

Abstract
The B cell–specific enzyme activation-induced cytidine deaminase (AID) has been shown to be essential for isotype switching and affinity maturation of antibody genes during the immune response. Conversely, AID activity has also been linked to autoimmunity and tumorigenesis. Determining how AID expression is regulated in vivo is therefore central to understanding its role in health and disease. Here we use phylogenetic footprinting and high-resolution histone acetylation mapping to accurately demarcate AID gene regulatory boundaries. Based on this strategy, we identify a novel, positive regulatory element required for AID transcription. Furthermore, we generate two AID indicator mouse strains using bacterial artificial chromosomes that faithfully recapitulate endogenous AID expression. The first strain uses a green fluorescent protein reporter to identify B cells that actively express AID during the immune response. In the second strain, AID transcription affects the permanent expression of a yellow fluorescent protein reporter in post–germinal center and terminally differentiated lymphocytes. We demonstrate the usefulness of these novel strains by resolving recent contradictory observations on AID expression during B cell ontogeny.
The elimination of antigens by B lymphocytes is performed by antibody molecules that contain an N-terminal variable domain followed by a C-terminal constant domain. During the immune response, B lymphocytes express the activation-induced cytidine deaminase (AID) enzyme, which induces two major alterations in Ig gene loci to enhance antibody and B cell function. First, the process of somatic hypermutation introduces single point mutations at variable genes, which can increase antibody affinity for antigens (1). Second, the mechanism of class switch recombination replaces the Ig heavy chain constant region Cμ for Cγ, Cɛ, or Cα, thereby controlling the antibody effector function (2).
The importance of AID in the humoral immune response is highlighted in AID-deficient patients and animals, which are highly susceptible to infection and exhibit gut flora–dependent hyperplasia of intestinal villi (3, 4). Conversely, complex diseases such as autoimmunity have long been associated with AID-dependent hypermutation (5). Moreover, untimely, ectopic, or elevated AID expression results in translocations and malignant transformation of B cells (6, 7) and T cells (8). These considerations emphasize the need to understand the molecular pathways that regulate AID expression during B cell ontogeny.
Initially, AID expression was thought to be limited to follicular germinal center (GC) B cells (9). Conversely, recent evidence suggests that AID is also active outside of the GC microenvironment. In mice carrying a highly reduced V gene repertoire, hypermutation and AID transcripts were reported in a small subset of immature bone marrow B cells (10). In a lupus-prone mouse, hypermutation and affinity maturation occur at the T cell zone–red pulp border (11). Likewise, in rheumatoid arthritis patients, somatic hypermutation has been shown to occur in B cells residing in nonlymphoid synovial tissue (12). In normal mice, AID transcription and class switch recombination might occur in the gut lamina propria outside of Peyer's patches (PPs) and isolated lymphoid follicles (ILFs) (13), although others have recently contradicted these findings (14). Large extrafollicular proliferating cells have also been shown to express AID in human lymphoid tissues (15, 16). Finally, recent data indicate that AID is induced in pre–B cells in response to retroviral infection, perhaps as part of an innate defense mechanism (17). These studies therefore underscore the need for experimental model systems to monitor AID expression in the context of the entire organism.
The development of indicator mouse strains using bacterial artificial chromosomes (BACs) has greatly facilitated the study of gene expression and the unraveling of gene regulatory elements in the past decade (18, 19). The success of gene reporter expression systems is based on the premise that transgene constructs must carry sufficient regulatory information to recapitulate tissue-specific and copy-dependent regulation of genes of interest. However, the in vivo demarcation of gene regulatory boundaries typically relies on the production of large cohorts of transgenic animals (20, 21), making this process expensive and time-consuming. We here provide an alternative approach that combines comparative genomic analysis with high-resolution histone H3 acetylation mapping. This strategy allowed us to predefine the minimal DNA segment capable of recapitulating the temporal and spatial expression patterns of endogenous AID and led to the development of two complementary AID reporter lines that efficiently track AID-expressing cells as well as their progeny. Our studies define AID expression during T cell–dependent and –independent immune responses as well as upon retroviral infection of immature B cells.
RESULTS
Mapping AID gene regulatory boundaries
To identify the cis-regulatory elements governing AID transcription in vivo, we used phylogenetic footprinting to compare one megabase of human, dog, and mouse genomic DNA containing the AID (Aicda) gene. Using a combination of global and local alignment algorithms (http://family.caltech.edu/ and http://www-gsd.lbl.gov/vista/index.shtml), we generated alignment plots that revealed the conserved protein-encoding sequences (exons) of three syntenic genes within the one megabase of genomic DNA analyzed: Kiaa1238, Mfap5, and Aicda (Fig. 1, red peaks). Additionally, we found 11 highly conserved noncoding sequences (CNSs) that exhibited on average ≥75% identity in all three species (Fig. 1, black peaks).
Figure 1.

Phylogenetic analysis of Aicda. AID locus in human chromosome 12. Homology “peaks and valleys” graph showing percent conservation between human, dog, and mouse AID genomic loci. The nucleotide sequence is plotted on the horizontal axis. The vertical axis depicts the percent identity between the three species. Only conserved regions containing at least one peak with percent identity >75% within a shifting window of 100 bp in all three species are represented. Red peaks correspond to gene exons, whereas black peaks denote CNSs (I–XI). Genes and their orientation are represented by their RefSeq name and by arrows, respectively. As in the human, the mouse and dog Mfap5 and Kiaa1238 orthologs are located 5′ of Aicda and form a synteny group within the 1 MB interval analyzed. Conversely, the mouse and dog Clecsf9 and Clecsf8 genes are located beyond the 1 MB sequence analyzed here, and no homology is therefore assigned to these genes. Alignment was obtained using Family Relations (http://family.caltech.edu/) and VISTA software (http://www-gsd.lbl.gov/vista/index.shtml).
