

Abstract
Viral mutational escape can reduce or abrogate recognition by the T cell receptor (TCR) of virus-specific CD8+ T cells. However, very little is known about the impact of cytotoxic T lymphocyte (CTL) epitope mutations on interactions between peptide–major histocompatibility complex (MHC) class I complexes and MHC class I receptors expressed on other cell types. Here, we analyzed a variant of the immunodominant human leukocyte antigen (HLA)-B2705–restricted HIV-1 Gag KK10 epitope (KRWIILGLNK) with an L to M amino acid substitution at position 6 (L6M), which arises as a CTL escape variant after primary infection but is sufficiently immunogenic to elicit a secondary, de novo HIV-1–specific CD8+ T cell response with an alternative TCR repertoire in chronic infection. In addition to altering recognition by HIV-1–specific CD8+ T cells, the HLA-B2705–KK10 L6M complex also exhibits substantially increased binding to the immunoglobulin-like transcript (ILT) receptor 4, an inhibitory MHC class I–specific receptor expressed on myelomonocytic cells. Binding of the B2705–KK10 L6M complex to ILT4 leads to a tolerogenic phenotype of myelomonocytic cells with lower surface expression of dendritic cell (DC) maturation markers and co-stimulatory molecules. These data suggest a link between CTL-driven mutational escape, altered recognition by innate MHC class I receptors on myelomonocytic cells, and functional impairment of DCs, and thus provide important new insight into biological consequences of viral sequence diversification.
Viral proteins are intracellularly degraded into small peptides and then loaded on MHC class I molecules, which are presented on the surface of infected cells. These peptide–MHC class I complexes serve as physiologic ligands for the TCR and, upon TCR engagement, trigger several lymphocyte effector functions. In addition, peptide–MHC class I complexes also serve as ligands for several alternative receptors, such as the killer-Ig receptors (KIRs) expressed on NK and T cells (1) or the Ig-like transcript (ILT) receptors predominantly expressed either on T cells (ILT2/leukocyte Ig receptor [LIR]1) (2, 3) or on macrophages, monocytes, and DCs (ILT4/LIR2) (4, 5). Binding of peptide–MHC class I complexes to KIRs (6) or ILT2 (7–9) receptors results in an impairment of lymphocyte effector functions, such as cytotoxic properties or cytokine secretion capacities, whereas binding to ILT4 can lead to the transformation of macrophages and DCs into tolerogenic cells with lower expression of co-stimulatory molecules and ineffective antigen-presenting characteristics (10, 11).
A series of recent studies has shown that HIV-1 can use its extraordinary genetic plasticity to evade host immune surveillance (12–19). This has been demonstrated particularly clearly in the context of CD8+ T cell responses, in which highly specific interactions between the peptide–MHC class I complex and the TCR can be affected by single amino acid variations in the antigenic peptide that reduce peptide binding to the restricting MHC molecule (12, 15), interfere with recognition by TCR contact residues (16, 20), or prevent intracellular peptide processing (21, 22). The interactions of peptide–MHC class I complexes with KIRs are believed to be also dependent on the sequence of the presented antigenic peptide (23–26). However, although artificial single amino acid changes in antigenic peptides can lead to a differential recognition by KIRs and may thus modify their inhibitory properties (27), evidence for a selection of HIV-1 mutational CTL escape variants affecting KIR recognition is currently lacking. The degree to which the binding of peptide–MHC class I complexes to ILT receptors depends on the sequence of the antigenic peptide is currently unknown, and it is uncertain if HIV-1 mutational escape can affect the interaction of peptide–MHC class I complexes with these receptors.
In this study, we performed a detailed analysis of the genetic evolution of the HLA-B2705–restricted HIV-1 cytotoxic T cell epitope KK10 (KRWIILGLNK), which is an extremely immunodominant epitope in HLA-B2705–expressing HIV-1–infected individuals. We show that a variant of this epitope with an L to M substitution at position 6 (L6M) frequently arises as a CTL escape variant. Furthermore, our data indicate that this mutation increases binding to ILT4 and by this way leads to enhanced functional inhibition of DCs. These data indicate that viral mutational escape does not only affect recognition by CD8+ T cells, but can also create better ligands for inhibitory MHC class I receptors on myelomonocytic cells, and thus contribute to a tolerogenic functional profile of these cells.
RESULTS
Lack of recognition of the KK10 L6M variant by KK10-specific CD8+ T cells during primary HIV-1 infection
The KK10 epitope is an extremely immunodominant target for HIV-1–specific CD8+ T cells in HLA-B2705–expressing individuals (28, 29) and has been associated with a slower HIV-1 disease progression (30, 31). The most frequent HLA-B2705–associated mutation in this epitope involves an L to M amino acid substitution at position 6 (L6M), which does not substantially affect the binding avidity to the restricting HLA molecule (29), peptide processing (29), or viral fitness (32), but may alter the interaction with TCR contact residues (27). Mutations at position 2 in this epitope, which typically occur late during the disease and subsequent to the L6M mutation, are also strongly associated with HLA-B2705 expression and have been shown to reduce binding to HLA-B2705 (29).
To determine if the KK10 L6M variant is associated with loss of CD8+ T cell recognition and thus represents a CTL escape variant, we tested the recognition of this variant in six HIV-1–infected HLA-B2705+ individuals identified during primary HIV-1 infection. All of these subjects mounted a strong CD8+ T cell response against the KK10 WT peptide that was present in their autologous viral sequence, as determined by population viral sequencing. Using interferon γ ELISPOT assays, we found that in addition to the KK10 WT peptide, KK10 variants with amino acid substitutions at position 2 (R2K or R2T) were still recognized by CD8+ T cells during primary infection, although the avidity of recognition decreased by ∼10-fold and might be weaker when the variant peptide is naturally presented (Fig. 1) (29). In contrast, the KK10 L6M variant and the variants with combined mutations at position 2 and 6 (R2K/L6M, R2T/L6M) were only very poorly recognized during primary HIV-1 infection compared with either the KK10 WT sequence or the KK10 variants with amino acid changes at position 2 only (Fig. 1). Thus, these data show a relative lack of recognition of the KK10 L6M variant in primary infection and suggest that the subsequent emergence of this viral mutation results from CD8+ T cell–mediated immune pressure.
Figure 1.

Consistent lack of recognition of the KK10 L6M variant by KK10-specific CD8+ T cells during primary HIV-1 infection. (A) Cross-reactivity of KK10-specific CD8+ T cells against KK10 variants with amino acid substitutions at positions 2, 6, or 2 and 6 as determined by interferon-γ ELISPOT with PBMC samples collected during primary infection. Data from one representative study individual infected with a virus harboring the KK10 WT epitope are shown. (B) Recognition of the KK10 WT and variant peptides by six HIV-1–infected individuals during primary HIV-1 infection.
De novo CD8+ T cell response against the KK10 L6M variant during chronic HIV-1 infection
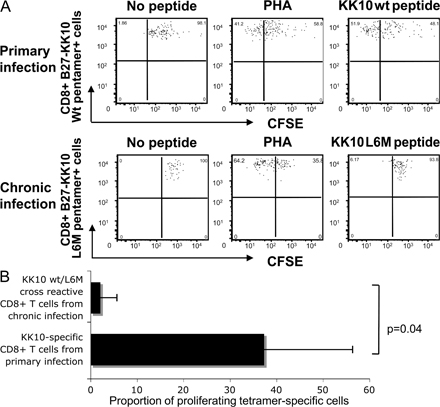
In three of the above-mentioned individuals, we had the opportunity to longitudinally analyze the evolution of the KK10 epitope after primary infection in the absence of antiretroviral treatment. In these longitudinal studies, we found that all three individuals developed the KK10 L6M mutation in the autologous viruses during the subsequent disease process, most likely resulting from the relative lack of recognition of this variant by the primarily mounted KK10-specific CD8+ T cell responses. This switch in the autologous viral KK10 sequence was associated with a strong increase in the CD8+ T cell population able to recognize the KK10 L6M variant (Fig. 2 A) and the recruitment of an alternative TCR α and β chain repertoire (Table I), indicating the de novo generation of a KK10 L6M variant-specific CD8+ T cell response. The recognition of the L6M variant in these individuals was at least partially mediated by newly generated KK10-specific CD8+ T cell clones with cross-reactive TCRs that had almost identical capacities to recognize the KK10 WT and the KK10 L6M variant (Fig. 2 B). A strong recognition of the KK10 L6M variant was also detected by cross-sectional analysis of five additional subjects with chronic HIV-1 infection harboring the L6M mutation in their autologous viral sequence (Fig. 2 C). Overall, these data show that the KK10 L6M mutation represents an escape variant from the initially recruited TCR repertoire of KK10-specific CD8+ T cells during primary infection, but is sufficiently immunogenic to elicit a variant-specific de novo CD8+ T cell response in chronic infection.
