol. Allergy Clin. North Am. 25:247–262. [DOI] [PubMed] [Google Scholar]
- 39.Sly, R.M. 1999. Changing prevalence of allergic rhinitis and asthma. Ann. Allergy Asthma Immunol. 82:233–248. [DOI] [PubMed] [Google Scholar]
- 40.Grayson, M.H., R.S. Hotchkiss, I.E. Karl, M.J. Holtzman, and D.D. Chaplin. 2003. Intravital microscopy comparing T lymphocyte trafficking to the spleen and the mesenteric lymph node. Am. J. Physiol. Heart Circ. Physiol. 284:H2213–H2226. [DOI] [PubMed] [Google Scholar]
- 41.Flynn, K.J., G.T. Belz, J.D. Altman, R. Ahmed, D.L. Woodland, and P.C. Doherty. 1998. Virus-specific CD8+ T cells in primary and secondary influenza pneumonia. Immunity. 8:683–691. [DOI] [PubMed] [Google Scholar]
- 42.Lyons, A.B. 2000. Analysing cell division in vivo and in vitro using flow cytometric measurement of CFSE dye dilution. J. Immunol. Methods. 243:147–154. [DOI] [PubMed] [Google Scholar]
- 43.Galli, S.J., A.M. Dvorak, J.A. Marcum, T. Ishizaka, G. Nabel, H. Der Simonian, K. Pyne, J.M. Goldin, R.D. Rosenberg, H. Cantor, and H.F. Dvorak. 1982. Mast cell clones: a model for the analysis of cellular maturation. J. Cell Biol. 95:435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gillespie, S.R., R.R. DeMartino, J. Zhu, H.J. Chong, C. Ramirez, C.P. Shelburne, L.A. Bouton, D.P. Bailey, A. Gharse, P. Mirmonsef, et al. 2004. IL-10 inhibits Fc epsilon RI expression in mouse mast cells. J. Immunol. 172:3181–3188. [DOI] [PubMed] [Google Scholar]
- 45.Gomez, G., C.D. Ramirez, J. Rivera, M. Patel, F. Norozian, H.V. Wright, M.V. Kashyap, B.O. Barnstein, K. Fischer-Stenger, L.B. Schwartz, et al. 2005. TGF-beta 1 inhibits mast cell Fc epsilon RI expression. J. Immunol. 174:5987–5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bleul, C.C., R.C. Fuhlbrigge, J.M. Casasnovas, A. Aiuti, and T.A. Springer. 1996. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J. Exp. Med. 184:1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}