To examine potential contributions of CNSs to AID transcription, we explored their ability to attract histone acetyltransferase activity in B cells stimulated with LPS and IL-4 in vitro. Hyperacetylation of core histones in transcribed genes results in part from the interaction of transcription factors with regulatory elements, such as promoters and enhancers (22). To monitor changes in chromatin acetylation at high resolution, we assessed the percentage of acetylated histone H3 by chromatin immunoprecipitation and real-time PCR analysis (Fig. 2 A). In mature resting B cells, which do not express AID, we found H3 acetylation across Aicda at low levels (<10%; Fig. 2 B, blue circles). This acetylation was comparable to the calculated H3 acetylation in the mammalian genome (8%; see Materials and methods) as well as in neighboring genes such as Mfap5 (Fig. 2 B), which is not appreciably transcribed in B cells (not depicted).
Figure 2.

H3 acetylation status at the AID locus. (A) Percent homology identity between the dog, human, and mouse genomes as a function of the mouse AID locus in chromosome 6. Homology peaks and genes are depicted as in Fig. 1. Black dots represent the position of quantitative PCR primers used to assess H3 acetylation by CHIP. AID exons are marked with yellow boxes. E, EcoRV; S, SalI; X, XbaI. (B) Absolute percentage of histone H3 acetylation from resting (blue circles) or LPS-activated (red squares) B cells. (C) DNaseI hypersensitivity at CNS VII and X. Nuclei prepared from LPS-activated mouse B cells were untreated (lane 1) or treated with increased concentrations of DNaseI (lanes 2–6). DNA was extracted, digested, and analyzed by Southern blot hybridization using VII- or X-specific probes (PVII and PX, respectively). Horizontal arrows indicate DNaseI hypersensitive sites that correlate with CNS-X. (D) Detailed map of mouse CNS X. Elements A and B (red boxes) depict two highly conserved sequences between mouse and humans that correlate with the Southern blot bands obtained by DNaseI hypersensitivity. Putative transcription factor binding sites in elements A and B, predicted using VISTA software, are shown.
In contrast to resting lymphocytes, B cells activated with LPS and IL-4, which induces AID transcription, showed a dramatic increase in H3 acetylation at the Aicda locus. Nearly all nucleosomes of AID exon 1 and its vicinity (corresponding to CNS VIII) were acetylated (Fig. 2 B, red squares). This first peak of acetylation included an intronic regulatory element shown to bind E proteins during AID expression (23). Acetylation was decreased at exons 2–5 (50–70%), and a further decrease to basal acetylation levels was observed near Aicda's polyA signal sequences. These results are consistent with the notion that acetylation signals are propagated over the chromatin of coding regions through interactions of histone acetyltransferases and transcription complexes (24).
Unlike CNS I–VII and XI, which exhibited no change in H3 acetylation upon B cell activation (Fig. 2 B and not depicted), a second acetylation peak occurred at CNS X. This element, located ∼7 kb downstream of mouse Aicda (Fig. 2 A) or ∼24 kb 3′ of human Aicda (Fig. 1), is composed of two elements highly conserved in humans and mice (Fig. 2 D). To investigate whether CNS X functions as a bona fide protein-binding site upon AID transcription, we performed DNaseI hypersensitivity assays on nuclei obtained from resting and activated B lymphocytes. In contrast to CNS VII, which is not hyperacetylated during B cell activation (Fig. 2 C, left), we found clear evidence of protein binding at CNS X (Fig. 2 C, right), strongly suggesting that this element functions as a regulator of AID transcription.
Monitoring in vivo AID expression using AID-GFP reporter mice
The above studies predicted that AID proximal promoter elements and CNS X might be sufficient to direct physiological expression of an AID reporter gene. To evaluate this hypothesis, we generated transgenic mice with a 45-kb BAC carrying the mouse AID gene, including CNS X (Fig. 2 D and Fig. 3 A, top). By means of homologous recombination (25), we modified the BAC by fusing the GFP gene to AID exon 5. To define the role of CNS X in AID expression, we also created a second transgene construct in which CNS X was deleted by recombination (AID-GFP-ΔX; Fig. 3 A, bottom). Two AID-GFP and three AID-GFP-ΔX transgenic mouse strains were generated with two to five transgenic copies per line.
Figure 3.

AID expression in AID-GFP and AID-GFP-ΔX transgenic mice. (A) Molecular schemes of AID-GFP and AID-GFP-ΔX transgenes. The position of CNS X is indicated. (B) FACS analysis of AID expression in wild-type (left) and transgenic mice (middle and right) immunized with NP-CGG and CFA. Cells were stained with CD95-PE and gated on B220 (PercP-Cy5.5). Apoptotic cells were excluded by To-Pro-3 or DAPI staining. The GC nature of GFP+ cells was confirmed by GL7-APC staining (not depicted). (C) Endogenous AID (gray bars), AID-GFP (green bars), and AID-GFP-ΔX (red bars) transcripts were measured by quantitative PCR from resting, LPS, or LPS plus IL-4 stimulated B lymphocytes. The diagram on the left depicts primer positions.