Figure 2.

De novo HIV-1–specific CD8+ T cell responses against the KK10 L6M variant during chronic HIV-1 infection. (A) Intra-individual comparison of the recognition of the KK10 WT (left panel) and the KK10 L6M (right panel) variant during primary (KK10 WT sequence in autologous virus) and chronic (KK10 L6M variant sequence in autologous virus) HIV-1 infection as measured by interferon-γ ELISPOT in three study subjects. (B) Cross-recognition of the KK10 WT and the KK10 L6M variant by two different KK10-specific CD8+ T cell clones isolated during chronic HIV-1 infection in the respective study individuals as measured by interferon-γ ELISPOT. Data from two clones using the indicated TCRs are shown. (C) Recognition of naturally occurring KK10 variants in eight study subjects with chronic HIV-1 infection harboring the KK10 L6M variant in their autologous virus.
Table I.
HLA-B2705–KK10-specific TCR α and β chain repertoire
|
|
|
KK10 WT in autologous virus
|
|
KK10 L6M in autologous virus
|
||
|---|---|---|---|---|---|---|
| Study patient | Time | TCR α chain | TCR β chain | Timea | TCR α chain | TCR β chain |
| 1 | Day 41 PP | n. d. | Vβ27-CASSQRTGELF-J2.2 (8/11) | Day 760 PP | n. d. | Vβ27-CASRVAEVNYEQY-J2.7 (4/15) |
| Vβ20.1-CSAWTSGGRADTQY-J2.3 (3/11) | Vβ5.6-CASSYSGSNYEQY-J2.7 (11/15) | |||||
| 2 | Day 0 PP | Vα19-CALGEANTGFQKLV-J8 (2/24) | Vβ5.4-CASSSTAPDTEAF-J1.1-(1/46) | Day 325 PP | Vα4-CLVVRMDSSYKLI-J12 (4/23) | Vβ6.6-CASSYSRGAGNTIY-J1.3 (1/43) |
| Vα19-CAPSEANTGFQKLV-J8 (1/24) | Vβ5.4-CASSGTAPAAEAF-J1.1 (1/46) | Vα4-CLVVWMDSSYKLI-J12 (1/23) | Vβ28-CASSLKDEQF-J2.1-(1/43) | |||
| Vα19-CALSEADTGFQKLV-J8 (1/24) | Vβ5.4-CASSLTAPDTEAF-J1.1 (32/46) | Vα1.1-CAVQSDYKLS-J20 (6/23) | Vβ24-CATGLPEGSQETQY-J2.5 (1/43) | |||
| Vα19-CALSEANTGFQKLV-J8 (LGTG) (1/24) | Vβ27-CASSRSTGELF-J2.2 (10/46) | Vα5-CAERGLMDTGRRALT-J5 (12/23) | Vβ19-CASSRRALRGYT-J1.2 (5/43) | |||
| Vα19-CALSEANTGFQKLV-J8 (FETG) (1/24) | Vβ20.1-CSARDQRDYQETQY-J2.5 (2/46) | Vβ7.2-CASSLGRGNEQF-J2.1-(2/43) | ||||
| Vα19-CALSEANTGFQKLV-J8 (18/24) | Vβ27-CASSQRTGELF-J2.2 (8/43) | |||||
| Vβ27-CASSPRTGELF J2.2 (5/43) | ||||||
| Vb20.1-CSARETSGAYNEQF J2.1(18/43) | ||||||
| 3 | Day 21 PP | Vα8.4-CAVTLLGTGGFKTI-J9 (9/18) | Vβ7.9-CANSLDGDQPGH-J1.5 (1/24) | Day 235 PP | Vα8.6-CAVSDPGFKTI-J9 (11/21) | Vβ20.1-CSAREGVEGYT-J1.2 (21/21) |
| Vα12.2-CAV NKVEFNAGGTSGKLT-J52 (9/18) | Vβ7.9-CASSLDGDQPQH-J1.5 (4/24) | Vα19-CALTDQQRAGNMLT-J39 (9/21) | ||||
| Vβ7.9-CASSLDRDEQF-J2.1 (1/24) | Vα19-CAQTDQQRAGNMLT-J39 (1/21) | |||||
| Vβ27-CASSKNQWEQY-J2.7 (7/24) | ||||||
| Vβ27-CASSPTSYEQY-J2.7 (1/24) | ||||||
| Vβ9-CASSSFDRANEQF-J2.1 (5/24) | ||||||
| Vβ20.1-CSAREGVEGYT-J1.2 (1/24) | ||||||
| Vβ20.1-CSARPGLAGELYEQY-J2.7 (2/24) | ||||||
| Vβ20.1-CSARAGLAGALYEQY-J2.7 (2/24) | ||||||
PP, days after presentation off therapy.
Time points were chosen based on sample availability and might not necessarily reflect the time when the L6M mutation first emerged.
Enhanced binding of the HLA-B2705–KK10 L6M complex to ILT4 on monocytes and DCs
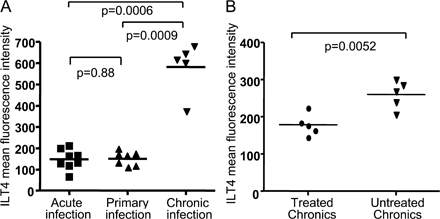
To determine if the L6M mutation in the KK10 epitope, in addition to altering binding to the TCR, also affects recognition by other HLA class I receptors expressed on lymphocytes, monocytes/macrophages, or DCs, we stained PBMCs from chronically HIV-1–infected, treatment-naive, and HLA-B2705− individuals with HLA-B2705 pentamers refolded with the KK10 WT or KK10 L6M peptides. In a total of 10 HLA-B2705− study individuals, no binding of either one of these pentamers to NK cells, T cells, or B cells was found, suggesting that TCR-independent interactions with lymphocellular MHC class I receptors, such as ILT2 or KIRs that were expressed in 5–15% and 5–35% of lymphocytes, respectively (2, 33), did not occur. However, in all 10 study subjects, we consistently found that both pentamers were clearly able to bind to peripheral blood CD14+ monocytes and CD11c+ HLA-DR+ lin− peripheral blood DCs. Interestingly, the binding of the HLA-B2705–KK10 L6M pentamer to these leukocellular subgroups was between two- and threefold stronger than that of the B2705 pentamer refolded with the WT peptide (Fig. 3, A and B), although no difference in binding intensity was seen between the two pentamers on cross-reactive HLA-B2705–KK10-specific CD8+ T cell populations (Fig. 3 A). More intense binding of the B2705–KK10 L6M pentamer to CD14+ monocytes compared with the B2705/KK10 WT pentamer was observed over a wide range of pentamer concentrations (Fig. 3 C). Binding of the pentamers to macrophages and DCs was abrogated by antibodies blocking ILT4, an inhibitory MHC class I receptor expressed on myelomonocytic cells (Fig. 3 B), which is up-regulated in chronic untreated HIV-1 infection compared with HIV-1–uninfected individuals (34), individuals with acute or primary HIV-1 infection (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20061865/DC1), or individuals with chronic treated HIV-1 infection (Fig. S1 B). In contrast, blocking antibodies directed against several other MHC class I receptors, including those against ILT2, had no effect on pentamer binding intensity to these macrophages or monocytes (not depicted). To confirm an antigenic peptide variant–specific binding mechanism between ILT4 and HLA-B27–KK10 complexes, these receptor–ligand interactions were further analyzed using recombinant proteins (ILT4-IgG dimers and HLA-B2705 KK10 WT/L6M tetramers) and surface plasmon resonance (SPR) experiments. These assays indicated an apparent steady-state equilibrium constant (KD) of 122 nM for the binding of the HLA-B27–KK10 L6M complex to an ILT4 dimer compared with a KD of 733 nM for the interaction between the WT HLA-B27–KK10 complex and the ILT4 dimer (Fig. 3 D). Although the use of recombinant ILT4 in a dimer form precludes assessment of the affinity between HLA-B2705 and naturally occurring, monomeric ILT4, this observation corresponds to a sixfold increase in apparent affinity for the interaction involving the L6M mutation relative to that of the WT. Overall, these data indicate that HLA-B2705 KK10 pentamers can bind to myelomonocytic cells via ILT4, and that the degree of binding is influenced by the amino acid sequence of the bound KK10 peptide.