Transgenic mice and wild-type littermates were immunized in the front footpads with the conjugated hapten nitrophenol (NP)-CGG. 5 d after immunization, we developmentally staged axillary lymph node B lymphocytes by flow cytometry using a panel of cell surface markers, including the B cell GC marker CD95. Unlike nontransgenic littermates (Fig. 3 B, left), nearly all CD95high GC B cells in AID-GFP mice exhibited green fluorescence (Fig. 3 B, middle). AID-GFP was absent in resting B220+ B cells (Fig. 3 B, middle, CD95low), bone marrow B cell populations (discussed below), and thymic or peripheral CD4+ or CD8+ T cells (not depicted). This GFP expression pattern matches the reported transcription profile of AID (9). The coordinate expression of AID-GFP and endogenous AID mRNA was validated by real-time PCR in resting and LPS plus IL-4–activated B lymphocytes (Fig. 3 C). In addition, RT-PCR analysis of seven mouse tissues revealed that AID expression is circumscribed to lymphoid organs both in wild-type and transgenic animals (Fig. S1). Analysis of the three transgenic AID-GFP-ΔX lines showed no green fluorescence in GC B cells in the absence of CNS X (Fig. 3 B). Furthermore, PCR analysis showed a five- to sevenfold decrease in AID-GFP transcripts in activated AID-GFP-ΔX lymphocytes compared with AID-GFP controls (Fig. 3 C). We conclude that our AID-GFP transgene recapitulates the spatiotemporal expression of endogenous AID and that CNS X is required for AID transcription during the immune response.
Activated B cells leaving the GC differentiate into plasma or memory B cells. To examine whether AID expression is maintained or down-regulated as B cells exit the GC microenvironment, we assessed green fluorescence in post-GC B cells during the NP response. We first analyzed pre-plasma memory B cells (NP+IgD−B220−CD138−) (26, 27) in AID-GFP mice immunized with NP-KLH. As described previously, these GC-derived B cells appear in the spleen 7–10 d after immunization, are somatically hypermutated, do not secrete antibody, and persist after primary challenge (27). We found that although NP+B220+CD95high GC cells express abundant AID-GFP (Fig. 4 A, right), pre-plasma memory lymphocytes lack AID-GFP expression (Fig. 4 A, middle) suggesting that AID is down-regulated as cells exit the GC microenvironment. To confirm this result, we assessed AID transcription in post-GC NP-binding cells recovered from the blood of nontransgenic animals. NP-specific IgG1+ B cells carrying somatic mutations appear in the blood as early as 7 d after immunization (28). To determine AID mRNA levels in this small compartment, we developed a single cell RT-PCR strategy (SC-RT-PCR; see Materials and methods). Consistent with previous observations (28), few NP-specific B cells were found in unimmunized mice (Fig. 4 B, day 0), whereas 7 d after NP-CGG immunization, B220+IgG1+NP+ B cells were clearly present in the blood (Fig. 4 B, day 7). We found that although nearly all (70 of 72) B220+IgG1+NP+CD95high lymph node GC cells were AID+ at day 7, AID transcripts were absent in B220+IgG1+NP+blood lymphocytes (Fig. 4 B). In agreement with these observations, no AID-GFP+ cells were observed in the blood of AID-GFP–immunized mice (not depicted).
Figure 4.

Dynamics of AID expression in T cell–dependent and –independent immune responses. (A) AID-GFP mice were immunized with NP-CGG, and CD4−CD8−IgD−PI (or DAPI)− lymphocytes recognizing NP were subdivided by B220 expression (left) and assessed for CD138 and AID-GFP expression 7–10 d after immunization (middle and right). Antibodies were IgD-biotin/Streptavidin-PE-Cy5, CD4-PE-Cy5, CD8-PE-Cy5, NP-APC, CD95-PE-Cy7, B220-APC-Cy7, and CD138-PE. (B) Left: NP staining of blood lymphocytes (IgM−IgD−Gr-1−F4/80−) recovered from unimmunized (day 0) and NP-CGG–immunized (day 7) wild-type mice. Staining was To-Pro-3, F4/80-Alexa 647, GR1-Alexa 647, IgM-APC, IgD-Alexa 647 (Dump channel) and NP-PE, IgG1-Biotin-Streptavidin-PercPCy5.5, and B220-FITC. Right: Analysis of AID transcription by SC-RT-PCR. NP+ cells were recovered by cell sorting from blood (72 cells analyzed) or lymph node GCs (72 cells) isolated from wild-type or AID−/− mice. (C) Left: AID-GFP animals carrying the pre-recombined B1-8hi heavy chain gene were immunized with NP-haptenated Ficoll and splenic B cells analyzed 5 d after immunization. AID-GFP expression in resting (B220+CD138−), T cell–independent GC (B220+CD138low), plasmablasts (B220+CD138high), and plasma cells (B220−CD138high) is indicated with histograms. Bottom histograms show CD95 expression in AID-GFP+ cells from population 3 or population 4. Right: Analysis of AID transcription by SC-RT-PCR in the indicated populations isolated from B1-8hi mice.
To determine AID expression in T cell–independent immune responses, we analyzed AID-GFP mice carrying the NP-specific heavy chain B1-8 (B1-8hi) (29). 5 d after NP-Ficoll intraperitoneal injection, B1-8hi mice develop T cell–independent GCs (B220+CD95highCD138low B cells) and large numbers of B220−CD138high plasma cells in the spleen (29). We found that GC cells express abundant AID-GFP after NP-Ficoll challenge (48% of B220+CD138low cells [Fig. 4 C], but nearly 100% of CD95high cells within that population [not depicted]). AID-GFP was, however, absent in terminally differentiated B220−CD138high plasma cells (Fig. 4 C, population 4), as determined both by flow cytometry and SC-RT-PCR analysis, a result consistent with the reported down-regulation of AID mRNA in plasma cells (30). Yet, a subset of B220+CD138high plasmablasts expressed AID mRNA, AID-GFP, and high levels of CD95 (Fig. 4 C, population 3). These lymphocytes might represent an intermediate stage between B220+CD138lowCD95high GC cells and fully differentiated B220+CD138high plasmacytes (31). Collectively, our data demonstrate that AID is expressed both in T cell–dependent and –independent GCs, but it is extinguished as B lymphocytes exit the GC microenvironment either as memory or plasma cells.