Figure 3.

Enhanced ILT4-mediated binding of the HLA-B2705–KK10 L6M complex to peripheral blood macrophages and DCs. (A) Histograms indicating the binding of the HLA-B2705–KK10 WT or KK10 L6M pentamers to macrophages, DCs, lymphocytes, or KK10-specific CD8 T cell clones in the presence or absence of ILT4-blocking antibodies. (B) Mean fluorescence intensity of the respective pentamers after gating on CD14+ macrophages or CD11c+ lin− HLA-DR+ DCs. Data from 10 HLA-B2705− chronically HIV-1–infected, treatment-naive subjects are shown. (C) Mean fluorescence intensity of the B2705–KK10 WT and L6M variant pentamers after staining with the indicated pentamer dilutions (x axis) on CD14+ macrophages. (D) SPR sensograms reflecting the control surface–subtracted interactions between immobilized HLA-B27–KK10 L6M (top) and B27–KK10 WT (bottom) with recombinant ILT4. ILT4 was injected in five serial twofold dilutions from a starting concentration of 74 μg/ml (1 μM). Inset plots show nonlinear regression analysis of maximal responses versus concentration. One representative experiment out of three is shown.
The HLA-B2705–KK10 L6M complex leads to ILT4-mediated functional impairment of DCs
Previous data have shown that agonistic ILT4 engagement by MHC class I molecules can transform the functional repertoire of DCs toward a tolerogenic profile with lower expression of co-stimulatory molecules and a decreased capacity for T cell expansion (10, 11). To test if the B2705–KK10 L6M complex preferentially increases the tolerogenic properties of DCs or myelomonocytic cells in comparison to the HLA-B2705–KK10 WT complex, we stimulated immature, monocyte-derived DCs (MDDCs) with a maturation-inducing cytokine cocktail in the presence or absence of the two B2705/KK10 pentamers and measured the ensuing up-regulation of DC maturation surface markers (HLA-DR, CD83, CD40, CD80, and CD86). For these experiments, monocyte-derived immature DCs with strongly detectable ILT4 surface expression were generated by a 5-d ex vivo culture with GM-CSF. As shown in Fig. 4 (A and C), we consistently observed that the up-regulation of such molecules was inhibited substantially when cells had been exposed to B2705–KK10 L6M pentamer complexes, whereas incubation with the B2705–KK10 WT pentamers showed no obvious inhibitory effect.
Figure 4.

Inhibition of the maturation of MDDCs by HLA-B2705–KK10 L6M complexes. (A and B) Histograms indicating the expression intensity of CD40, CD86, and HLA-DR of myelomonocytic cells after cytokine-induced maturation in the presence or absence of HLA-B2705–KK10 WT or B2705–KK10 L6M pentamers (A) or autologous HLA-B2705–expressing DCs pulsed with the KK10 WT or KK10 L6M variant peptide (B). One example is shown. (C) Mean and standard deviation of the fluorescence intensity of DC maturation markers and co-stimulatory molecules after cytokine cocktail–mediated maturation in the presence or absence of the B2705–KK10 WT or L6M pentamers. Experiments were independently performed four times. (D) Assessment of HLA-B27–KK10 L6M–mediated functional inhibition of DCs by mixed lymphocyte reactions. Dot plots indicate the proportion of allogenic CD4 or CD8 T cells after a 6-d exposure to MDDCs in the presence of HLA-B27–KK10 WT or L6M pentamers. One experiment out of three in three different study subjects is shown.
Because stimulation with L6M pentamers had an inhibitory effect on DC maturation, we subsequently determined whether peptide-loaded APCs would have a similar effect. Therefore, we generated ILT4-expressing DCs from the PBMCs of HIV-1–uninfected HLA-B2705–expressing donors, which were subsequently divided into two populations. The first population (“presenters”) was labeled with CFSE, matured with cytokine cocktail, and loaded with KK10 WT or KK10 L6M peptide, and the second population (“responders”) was initially left immature, mixed with the autologous presenters for 4 h, and then matured with cytokine cocktail. After 16 h, the degree of maturation of the responders was determined by flow cytometry, whereas the CFSE-labeled presenters were excluded from the analysis. As shown in Fig. 4 B, presenter cells loaded with the KK10 L6M peptide, but not with KK10 WT peptide or without peptide, were able to inhibit the up-regulation of CD86, CD40, and HLA-DR on the responder DC population. This indicates that in addition to pentamers, HLA-B2705 complexes on the surface of live APCs can also mediate peptide-specific inhibitory effects in trans.
To determine if the B2705–KK10 L6M–mediated inhibition of DC maturation would translate into functional impairments of these cells, we next used MDDCs matured in the presence of HLA-B2705–KK10 WT or L6M pentamers to perform mixed lymphocyte reactions with CFSE-labeled allogenic T cells. These experiments indicated a substantially reduced proportion of proliferating allogenic T cells after exposure to MDDCs matured with B2705–KK10 L6M pentamers compared with those matured in the presence of the WT pentamer (Fig. 4 D). Overall, these data indicate that myelomonocytic binding of the B2705–KK10 L6M complex can result in inhibition of DC maturation and function.
To determine whether inhibition of myelomonocytic cell maturation by HLA-B2705–KK10 L6M complexes required ILT4, we knocked down ILT4 expression on MDDCs using targeted small interfering RNA (siRNA). As shown in Fig. 5 A, an siRNA pool specific for the ILT4 message, but not an siRNA pool specific for ILT2 message, led to a substantial reduction in ILT4 surface expression on MDDCs. We next tested whether the inhibition of DC maturation mediated by HLA-B2705–KK10 pentamers or KK10 peptide–pulsed B2705+ presenting cells could be reversed by ILT4 knockout. Fig. 5 (B and C) shows that the inhibition of MDDC maturation by the HLA-B2705–KK10 L6M complexes either in pentamer form or on the surface of live APCs was abrogated by ILT4-specific siRNA in a dose-dependent fashion. Thus, these data indicate that HLA-B2705–KK10 L6M complexes inhibit myelomonocytic cell maturation and co-stimulatory molecule surface expression via antigenic peptide variant-specific binding to ILT4.
Figure 5.

Reversion of the HLA-B2705–KK10 L6M–mediated inhibitory effects after siRNA-induced down-regulation of ILT4. (A) Flow cytometric assessment of ILT4 surface expression on MDDCs after electroporation with ILT4- or ILT2-specific siRNA. (B and C) Histograms indicating the surface expression of co-stimulatory molecules or DC maturation markers after exposure of ILT4 or ILT2 siRNA–transfected myelomonocytic cells to HLA-B2705–KK10 WT or L6M pentamers (B) or to autologous HLA-B2705–expressing DCs pulsed with the KK10 WT or KK10 L6M variant peptide (C). Low dose siRNA concentration, 0.5 nmol/million cells; high dose siRNA concentration, 1 nmol/million cells.
Frequent antigenic peptide dependency of MHC class I recognition by ILT4
In our final series of experiments, we tested whether a peptide-specific HLA class I binding to ILT4 also occurs in the context of MHC class I molecules other than HLA-B2705. To analyze this, PBMCs from 10 chronically HIV-1–infected untreated study subjects were stained with a series of HLA-A2, -A3, -B7, -B8, or -B40 pentamers refolded with a variety of epitopic peptides derived from HIV-1, HCV, CMV, or EBV. These experiments indicated that in the context of a shared HLA allele, pentamer binding intensity to CD14+ peripheral blood macrophages depended dramatically on the antigenic peptide presented by the MHC class I complex (Fig. 6 A). This was particularly true for HLA-A2 molecules, which bound strongly to macrophages when presenting the HIV-1–derived YV9 epitope, but showed almost no binding when presenting the epitopes RV9 and IV9. Again, the binding of pentamers to macrophages was abrogated by ILT4 blocking antibodies (Fig. 6 B). Pentamer binding to respective antigen-specific CD8+ T cell populations revealed no similar differences in the binding intensity (not depicted), indicating that the observed peptide-specific differences between pentamer binding to monocytes were not due to inherent fluorescence emission characteristics of these reagents. Overall, these data show that the sequence of the presented antigenic peptide influences the ILT4-mediated binding strength of HLA class I molecules to monocytes.
Figure 6.

Peptide-dependent binding of HLA class I molecules to ILT4 on CD14+ peripheral blood monocytes. (A) Data indicate the mean fluorescence intensity of the HLA class I pentamers refolded with the indicated epitopic peptides in 10 chronically HIV-1–infected, treatment-naive study subjects. (B) Pentamer binding in the presence (open symbols) or absence (solid symbols) of ILT4 blocking antibodies from five individuals for six epitopes is shown.
DISCUSSION
Numerous studies have indicated that HIV-1 uses its extraordinary genetic flexibility to evade CD8+ T cell–mediated immune responses (12–19). Although viral escape mutations can directly reduce the recognition by CD8+ T cells, almost nothing is known about how these viral gene alterations affect the interaction with innate inhibitory MHC class I receptors expressed on NK cells, T cells, and myelomonocytic cells. In this study, we show that HIV-1 mutations arising from CD8+ T cell–mediated immune pressure can substantially enhance the binding of viral peptide–MHC class I complexes to the inhibitory ILT4 receptor on peripheral blood monocytes and DCs and may, in this way, have a critical impact on their functional profile, specifically with regard to their ability for maturation and co-stimulatory molecule up-regulation. Thus, our data suggest HIV-1 gene diversification leading to enhanced ILT4-mediated inhibition of monocyte and DC function as a novel mechanism of viral immune evasion.
Although our data clearly suggest that the interaction between ILT4 and peptide–MHC class I complexes occurs in a peptide-specific fashion, it is presently unclear how this peptide specificity is mediated at the molecular level. Crystal structure data have indicated that the two distal extracellular domains (D1 and D2) of ILT2, an MHC class I–specific inhibitory receptor closely related to ILT4, bind MHC class I molecules at the intersection of the α 3 domain, which is relatively conserved among different HLA alleles, and the nonpolymorphic β 2 microglobulin (35). Although the structural binding positions of ILT4 to HLA-B27 have not been identified, it has been shown that ILT4 binds HLA-G also in the α 3 domain (36). Yet, recent data have indicated that binding intensities between ILT2 and a CMV-encoded HLA homologue can clearly be determined by polymorphisms in the α 1 region (37), which suggests that the conformation of the antigenic peptide-presenting α 1 domain can at least indirectly impact on the binding of HLA molecules to myelomonocytic MHC class I receptors. The peptide-dependent binding of peptide–MHC class I complexes to ILT4 shown here therefore similarly suggests that the structural conformation of the putative ILT4 binding site in the α 3 domain of HLA-B27 may be shaped indirectly by the type of antigenic peptide presented by the MHC, or that additional interactions exist between ILT4 and MHC class I molecules, potentially involving the two proximal extracellular domains of ILT4 (D3 and D4).
DC dysfunction in chronic HIV-1 infection has been demonstrated in a variety of previous investigations, and the functional defects of these cells might be causally linked to the functional impairment of HIV-1–specific CD8+ T cells that has been reported in several previous studies (38–41). Factors contributing to DC dysfunction in chronic HIV-1 infection include, among others, the down-modulation of co-stimulatory molecules by HIV-1 gene products such as vpr (42, 43), the direct infection of DCs by HIV-1 (44), and the gp120-mediated inhibition of IL-12 secretion by DCs (45). This study extends these findings by showing that viral CTL escape mutations can lead to active suppression of DC maturation by inhibitory MHC class I receptors expressed on these cells. As recent data have suggested that immature DCs induce T cells with suppressive activity upon presentation of antigen to naive T lymphocytes (46), the maturation defect mediated by ILT4 binding of the B2705–KK10 L6M complex is likely to have important consequences for the functionality of T cell responses generated. Moreover, our observation strengthens previous findings indicating that viruses can use the immunomodulatory properties of MHC class I interactions with ILT2/ILT4 as a means to decrease overall antiviral immune activities. For instance, it has been recently shown that CMV encodes for an MHC class I homologue molecule with substantially enhanced affinity to ILT2 compared with regular MHC class I molecules, thus suggesting that increasing ILT2-mediated inhibitory signals might be an active viral mechanism to reduce antiviral immune surveillance (47). Given the powerful tolerogenic effects that ILT4 can exert for the prevention of semiallogeneic or allogeneic cell rejection (10), it is likely that increased binding between ILT4 and the HLA-B27–KK10 L6M complexes can substantially contribute to the impairment of DCs in chronic HIV-1 infection. However, for a complete understanding of the biological effects of the KK10 WT and L6M peptides on DC function, it will be necessary to analyze how the KK10 WT form and its variants affect recognition of HLA-B2705 by the other myelomonocytic MHC class I receptors that have been described, including stimulatory receptors such as LIR6, which appears to have a particular capacity for recognizing HLA-B2705 (48, 49).
The physiological consequences of the increased ILT4 binding interaction with B2705–KK10 L6M in vivo are likely to depend on the expression intensity of ILT4 on monocytes and DCs, and HLA class I molecules on HIV-1 target cells. The prominent ILT4 up-regulation during chronic HIV-1 infection described here and elsewhere (34) therefore suggests that ILT4-mediated inhibition of functional properties of APCs occurs later during the disease process, and may contribute to the failure of HIV-1 patients to mount effective pathogen-specific T cell responses during the chronic phase of their disease. From this perspective, it appears surprising that we observed a de novo generation of a KK10 L6M–specific CD8+ T cell response despite immunosuppressive effects resulting from the interaction between the B27–KK10 L6M complex with ILT4. However, it is important to mention that the KK10 L6M variant seems to be highly immunogenic, and that there is only limited surface expression of ILT4 during the early stages of HIV-1 infection, at the time when the variant-specific CD8+ T cell response was generated (Fig. S1 A). Moreover, it is well conceivable that the ILT4-mediated tolerogenic functional profile of DCs does not abrogate their ability for the physical generation of new HIV-1–specific CD8+ T cells, but instead only induces a defective functional profile of these immune responses. Indeed, dysfunctional HIV-1–specific CD8+ T cells have been repeatedly reported in chronic HIV-1 infection (38, 40, 41), and we similarly observed that the proportion of proliferating KK10 L6M/WT cross-reactive CD8+ T cells in chronic infection (n = 3) was significantly lower than that of KK10 WT–specific CD8+ T cells in primary infection (n = 3; mean of 2 vs. 37%, P = 0.04) (Fig. S2). Finally, similar to other MHC class I receptors such as KIRs, the sequence of ILT4 can be polymorphic, and the identified ILT4 polymorphisms are likely to change the interaction with MHC class I receptors; therefore, consequences of increased binding of MHC class I complexes to ILT4 that result from CTL escape mutations will have to be evaluated in the context of ILT4 gene sequence alterations (50).