Genetic labeling of B lymphocytes after AID expression
As stated above, the AID-GFP transgene and endogenous AID are activated in GC cells undergoing switching and/or hypermutation, but turned off during plasma and memory B cell differentiation. To permanently tag the progeny of AID-expressing B cells with a fluorescent reporter protein, we introduced the bacterial cre recombinase gene in lieu of AID exon 1 (Fig. 5 A). AID-Cre transgenic mice were bred to the ROSA26-EYFP reporter strain, in which the yellow fluorescent protein (YFP) is expressed indelibly upon Cre-mediated deletion of a floxed neomycin gene (Fig. 5 A) (32). To evaluate the interplay of the two transgenes in vivo, we measured yellow fluorescence in different B cell subsets from AID-Cre-YFP double transgenic mice. Six different founder lines with approximately one to five transgene copies were analyzed. We found no YFP expression in immature bone marrow B cells (see below) or other hematopoietic lineage cells (not depicted). In addition, the spleen or axillary lymph nodes of nonimmunized mice, which have few GCs, showed a small number of lymphocytes expressing YFP (Fig. 5 B, left). In contrast, we found that PPs, which harbor constitutive GCs due to persistent B cell stimulation with gut antigens, had large numbers (∼20% of B220+) of CD95highYFP+ GC cells (Fig. 5 B, right), demonstrating that AID transcription promotes Cre expression in activated B cells. Consistent with this idea, virtually all lamina propria CD138+ plasma cells, which originate in PPs and possibly other sites of B cell activation, were YFP+ in AID-Cre-YFP mice (Fig. 5 C).
Figure 5.

Permanent labeling of B cells expressing AID. (A) Upper schematics: molecular maps of the AID-Cre (top) and Rosa-Neo-EYFP (bottom) transgenes (S, SalI; P, PacI; E, EcoRI). Lower Southern blot: three independent AID-Cre transgenic lines were generated, with copy number ranging between 1 to 5 copies. (B) FACS pseudocolor plots showing CD95-PE and YFP expression in B cells (gated on B220 (PercP-Cy5.5)) from AID-Cre-YFP compound mice. Left panel: axillary lymph node cells from nonimmunized mice; right panel: PPs. (C) Immunofluorescence analysis of gut lamina propria CD138+ plasma cells from AID-Cre-YFP mice. Scale bar = 20μm. (D) Mutation analysis of the JH4 intron from CD95lowYFP+, CD95highGFP+, CD95highGL7+ lymph node cell sorted from immunized AID-Cre-YFP, AID-GFP+, and AID−/− mice, respectively. Segments in pie charts are sized in proportion to the number of sequences carrying mutations as indicated. The number of sequences analyzed is given in the center of the pie charts. Mutation frequency and P values are indicated below. Statistical significance was assessed by a two-tailed t test assuming unequal variance and comparing values to mutation background determined in AID−/− sequences.
In both axillary lymph nodes and PPs, we consistently found resting B220+CD95low B cells expressing YFP (Fig. 5 B). These lymphocytes likely include post-GC memory and plasma cells that have undergone affinity maturation. To test this prediction, we evaluated the presence of somatic hypermutation in these lymphocytes by cloning and sequencing their JH4 intron. We found that B220+CD95lowYFP+ lymphocytes carry a similar amount of mutations to that measured in GC B cells from AID-GFP mice (Fig. 5 D). This result indicates that this population largely derives from activated follicular B cells. Collectively, our results demonstrate that the AID-Cre and ROSA-Neo-YFP transgenes efficiently tag the progeny of AID-expressing B cells.
AID in the GALT
IgA, together with innate mucosal immunity, provides the first line of defense against bacterial antigens in the GALT. AID expression and class switching to IgA have been proposed to take place either in situ in the gut lamina propria (13) or within GCs in PPs and ILFs of the small intestine (14). To ascertain the site of AID expression in mucosal surfaces, we determined green fluorescence in AID-GFP lymphocytes isolated from small intestine PPs and large intestine lamina propria. Under specific pathogen-free conditions, the large intestine (which normally lacks PPs) fails to develop ILF GCs (unpublished data). We found little AID-GFP expression in B220+IgM+ PP B cells (Fig. 6 A, middle). In contrast, around 65% of all B220+IgA+ PP cells expressed AID and high levels of CD95 (Fig. 6 A, right panel and top histogram, respectively), indicating that PP GCs are primarily populated by IgA+ lymphocytes. As in PPs, B220+IgM+ B cells of the large intestine lamina propria displayed little green fluorescence (Fig. 6 B, left). However, a population of B220+IgA+ B lymphocytes expressed AID-GFP and high levels of CD95 in the large intestine (Fig. 6 B, right, and Fig. 6 A, bottom histogram, respectively). IgA+CD95highAID-GFP+ lamina propria B cells may represent lymphocytes stimulated in situ perhaps by gut antigens as proposed previously (13). Alternatively, these lymphocytes could be recently emigrated PP or ILF B cells carrying residual AID-GFP protein. To evaluate these possibilities, we determined green fluorescence in the lamina propria of AID-GFP-OcaB−/− mice. In the absence of OcaB, the capacity of B lymphocytes to form pauciclonal aggregates and differentiate into plasmacytes in T cell–rich areas of lymphoid organs is intact, whereas B cell translocation to follicles and GC formation is impaired (33–35). In consequence, OcaB−/− mice lack both PPs, ILFs in the gut, and GCs in mesenteric lymph nodes (Fig. 6 C, micrographs, and not depicted) (33–35). We found that OcaB−/− AID-GFP mice, although unable to form GCs, displayed IgA+CD95highAID-GFP+ B cells in the large intestine lamina propria (Fig. 6 C, right), although at lower levels compared with wild-type animals (Fig. 6 B, right). To determine whether AID-GFP+ B cells showed ongoing AID transcription, we performed SC-RT-PCR. We found evidence of AID mRNA in nearly all CD95high lymphocytes analyzed, whereas AID was rarely found in CD95low cells (Fig. 6 C, bottom). Collectively, our results demonstrate that gut lamina propria B cells can express AID outside of PPs and ILF GCs.