The data shown here do not provide evidence for an active selection of viral variants with higher binding intensity to ILT4, and in this way they are clearly different from HIV-1 gene mutations leading to CD8+ T cell escape, for which active selection of escape variants has been documented in a variety of cases. The KK10 L6M variant is selected for almost exclusively in HLA-B27–expressing individuals and therefore appears to be clearly related to HIV-1–specific CD8+ T cell–mediated immune pressure. However, instead of an active selection of viral variants with increased binding to ILT4, it appears possible that the type of CTL escape mutations that are selected could not only depend on the structural tolerance of the virus and the ensuing fitness costs, but might also be influenced by its effects on its binding intensity to ILT4. Future work will therefore be necessary to assess the exact role that binding to ILT4 plays in shaping viral evolution and how peptide-specific binding properties of MHC class I complexes to ILT4 can be taken advantage of for the design of HIV-1 vaccines and immunogens.
In summary, the data presented here provide evidence that HIV-1 CTL escape mutations can increase recognition of peptide MHC class I complexes by the inhibitory myelomonocytic MHC class I receptor ILT4, and that this increase is functionally relevant for impairing mitogen-induced myelomonocyte maturation and co-stimulatory molecule up-regulation. By indicating an association between CTL-mediated mutational escape, altered recognition by inhibitory innate immune receptors, and ensuing functional defects of professional APCs, these data contribute substantially to the understanding of biological events leading to and resulting from HIV-1 sequence evolution and will be relevant for the targeted manipulation of adaptive immune responses against HIV-1.
MATERIALS AND METHODS
Subjects.
HIV-1–infected individuals with acute, primary, or chronic HIV-1 infection participating in this study were recruited from the Massachusetts General Hospital or the Fenway Community Health Care Center in Boston. Individuals with acute HIV-1 infection had detectable HIV-1 RNA and a negative HIV-1 ELISA. Primary HIV-1 infection was defined by absent or incomplete (<3 bands) HIV-1 Western blot reactions and/or a documented HIV-1− test <1 yr before assessment. Chronically infected subjects were infected for at least 3 yr and were taking no antiretroviral therapy at the time of study participation, unless otherwise indicated. All subjects gave written informed consent to participate, and the study was approved by the Massachusetts General Hospital Institutional Review Board.
Synthetic peptides.
Peptides corresponding to described optimal HIV-1 CD8+ T cell epitopes and their variants (http://hiv-web.lanl.gov) were synthesized at the MGH Peptide Core Facility on an automated peptide synthesizer using F-moc technology.
ELISPOT assay.
ELISPOT assays were performed as described previously (51). In brief, PBMCs were plated in 96-well polyvinylidene plates that had been precoated with 0.5 μg/ml of an anti–human interferon-γ mAb (Mabtech). PBMCs were added at a concentration of 100,000 cells per well in a volume of 100 μl RPMI 1640 medium supplemented with 10% FCS, 10 mM Hepes buffer, 2 mM l-glutamine, and 50 U/ml penicillin-streptomycin. Plates were incubated overnight at 37°C, 5% CO2, and developed on the next day as described elsewhere (51). Wells containing PBMCs and medium with phytohemagglutinin or without any peptide were used as positive or negative controls, respectively, and run in triplicate on each plate. To calculate the number of specific T cells, the number of spots in the negative control wells was subtracted from the counted number of spots in each experimental well. Responses were considered positive if there were >50 spot-forming cells/106 PBMCs and at least three times the mean number of spot-forming cells in the experimental wells compared with the three control wells.
Sorting of tetramer+ HIV-1–specific CD8+ T cell populations.
Fresh or frozen PBMC samples were stained with PE-labeled MHC class I pentamers refolded with epitopic HIV-1 peptides (ProImmune) and fluorophore-labeled CD8+ antibodies, followed by decontamination with 1:100 dilution of fixation solution A (Caltag). Tetramer+ CD8+ cells were sorted on a FACS Aria cell sorter (BD Biosciences) at 70 pounds per square inch. The purity of sorted cell populations was consistently >98%.
TCR α and β chain sequencing.
mRNA was extracted from tetramer+ CD8+ T cells using the RNeasy mini kit (QIAGEN). Anchored RT-PCR was then performed using a modified version of the SMART (switching mechanism at 5′ end of RNA transcript) procedure and a TCR α or β chain constant region 3′ primer to obtain PCR products containing the Vα or β chain in addition to the CDR3 region, the Jα/β region, and the beginning of the Cα/β region. In brief, reverse transcription was performed at 42°C for 90 min with primers provided for the 5′-RACE reaction in a SMART-RACE PCR kit (BD Biosciences). First and second round PCR were then performed using a universal 5′-end primer (5′-CTAATACGACTCACTATAGGGC-3′) and nested gene-specific 3′-end primers annealing to the constant region of the TCR α or β chain (Cα outer, GTCCATAGACCTCATGTCTAGCACAG; Cα inner, ATACACATCAGAATCCTTACTTTG; Cβ outer, 5′-TGTGGCCAGGCACACCAGTGTGGCC-3′; Cβ inner, 5′-GGTGTGGGAGATC-TCTGCTTCTGA-3′). PCR reaction conditions were as follows: first run: 95°C for 30 s and 72°C for 2 min for 5 cycles; 95°C for 30 s, 70°C for 30 s, and 72°C for 2 min for 5 cycles; and 95°C for 30 s, 60°C for 30 s, and 72°C for 1 min for 25 cycles; second run: 95°C for 30 s, 60°C for 30 s, and 72°C for 1 min for 30 cycles. The PCR product was ligated into the TOPO TA cloning vector (Invitrogen) and used to transform Escherichia coli (Mach 1; Invitrogen). Colonies were selected, amplified by PCR with M13 primers, and sequenced by T7 or T3 primers on an ABI 3100 PRISM automated sequencer. Sequences were edited and aligned using Sequencher (Gene Codes Corp.) and Se-Al (University of Oxford, Oxford, UK) and compared with the human TCR genes database (http://imgt.cines.fr/textes/IMGTrepertoire/Proteins/). The TCR Vα/β chain classification system used is that of the international ImMunoGeneTics database (52).
Generation of CTL clones.
CTL clones were isolated by limiting dilution as described previously (53) using the CD3-specific mAb 12F6 as a stimulus for T cell proliferation. Developing clones were screened for HIV-1–specific CTL activity by a chromium-51 release assay against autologous B lymphoblastoid cell lines pulsed with the target peptide. HIV-1–specific clones were maintained by stimulation every 14–21 d with an anti-CD3 mAb and irradiated allogeneic PBMCs.
Sequencing of autologous virus.
Nested PCR for Gag on proviral DNA or plasma viral RNA was performed as described previously (14). PCR fragments were population sequenced to identify regions of sequence variation. All fragments were sequenced bidirectionally on an ABI 3100 PRISM automated sequencer (Applied Biosystems). If the height of the secondary peak at a given residue in the chromatogram was >25% of the dominant peak, a mixed base was considered present at that position. Sequencher (Gene Codes Corp.) and MacVector 4.1 (Oxford Molecular) were used to edit and align sequences.
Flow cytometry.
Fresh or frozen PBMCs were stained with pentamers (ProImmune) for 20 min at room temperature. After one wash with PBS, cells were incubated with PerCP-labeled CD14 antibodies or PE-Cy7–labeled HLA-DR antibodies, APC-labeled CD11c antibodies, and a cocktail of PerCP-labeled lineage antibodies (CD3, CD8, CD14, CD56, and CD19). For ILT4 surface assessments, cells were stained with an ILT4-specific antibody (clone 42D1). After 30 min of staining at room temperature, cells were resuspended in PBS containing 2% paraformaldehyde. Cells were acquired on a FACSCalibur (BD Biosciences) instrument. Compensation was performed with cells stained separately with individual antibodies used in the test samples. To block pentamer binding, PBMCs were incubated with an ILT4 antibody (clone 287219; R&D Systems). An array of alternative antibodies, including those blocking the interaction to KIR3DL1 (clone DX9; BioLegend) or ILT2 (clone HP-F1; provided by M. Lopez-Botet, Universitat Pompeu Fabra, Barcelona, Spain) were also tested for their ability to block pentamer binding to cells.