Figure 6.

AID expression in the GALT. AID expression in IgM (APC) or IgA (biotin/Streptavidin PercP-Cy5.5) B lymphocytes isolated from PPs (A) and lamina propria (LP; B) from AID-GFP mice (B220 was APC-Cy7 conjugated). Histograms show CD95 (PE) expression in the indicated populations. (C) Left micrographs: PNA staining of mesenteric lymph nodes (MLNs) isolated from OcaB+/+ and OcaB−/− mice (bar, 150 μm). Right density plot shows AID-GFP expression of IgA+ cells isolated from the lamina propria of AID-GFP-OcaB−/− mice. Bottom gel shows analysis of AID expression by SC-RT-PCR in the indicated B cell populations recovered from OcaB−/− mice as indicated.
AID expression in immature B lymphocytes
Hypermutation in “GALT species,” including rabbit, sheep, and cattle, diversifies the preimmune repertoire before antigen B cell stimulation (36). In contrast, V(D)J hypermutation in normal mice and humans is widely believed to be restricted to the GC microenvironment. This view has been recently challenged by findings showing AID expression and hypermutation in immature bone marrow B cells in mice expressing a highly restricted Ig repertoire (quasimonoclonal [QM] mice) (10). To determine whether AID is present in developing lymphocytes from wild-type mice, we characterized bone marrow B cells from AID-GFP and AID-Cre-YFP mice. We found that AID-GFP was absent from pro–/pre–B (B220lowIgM−), transitional (B220lowIgM+), and mature recirculating bone marrow B cells (B220highIgM+) recovered from AID-GFP transgenics (Fig. 7 A, left, pseudo-color plot, and middle). Likewise, YFP was undetectable in immature and transitional B cells (Fig. 7 A, right, 1 and 2). However, in all mice analyzed, we found a subset of mature recirculating B cells expressing YFP, which might represent post-GC lymphocytes (Fig. 7 A, right, 3). To investigate whether the apparent absence of AID expression in immature B cells was due to low expression of fluorescent proteins in our reporter animals, we quantified AID transcription in sorted bone marrow B cell populations using real-time PCR. Background levels of AID mRNA in pro–/pre–B cells corresponded to a 10−3 dilution of PCR template prepared from B220+CD95high GC B lymphocytes isolated by cell sorting from the lymph nodes of immunized mice (Fig. 7 B). In agreement with published semiquantitative PCR analysis (10), AID transcripts were consistently detected in CD93+IgM+ bone marrow lymphocytes and splenic mature B cells, but AID expression levels in these cells were 100–500-fold lower than those measured in GC lymphocytes (Fig. 7 B), and this transcription was not sufficient to promote efficient Cre-mediated recombination or GFP expression in our AID-Cre-YFP or AID-GFP indicator mice, respectively. Thus, unmanipulated mice show little AID expression in immature B cells under normal conditions.
Figure 7.

AID expression in immature, bone marrow B lymphocytes. (A) AID expression determined by flow cytometry from bone marrow B220lowIgM− pro–/pre–B (population 1), transitional B220lowIgM+ (population 2), and recirculating B220highIgM+ (population 3) B lymphocytes obtained from AID-GFP (left histograms) or AID-Cre-YFP (right histograms) mice. All immature B cells were also CD93+ (not depicted). Antibodies were IgM-APC, B220-PercP-Cy5.5, CD93-biotin/Streptavidin-PE, and To-pro-3. (B) Real-time PCR analysis of AID transcripts in pro–, pre–, transitional, mature, and PP B lymphocytes cell sorted from wild-type mice. For comparative purposes, cDNA synthesized from GL7+CD95high GC B cells was serially diluted, as indicated below each column. cDNA was amplified using PCR primers annealing to AID exons 2 and 3. All B cell populations were sorted to >95% purity before RNA extraction and cDNA synthesis. Vertical bars represent the standard deviation of three independent experiments. (C) Bone marrow cells from AID-Cre-YFP mice were infected with Ab-MLV in vitro. The appearance of YFP+ cells was monitored in the infected cultures as a function of time (panels 1–4; only days 1 and 7 after infection are shown). The images are representative of three independent experiments. Panel 5 represents bone marrow B220+ cells isolated from AID-Cre-YFP mice and cultured for 7 d with IL-7 and in the presence of S17 cells. The following antibodies were used: CD43-PE, IgM-APC, B220-PercP-Cy5.5, and DAPI. Bar graph depicts the relative AID expression assessed by real-time PCR in Ab-MLV cultures.