MDDC maturation assays.
MDDC culture was performed in RPMI 1640 medium supplemented with penicillin, streptomycin, l-glutamine, Hepes buffer, and either 5% pooled human serum (Sigma-Aldrich) or 1% heparinized normal human plasma. Freshly isolated PBMCs were washed several times in RPMI medium to remove platelets, plated into Corning six-well plates in 5% pooled human serum medium, and incubated for 60 min at 37°C to adhere monocytes. Adherent monocytes were propagated in the presence of 50 μg/ml GM-CSF (Amgen) for 5 d in 1% human plasma medium. In accordance with results reported by Ristich et al. (11), ILT4 expression was detectable on MDDCs generated in the presence of GM-CSF and in the absence of IL-4 (unpublished data). On day 5, immature myelomonocytic cells were harvested using Hanks-based Cell-disassociation buffer (Invitrogen), incubated with B2705/KK10 WT or B2705/KK10 L6M pentamers (ProImmune) for 4 h, and then matured using a previously described cocktail containing IL-1β, TNF-α, PGE-2, and IL-6 (54). Alternatively, CFSE-labeled peptide-pulsed (60 min at 37°C) matured MDDCs were added instead of pentamers. After 16 h, matured MDDCs were harvested, stained with antibodies against CD83, CD40, CD80, HLA-DR, as well as CD86, and processed to flow cytometric analysis. To calculate the relative pentamer-mediated inhibition in the up-regulation of a specific surface molecule during MDDC maturation, the difference between the mean fluorescence intensity of that molecule in the presence or absence of the pentamer was divided by the difference of the corresponding mean fluorescence intensity before and after maturation in the absence of pentamers.
Mixed lymphocyte reactions.
MDDCs generated as described above were matured for 16-20 h in the presence of HLA–B27 KK10 WT or L6M pentamers and then mixed with allogenic CFSE-labeled PBMCs. After 6 consecutive days of culture in R10 medium supplemented with 10% FCS, 10 mM Hepes buffer, 2 mM l-glutamine, and 50 U/ml penicillin-streptomycin, cells were stained with monoclonal CD4 and CD8 antibodies and processed to flow cytometric analysis using a FACSCalibur instrument.
ILT4 knockdown.
siRNA pools specific for ILT4 message (LILRB2 ON-TARGETplus SMARTpool; Dharmacon Technologies) or ILT2 message (LILRB1 ON-TARGETplus SMARTpool; Dharmacon) were used at concentrations of 1 nmol/million cells and 0.5 nmol/million cells. MDDCs were harvested on day 4 of culture and washed twice with Optimem (Invitrogen). 106 MDDCs were suspended in 300 ul Optimem in the presence of the indicated amount of siRNA and transferred to a 4-mm electroporation cuvette (Bio-Rad Laboratories). Cells were left on ice for 5 min and then electroporated (900 V, 0.75 msec square wave; Bio-Rad Genepulser Xcell) and transferred back to culture medium for another 16 h before analysis.
SPR experiments.
To characterize the binding affinity between HLA–B27 KK10 WT or L6M complexes and ILT4 in a cell-free system, SPR experiments were performed using a BIAcore 3000 SPR instrument (BIAcore) in 10 mM Hepes buffer containing 150 mM sodium chloride, 3.4 mM EDTA, and 0.005% (vol/vol) surfactant P-20 at 25°C. Recombinant HLA–B27 KK10 WT or L6M tetramers (obtained from the National Institutes of Health [NIH]/National Institute of Allergy and Infectious Diseases tetramer core facility) at a concentration of 2mg/ml in 10 mM sodium acetate, pH 4.6, were individually immobilized (600 resonance units) to a CM5 sensor chip (BIAcore) using standard amine coupling methods. Toxic shock syndrome toxin (TSST)-1 (55) in an equivalent surface density was used as the control surface, as there is no specific binding between ILT4 and TSST-1. Serial twofold dilutions (74 μg/ml [1 μM] to 4.6 μg/ml [62.5 nM]) of a recombinant protein dimer containing two extracellular domains of human ILT4 fused to the Fc part of human IgG (R&D Systems) were injected over the sensor chip. All of the binding experiments were performed at a flow rate of 25 μl/min, and pulses of 10 mM HCl were used to regenerate both surfaces between injections. After subtraction of background binding between ILT4 and TSST-1, the apparent binding affinity (K D) was determined by nonlinear regression analysis of SPR responses to the various concentrations of injected ILT4 using the BiaEvaluation 4.1 software program (BIAcore).
Proliferation assays.
PBMCs (106/ml) were stained with 0.25 μM CFSE and incubated for 6 d in the presence of the KK10 WT or L6M peptide (38). Afterward, cells were harvested, stained with PE-labeled B2705 KK10 or L6M pentamers and CD8 APC antibodies, and subjected to flow cytometric analysis using a FACSCalibur instrument.
Statistical analysis.
Data are presented as means and standard deviation or medians and range. Statistical analysis was based on Student's t tests. P < 0.05 was considered significant.
Online supplemental material.
Fig. S1 shows ILT4 surface expression intensity on CD14+ monocytes from individuals with acute (n = 8), primary (n = 7), chronic untreated, or HAART-treated (n = 5) HIV-1 infection. Fig. S2 shows proliferative activity of KK10 WT–specific CD8+ T cells during primary and KK10 L6M–specific CD8+ T cells during chronic HIV-1 infection in three HIV-1–infected patients each. Figs. S1 and S2 are available at http://www.jem.org/cgi/content/full/jem.20061865/DC1.
Supplemental Material
Acknowledgments
The authors would like to acknowledge Dr. Amy Stout and the NIH tetramer core facility for generously providing custom HLA class I tetramers.
This study was supported by the NIH (to X.G. Yu, D.G. Kavanagh, and B.D. Walker), the Claflin Distinguished Scholar Award (to X.G. Yu), the Doris Duke Charitable Foundation (to X.G. Yu), and the Howard Hughes Medical Institute (to B.D. Walker).
The authors have no conflicting financial interests.
Abbreviations used: ILT, Ig-like transcript; KIR, killer-Ig receptor; LIR, leukocyte Ig receptor; MDDC, monocyte-derived DC; siRNA, small interfering RNA; SPR, surface plasmon resonance.