Recent data indicate that AID transcription can be induced in pro– and pre–B cells upon infection with Abelson murine leukemia virus (Ab-MLV) (17), perhaps as part of an innate defense mechanism against viruses, as has been shown for other APOBEC protein family members (37, 38). To investigate whether retroviral infection leads to AID expression in immature B cells, we monitored YFP fluorescence in AID-Cre-YFP transgenic bone marrow cells infected with Ab-MLV in vitro. In these cultures, as in vivo, Ab-MLV infects with higher tropism a subset of developing B220lowCD43+IgM− B lymphocytes, leading to preferential cell proliferation and transformation (Fig. 7 C, 1 and 2) (39). We found little or no YFP expression in day 1–infected CD43+ bone marrow B cells (Fig. 7 C, 3). However, between 5 and 10% of the cultured cells expressed YFP 7 d after infection (Fig. 7 C, 4; three independent experiments). The presence of YFP+ lymphocytes is unlikely due to preferential expansion of rare YFP+ clones, as few immature B cells expressed YFP after IL-7-S17 stromal cell culture (Fig. 7 C, 5). To confirm induction of AID expression upon infection, we measured endogenous AID mRNA levels in Ab-MLV/AID-Cre-YFP cultures by real-time PCR. We found that compared with uninfected controls, AID transcripts were present in B220+CD43+YFP+ and B220+CD43+YFP− B cells at levels comparable to those found in GC lymphocytes (Fig. 7 C, bar graph). We conclude that in wild-type bone marrow B cells, AID is not appreciably transcribed, but AID expression can be induced upon retroviral infection.
DISCUSSION
Here we have described a rapid method to generate indicator transgenic mice that efficiently recapitulate endogenous expression of a gene of interest. This strategy, which relies on phylogenetic footprinting and high-resolution histone acetylation mapping, delineates gene regulatory boundaries with great accuracy. In general, using chromatin acetylation would seem an obvious approach when designing reporter transgenic constructs, although it is rarely used to its full potential. One possible reason for its limited use might be the need to scan large segments of genomic DNA by chromatin immunoprecipitation and PCR, as cis-regulatory elements can be far removed from the genes they control (40). Such limitations could be easily overcome by high-throughput methods based on genomic DNA microarrays or similar ChiP-on-Chip technologies recently described (41, 42).
In this study, histone H3 acetylation mapping uncovered two elements at the mouse AID locus that undergo nearly complete acetylation upon B cell activation. The first element corresponds to Aicda proximal promoter sequences, including transcription-enhancing E2A binding sites (23). High levels of acetylation at these sites correlate with the reported ability of E-box proteins to recruit p300 acetyltransferase activity to chromatin (43). Because evidence indicates that p300 and E2A collaborate in B cell ontogeny (44), it will be important to determine whether the p300–E2A complex regulates AID gene transcription in activated B lymphocytes. The second hyperacetylation element, termed CNS-X, is a putative transcriptional enhancer, and its deletion nearly entirely abolishes AID expression both in GC as well as in in vitro–activated B cells. DNaseI hypersensitivity assays demonstrate protein binding activity at CNS-X in activated B cells. Additional studies will be required to determine the nature of these factors and the mechanism by which they regulate AID gene transcription upon B cell activation.
Our reporter strains show that AID expression is induced in GC B cells both during T cell–dependent and –independent immune responses, and that memory and/or plasma cell differentiation leads to AID transcriptional down-regulation. These results are consistent with previous studies showing that the transition of GC cells to the terminally differentiated plasma cell stage is regulated by mutually exclusive transcription programs (30). These gene regulatory networks, which depend on the activity of Pax5, Bcl6, IRF-4, and Blimp1, directly control AID gene expression as B cells differentiate out of the GC microenvironment (45). However, as shown in mice that lack or have poorly developed PPs and ILFs, GCs are clearly not the only location for terminal B cell differentiation (46, 47). Molecular characterization of lamina propria B cells indicates active, ongoing class switch recombination outside of GCs (13). Our results using wild-type and OcaB−/−AID-GFP mice support the idea that AID expression is induced in situ in the GALT.
We find that under physiological conditions, bone marrow–developing B cells do not significantly express AID. Previous experiments with animals expressing a limited repertoire (QM mice) suggested that AID may function in immature B cells (10). Similar conclusions were reached studying mMT−/− mice (48), where Igμ−δ− bone marrow B cells are arrested at the pro–B cell stage (49). In these animals, a small fraction of lymphocytes bypass the developmental block via class switch recombination, implying that AID is active at early stages of B cell development. However, both QM and mMT−/− differ from wild-type mice in that B lymphopoiesis occurs under strong selective pressure. Consequently, the physiological significance of bone marrow AID activity under wild-type conditions was unclear. Our reporter strains show no evidence of AID expression in the bone marrow of animals with a normal antibody repertoire, and sensitive real-time PCR analysis only detected AID transcripts in transitional CD93+IgM+ bone marrow B cells at levels 100–500-fold lower than those found in GC lymphocytes. Although we cannot rule out that under unique circumstances AID may play a role in early B cell lymphopoiesis, low levels of AID mRNA in the bone marrow of wild-type mice may be simply reminiscent of a common evolutionary past between mice and “GALT species” (36).
Although unperturbed bone marrow B cells show little AID expression, infection with a transforming retrovirus readily activates AID transcription in developing lymphocytes (17 and this study). Likewise, B cell infection with hepatitis C (50), Epstein-Barr virus (51, 52), or Moloney murine leukemia virus (53) leads to AID up-regulation, suggesting that AID induction is part of a general innate antiviral host response. It will be important to determine whether non–B cells upon viral infection can also trigger AID expression.