M. Lichterfeld and D.G. Kavanagh contributed equally to this work.
References
- 1.Parham, P. 2005. MHC class I molecules and KIRs in human history, health and survival. Nat. Rev. Immunol. 5:201–214. [DOI] [PubMed] [Google Scholar]
- 2.Colonna, M., F. Navarro, T. Bellon, M. Llano, P. Garcia, J. Samaridis, L. Angman, M. Cella, and M. Lopez-Botet. 1997. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J. Exp. Med. 186:1809–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cosman, D., N. Fanger, L. Borges, M. Kubin, W. Chin, L. Peterson, and M.L. Hsu. 1997. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity. 7:273–282. [DOI] [PubMed] [Google Scholar]
- 4.Colonna, M., J. Samaridis, M. Cella, L. Angman, R.L. Allen, C.A. O'Callaghan, R. Dunbar, G.S. Ogg, V. Cerundolo, and A. Rolink. 1998. Human myelomonocytic cells express an inhibitory receptor for classical and nonclassical MHC class I molecules. J. Immunol. 160:3096–3100. [PubMed] [Google Scholar]
- 5.Allan, D.S., M. Colonna, L.L. Lanier, T.D. Churakova, J.S. Abrams, S.A. Ellis, A.J. McMichael, and V.M. Braud. 1999. Tetrameric complexes of human histocompatibility leukocyte antigen (HLA)-G bind to peripheral blood myelomonocytic cells. J. Exp. Med. 189:1149–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moretta, L., C. Bottino, G. Ferlazzo, D. Pende, G. Melioli, M.C. Mingari, and A. Moretta. 2003. Surface receptors and functional interactions of human natural killer cells: from bench to the clinic. Cell. Mol. Life Sci. 60:2139–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saverino, D., M. Fabbi, F. Ghiotto, A. Merlo, S. Bruno, D. Zarcone, C. Tenca, M. Tiso, G. Santoro, G. Anastasi, et al. 2000. The CD85/LIR-1/ILT2 inhibitory receptor is expressed by all human T lymphocytes and down-regulates their functions. J. Immunol. 165:3742–3755. [DOI] [PubMed] [Google Scholar]
- 8.Dietrich, J., M. Cella, and M. Colonna. 2001. Ig-like transcript 2 (ILT2)/leukocyte Ig-like receptor 1 (LIR1) inhibits TCR signaling and actin cytoskeleton reorganization. J. Immunol. 166:2514–2521. [DOI] [PubMed] [Google Scholar]
- 9.Ince, M.N., B. Harnisch, Z. Xu, S.K. Lee, C. Lange, L. Moretta, M. Lederman, and J. Lieberman. 2004. Increased expression of the natural killer cell inhibitory receptor CD85j/ILT2 on antigen-specific effector CD8 T cells and its impact on CD8 T-cell function. Immunology. 112:531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang, C.C., R. Ciubotariu, J.S. Manavalan, J. Yuan, A.I. Colovai, F. Piazza, S. Lederman, M. Colonna, R. Cortesini, R. Dalla-Favera, and N. Suciu-Foca. 2002. Tolerization of dendritic cells by T(S) cells: the crucial role of inhibitory receptors ILT3 and ILT4. Nat. Immunol. 3:237–243. [DOI] [PubMed] [Google Scholar]
- 11.Ristich, V., S. Liang, W. Zhang, J. Wu, and A. Horuzsko. 2005. Tolerization of dendritic cells by HLA-G. Eur. J. Immunol. 35:1133–1142. [DOI] [PubMed] [Google Scholar]
- 12.Friedrich, T.C., E.J. Dodds, L.J. Yant, L. Vojnov, R. Rudersdorf, C. Cullen, D.T. Evans, R.C. Desrosiers, B.R. Mothe, J. Sidney, et al. 2004. Reversion of CTL escape-variant immunodeficiency viruses in vivo. Nat. Med. 10:275–281. [DOI] [PubMed] [Google Scholar]
- 13.Goulder, P.J., and B.D. Walker. 1999. The great escape -AIDS viruses and immune control. Nat. Med. 5:1233–1235. [DOI] [PubMed] [Google Scholar]
- 14.Allen, T.M., M. Altfeld, X.G. Yu, K.M. O'Sullivan, M. Lichterfeld, S. Le Gall, M. John, B.R. Mothe, P.K. Lee, E.T. Kalife, et al. 2004. Selection, transmission, and reversion of an antigen-processing cytotoxic T-lymphocyte escape mutation in human immunodeficiency virus type 1 infection. J. Virol. 78:7069–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barouch, D.H., J. Kunstman, M.J. Kuroda, J.E. Schmitz, S. Santra, F.W. Peyerl, G.R. Krivulka, K. Beaudry, M.A. Lifton, D.A. Gorgone, et al. 2002. Eventual AIDS vaccine failure in a rhesus monkey by viral escape from cytotoxic T lymphocytes. Nature. 415:335–339. [DOI] [PubMed] [Google Scholar]
- 16.Jones, N.A., X. Wei, D.R. Flower, M. Wong, F. Michor, M.S. Saag, B.H. Hahn, M.A. Nowak, G.M. Shaw, and P. Borrow. 2004. Determinants of human immunodeficiency virus type 1 escape from the primary CD8+ cytotoxic T lymphocyte response. J. Exp. Med. 200:1243–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bradney, A.P., S. Scheer, J.M. Crawford, S.P. Buchbinder, and D.C. Montefiori. 1999. Neutralization escape in human immunodeficiency virus type 1-infected long-term nonprogressors. J. Infect. Dis. 179:1264–1267. [DOI] [PubMed] [Google Scholar]
- 18.Richman, D.D., T. Wrin, S.J. Little, and C.J. Petropoulos. 2003. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc. Natl. Acad. Sci. USA. 100:4144–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang, W., J.P. Bielawski, and Z. Yang. 2003. Widespread adaptive evolution in the human immunodeficiency virus type 1 genome. J. Mol. Evol. 57:212–221. [DOI] [PubMed] [Google Scholar]
- 20.Leslie, A.J., K.J. Pfafferott, P. Chetty, R. Draenert, M.M. Addo, M. Feeney, Y. Tang, E.C. Holmes, T. Allen, J.G. Prado, et al. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 10:282–289. [DOI] [PubMed] [Google Scholar]
- 21.Draenert, R., S. Le Gall, K.J. Pfafferott, A.J. Leslie, P. Chetty, C. Brander, E.C. Holmes, S.C. Chang, M.E. Feeney, M.M. Addo, et al. 2004. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J. Exp. Med. 199:905–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yokomaku, Y., H. Miura, H. Tomiyama, A. Kawana-Tachikawa, M. Takiguchi, A. Kojima, Y. Nagai, A. Iwamoto, Z. Matsuda, and K. Ariyoshi. 2004. Impaired processing and presentation of cytotoxic-T-lymphocyte (CTL) epitopes are major escape mechanisms from CTL immune pressure in human immunodeficiency virus type 1 infection. J. Virol. 78:1324–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peruzzi, M., N. Wagtmann, and E.O. Long. 1996. A p70 killer cell inhibitory receptor specific for several HLA-B allotypes discriminates among peptides bound to HLA-B*2705. J. Exp. Med. 184:1585–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peruzzi, M., K.C. Parker, E.O. Long, and M.S. Malnati. 1996. Peptide sequence requirements for the recognition of HLA-B*2705 by specific natural killer cells. J. Immunol. 157:3350–3356. [PubMed] [Google Scholar]
- 25.Rajagopalan, S., and E.O. Long. 1997. The direct binding of a p58 killer cell inhibitory receptor to human histocompatibility leukocyte antigen (HLA)-Cw4 exhibits peptide selectivity. J. Exp. Med. 185:1523–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zappacosta, F., F. Borrego, A.G. Brooks, K.C. Parker, and J.E. Coligan. 1997. Peptides isolated from HLA-Cw*0304 confer different degrees of protection from natural killer cell-mediated lysis. Proc. Natl. Acad. Sci. USA. 94:6313–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stewart-Jones, G.B., K. di Gleria, S. Kollnberger, A.J. McMichael, E.Y. Jones, and P. Bowness. 2005. Crystal structures and KIR3DL1 recognition of three immunodominant viral peptides complexed to HLA-B*2705. Eur. J. Immunol. 35:341–351. [DOI] [PubMed] [Google Scholar]
- 28.Kelleher, A.D., C. Long, E.C. Holmes, R.L. Allen, J. Wilson, C. Conlon, C. Workman, S. Shaunak, K. Olson, P. Goulder, et al. 2001. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J. Exp. Med. 193:375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goulder, P.J., C. Brander, Y. Tang, C. Tremblay, R.A. Colbert, M.M. Addo, E.S. Rosenberg, T. Nguyen, R. Allen, A. Trocha, et al. 2001. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature. 412:334–338. [DOI] [PubMed] [Google Scholar]