In conclusion, our AID indicator strains efficiently track B cells expressing AID and their progeny in the context of the humoral immune response. These animals along with similar B cell activation reporter mice, such as Blimp-EGFP (54) and Cγ1-Cre (55) transgenics, will further our knowledge of the late stages of B cell differentiation both in the normal immune response as well as during autoimmunity and B cell tumorigenesis.
MATERIALS AND METHODS
Phylogenetic footprinting and histone H3 acetylation.
Resting splenic B lymphocytes were isolated by negative selection using CD43 MACS beads and processed directly for chromatin immunoprecipitation or cultured at 7 × 105 cells ml−1 in the presence of 25 μg ml−1 LPS (Escherichia coli 0111:B4; Sigma-Aldrich) for 3 d.
To immunoprecipitate acetylated histone H3–DNA complexes from resting and activated B cells, we used anti–acetyl-histone H3 antibodies (06-599; Upstate Biotechnology) according to the manufacturer's instructions. On average, 85 ng of acetylated DNA was recovered per 106 cells. To calculate the absolute percentage of acetylated histone H3 for a given genomic target, we amplified 4 ng anti–acetyl-histone H3 immunoprecipitated (IM) and input (IN) DNA (normalized using PicoGreen, P7589; Invitrogen) in the presence of SYBR Green (4309155; Applied Biosystems) using an iCycler (Bio-Rad Laboratories). Primers were designed to scan the mouse AID locus at ∼500-bp intervals. PCR products were between 100 and 150 bp in length. The fold difference between the two sets of samples for any given target sequence was calculated using the following PCR equation:
 |
(1) |
where X0 and X represent the DNA concentration before and after PCR amplification, respectively, E denotes the PCR efficiency and n represents the number of cycles necessary to achieve a specified threshold, which we set to cross a point at which amplification was linear (110 RFU using software from Bio-Rad Laboratories). Under these conditions, X becomes a constant and the ratio of the initial DNA abundance between the IM and the IN fractions can be calculated as follows:
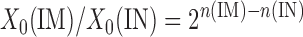 |
(2) |
Thus, the fold difference between the immunoprecipitated and input samples is determined as the difference in number of cycles each fraction takes to reach the specified threshold and taking the resulting power of 2. The fold difference X0(IM)/X0(IN) is also proportional to the percentage of acetylated histone H3 in the immunoprecipitated sample (%AcIM) over that of the input (%AcIN):
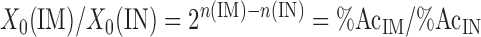 |
(3) |
Thus, the absolute percentage of histone H3 at any given genomic site can be assessed if %AcIN is known. To empirically determined %AcIN, we measured the total fraction (f) of the genome that can be pulled down by immunoprecipitation in the presence of an excess of anti-acetylated H3 antibody as follows:
 |
(4) |
where DNAIN and DNAIM are the amount of DNA measured before and after immunoprecipitation, respectively, and B represents nonspecific DNA pulled down by the antibody, which is constant when the immunoprecipitating antibody is in excess. To determine f, we performed several dilutions of cross-linked DNA obtained from B lymphocytes and measured, using PicoGreen, the absolute concentration of DNA before and after immunoprecipitation (performed in the presence of 100 μl anti-acetylated H3 antibody). By plotting DNAIM versus DNAIN and using linear regression analysis, we determined the slope f (0.08, Fig. 2 B) and concluded that 8% of all histone H3 is acetylated at any one point in time in the mammalian genome. This measurement is comparable to what has been previously determined for chicken cells (7%) (22). Therefore, by using the fold difference calculated by real-time PCR analysis, we can now assess the percentage of histone H3 acetylated at any given target in mammalian cells:
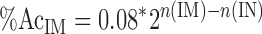 |
(5) |
DNaseI hypersensitivity.
B lymphocytes were isolated using CD43 MACS beads and cultured in the presence of 25 μg ml−1 LPS or 1 μg/ml anti-CD40 (HM40-3; BD Biosciences) and 25 ng ml−1 IL-4 (404-ML; R&D Systems) for 36 h. Resting and activated B cells were then washed, and the nuclei were isolated followed by exposure for 20 min at 37°C to increasing amounts of 10 U μl−1 DnaseI (diluted 1/160, 1/320, 1/640, 1/1280, 1/2560, 1/5120 and 1/10240, respectively; 92877720; Roche). The reaction was terminated with EDTA. Next, the nuclei were treated for 30 min at 37°C with RNase A (0.5 mg ml−1 DNase-free RNase A in TE) followed by incubation overnight with proteinase K at 55°C. The DNA was precipitated, resuspended in 20 μl TE, digested overnight with EcoRV or XbaI, and fractionated on Southern blots probed with 32P-labeled DNA fragments designated PI and PII. These probes were generated by PCR amplification using primer pairs 2B-5′ (TGTGTTGAGGTTTCATATCGCC) and 2B-3′ (CCAGCACTTGGGAGGCAAAG), and 7B-5′ (TCTCCAGCCCTGGAAACTTATG) and 7B-3′ (CTTGGTCACCTACAAGGAACTC), respectively.
Generation of AID-GFP, AID-GFPΔX, and AID-Cre transgenic mice.