- 30.Goulder, P.J., R.E. Phillips, R.A. Colbert, S. McAdam, G. Ogg, M.A. Nowak, P. Giangrande, G. Luzzi, B. Morgan, A. Edwards, et al. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat. Med. 3:212–217. [DOI] [PubMed] [Google Scholar]
- 31.Feeney, M.E., Y. Tang, K.A. Roosevelt, A.J. Leslie, K. McIntosh, N. Karthas, B.D. Walker, and P.J. Goulder. 2004. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term-nonprogressing child. J. Virol. 78:8927–8930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneidewind, A., M.A. Brockman, R. Yang, R.I. Adam, B. Li, S. Le Gall, C.R. Rinaldo, S.L. Craggs, R.L. Allgaier, K.A. Power, et al. 2007. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J. Virol. 81:12382–12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raulet, D.H., R.E. Vance, and C.W. McMahon. 2001. Regulation of the natural killer cell receptor repertoire. Annu. Rev. Immunol. 19:291–330. [DOI] [PubMed] [Google Scholar]
- 34.Vlad, G., F. Piazza, A. Colovai, R. Cortesini, F. Della Pietra, N. Suciu-Foca, and J.S. Manavalan. 2003. Interleukin-10 induces the upregulation of the inhibitory receptor ILT4 in monocytes from HIV positive individuals. Hum. Immunol. 64:483–489. [DOI] [PubMed] [Google Scholar]
- 35.Willcox, B.E., L.M. Thomas, and P.J. Bjorkman. 2003. Crystal structure of HLA-A2 bound to LIR-1, a host and viral major histocompatibility complex receptor. Nat. Immunol. 4:913–919. [DOI] [PubMed] [Google Scholar]
- 36.Shiroishi, M., K. Kuroki, L. Rasubala, K. Tsumoto, I. Kumagai, E. Kurimoto, K. Kato, D. Kohda, and K. Maenaka. 2006. Structural basis for recognition of the nonclassical MHC molecule HLA-G by the leukocyte Ig-like receptor B2 (LILRB2/LIR2/ILT4/CD85d). Proc. Natl. Acad. Sci. USA. 103:16412–16417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cerboni, C., A. Achour, A. Warnmark, M. Mousavi-Jazi, T. Sandalova, M.L. Hsu, D. Cosman, K. Karre, and E. Carbone. 2006. Spontaneous mutations in the human CMV HLA class I homologue UL18 affect its binding to the inhibitory receptor LIR-1/ILT2/CD85j. Eur. J. Immunol. 36:732–741. [DOI] [PubMed] [Google Scholar]
- 38.Lichterfeld, M., D.E. Kaufmann, X.G. Yu, S.K. Mui, M.M. Addo, M.N. Johnston, D. Cohen, G.K. Robbins, E. Pae, G. Alter, et al. 2004. Loss of HIV-1–specific CD8+ T cell proliferation after acute HIV-1 infection and restoration by vaccine-induced HIV-1–specific CD4+ T cells. J. Exp. Med. 200:701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Migueles, S.A., A.C. Laborico, W.L. Shupert, M.S. Sabbaghian, R. Rabin, C.W. Hallahan, D. Van Baarle, S. Kostense, F. Miedema, M. McLaughlin, et al. 2002. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat. Immunol. 3:1061–1068. [DOI] [PubMed] [Google Scholar]
- 40.Appay, V., D.F. Nixon, S.M. Donahoe, G.M. Gillespie, T. Dong, A. King, G.S. Ogg, H.M. Spiegel, C. Conlon, C.A. Spina, et al. 2000. HIV-specific CD8+ T cells produce antiviral cytokines but are impaired in cytolytic function. J. Exp. Med. 192:63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Betts, M.R., M.C. Nason, S.M. West, S.C. De Rosa, S.A. Migueles, J. Abraham, M.M. Lederman, J.M. Benito, P.A. Goepfert, M. Connors, et al. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 107:4781–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muthumani, K., D.S. Hwang, A.Y. Choo, S. Mayilvahanan, N.S. Dayes, K.P. Thieu, and D.B. Weiner. 2005. HIV-1 Vpr inhibits the maturation and activation of macrophages and dendritic cells in vitro. Int. Immunol. 17:103–116. [DOI] [PubMed] [Google Scholar]
- 43.Majumder, B., M.L. Janket, E.A. Schafer, K. Schaubert, X.L. Huang, J. Kan-Mitchell, C.R. Rinaldo Jr., and V. Ayyavoo. 2005. Human immunodeficiency virus type 1 Vpr impairs dendritic cell maturation and T-cell activation: implications for viral immune escape. J. Virol. 79:7990–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steinman, R.M., A. Granelli-Piperno, M. Pope, C. Trumpfheller, R. Ignatius, G. Arrode, P. Racz, and K. Tenner-Racz. 2003. The interaction of immunodeficiency viruses with dendritic cells. Curr. Top. Microbiol. Immunol. 276:1–30. [DOI] [PubMed] [Google Scholar]
- 45.Fantuzzi, L., C. Purificato, K. Donato, F. Belardelli, and S. Gessani. 2004. Human immunodeficiency virus type 1 gp120 induces abnormal maturation and functional alterations of dendritic cells: a novel mechanism for AIDS pathogenesis. J. Virol. 78:9763–9772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vlad, G., R. Cortesini, and N. Suciu-Foca. 2005. License to heal: bidirectional interaction of antigen-specific regulatory T cells and tolerogenic APC. J. Immunol. 174:5907–5914. [DOI] [PubMed] [Google Scholar]
- 47.Chapman, T.L., A.P. Heikeman, and P.J. Bjorkman. 1999. The inhibitory receptor LIR-1 uses a common binding interaction to recognize class I MHC molecules and the viral homolog UL18. Immunity. 11:603–613. [DOI] [PubMed] [Google Scholar]
- 48.Allen, R.L., T. Raine, A. Haude, J. Trowsdale, and M.J. Wilson. 2001. Leukocyte receptor complex-encoded immunomodulatory receptors show differing specificity for alternative HLA-B27 structures. J. Immunol. 167:5543–5547. [DOI] [PubMed] [Google Scholar]
- 49.Belkin, D., M. Torkar, C. Chang, R. Barten, M. Tolaini, A. Haude, R. Allen, M.J. Wilson, D. Kioussis, and J. Trowsdale. 2003. Killer cell Ig-like receptor and leukocyte Ig-like receptor transgenic mice exhibit tissue- and cell-specific transgene expression. J. Immunol. 171:3056–3063. [DOI] [PubMed] [Google Scholar]
- 50.Papanikolaou, N.A., E.R. Vasilescu, and N. Suciu-Foca. 2004. Novel single nucleotide polymorphisms in the human immune inhibitory immunoglobulin-like T cell receptor type 4. Hum. Immunol. 65:700–705. [DOI] [PubMed] [Google Scholar]
- 51.Lichterfeld, M., X.G. Yu, D. Cohen, M.M. Addo, J. Malenfant, B. Perkins, E. Pae, M.N. Johnston, D. Strick, T.M. Allen, et al. 2004. HIV-1 Nef is preferentially recognized by CD8 T cells in primary HIV-1 infection despite a relatively high degree of genetic diversity. AIDS. 18:1383–1392. [DOI] [PubMed] [Google Scholar]
- 52.Lefranc, M.P., C. Pommie, M. Ruiz, V. Giudicelli, E. Foulquier, L. Truong, V. Thouvenin-Contet, and G. Lefranc. 2003. IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev. Comp. Immunol. 27:55–77. [DOI] [PubMed] [Google Scholar]
- 53.Yu, X.G., H. Shang, M.M. Addo, R.L. Eldridge, M.N. Phillips, M.E. Feeney, D. Strick, C. Brander, P.J. Goulder, E.S. Rosenberg, et al. 2002. Important contribution of p15 Gag-specific responses to the total Gag-specific CTL responses. AIDS. 16:321–328. [DOI] [PubMed] [Google Scholar]
- 54.Kavanagh, D.G., D.E. Kaufmann, S. Sunderji, N. Frahm, S. Le Gall, D. Boczkowski, E.S. Rosenberg, D.R. Stone, M.N. Johnston, B.S. Wagner, et al. 2006. Expansion of HIV-specific CD4+ and CD8+ T cells by dendritic cells transfected with mRNA encoding cytoplasm- or lysosome-targeted Nef. Blood. 107:1963–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moza, B., R.A. Buonpane, P. Zhu, C.A. Herfst, A.K. Rahman, J.K. McCormick, D.M. Kranz, and E.J. Sundberg. 2006. Long-range cooperative binding effects in a T cell receptor variable domain. Proc. Natl. Acad. Sci. USA. 103:9867–9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}