BAC transgenes were constructed by modification of a 200-kb mouse BAC containing mouse Aicda (RPCI24-68I7). Deletions and insertions were generated using homologous recombination in bacteria as described previously (25). In brief, 1–2-kb constructs carrying two arms of homology (∼500 each) were ligated into the PsVI shuttle vector that was transformed into DH10B-competent cells expressing the RPCI24-68I7 BAC. The proper insertion of the respective modifications and final bacterial selection in chloramphenicol/fusaric acid plates were monitored by PCR using primers internal and external to the homologous constructs. By this method, two deletions were performed flanking the Aicda gene within the RPCI24-68I7 BAC. The 3′ deletion consisted of a 110.7-kb DNA fragment starting 13,717 bp 3′ of AID exon 5 and ending at the T7 promoter of the pTARBAC backbone vector of RPCI24-68I7. The 5′ deletion consisted of ∼37 kb DNA starting 25,202 bp 5′ of AID exon 1 and ending at the SP6 promoter of pTARBAC. Finally, the GFP gene was introduced in such a way as to create a fusion to AID exon 5. To generate the AID-GFPΔX transgenic construct, a 6-kb XbaI fragment containing CNS-X (3,905–9,917 bp downstream of mouse AID exon 5) was deleted from the trimmed BAC. Alternatively, the bacterial Cre recombinase gene was inserted in lieu of AID exon 1 so that only the recombinase (and not AID) would be expressed from the transgene. As an example of the strategy used to clone and select the aforementioned constructs, primers used to amplify the 5′ arm of AID-Cre were: 5′(1) aagtcgacCCAATGTGAGAAGTGTCCAGTG and 5′(2) GTGTACGGTCAGTAAATTGGACATatcggtctccagcgtgactttc; to amplify the 3′ arm: 3′(1) CTGCTGGAAGATGGCGATTAGGGACAGgtaacaagacagtc and 3′(2) gtcgacagtaAGGAGTTGCTACGACCTC; and the Cre gene was amplified with Cre1 gaaagtcacgctggagaccgatATGTCCAATTTACTGACCGTACAC and Cre2 gactgtcttgttacCTGTCCCTAATCGCCATCTTCCAGCAG. To create the final homologous recombination construct, 10 ng of each of the DNA fragments was fused in a PCR reaction using primers 5′(1) and 3′(2). The DNA piece was then extracted, sequenced, digested with SalI, and ligated to PsVI. To assess recombination of the construct's right arm we used primers Cre5′(1) CACACTCACAGAGCTCATTATCATG and Cre5′(2) GTTGCATCGACCGGTAATGCAG, whereas left arm recombinations were determined with Cre3′(1) ATCCGTAACCTGGATAGTGAAACAG and Cre3′(2) CCATGCGAGTCTTAAGATGTTGG.
All animal experiments were performed according to the NIH guidelines for laboratory animals and were approved by the Scientific Committee of the NIAMS Animal Facility.
RT-PCR and SC-RT-PCR.
To discriminate and compare endogenous AID and transgenic AID-GFP transcripts, splenic B cells from AID-GFP (and AID-GFP-ΔX) mice were isolated with CD43 microbeads and cultured in the presence of LPS or LPS plus IL-4 for 48 h. RNA was extracted with RNAqueous-Micro (Ambion) and treated twice with DNaseI. cDNA was generated, and the level of endogenous AID transcripts were monitored by real-time PCR with primers: AIDWT-RT5′: CCCTTGTACGAAGTCGATGAC, AIDWT-RT3′: ATCACGTGTGACATTCCAGGAG. AID-GFP transcripts were amplified with: AID-GFP-RT5′: GACTTGCGAGATGCATTTCGTATG and AID-GFP-RT3′: GCTGAACTTGTGGCCGTTTAC. To determine the fold difference between different samples, cDNA obtained from AID-GFP–activated B cells was diluted twofold, fivefold, 10-fold, 50-fold, and 100-fold, and both endogenous AID and AID-GFP were amplified by real-time PCR. We normalized cDNA between samples by amplifying from each cDNA reaction the DNA-PK subunit Ku70 mRNA with the following primers: Ku70-5′: TGCCCTTTACTGAGAAGGTGAC and Ku70-3′: TGCTGCAGGACTGGATTCTC.
To quantify AID transcription in different B cell compartments (Fig. 4), mouse AID primer sequences were obtained from PrimerBank (http://pga.mgh.harvard.edu/primerbank/index.html): PrimerBank-mAID5′: GCCACCTTCGCAACAAGTCT and PrimerBank-mAID3′: CCGGGCACAGTCATAGCAC.
To analyze AID expression at the single cell level, B lymphocytes were stained as indicated and individual cells were sorted using a MoFlo (DakoCytomation) instrument into iCycler 96-well plates (Bio-Rad Laboratories) containing 10 μl of 10 mM RNAase-Free Tris buffer and 1 U/μl of RNase inhibitor (Promega). RT-PCR was set up by adding to each well 15 μl of QIAGEN's OneStep RT-PCR master mix plus 0.6 μM of PrimerBank primers (described above). The plates were incubated (iCycler; Bio-Rad Laboratories) at 50°C for 30 min, followed by a 15-min cycle at 95°C and 40 cycles of 10 s at 95°C and 30 s at 55°C. Samples were run in a 2% agarose gel.
Hypermutation analysis.
GC or activated B cells were sorted using a MoFlo high-speed cell sorter (DakoCytomation). Genomic DNA was isolated, and the JH4 intron was amplified by nested PCR using KOD DNA polymerase (Novagen) and primers V1: AGCCTGACATCTGAGGAC and V2: TAGTGTGGAACATTCCTCAC for 30 cycles at 55°C. The second amplification step was performed with primers V3: CTGACATCTGAGGACTCTGC and V4: GCTGTCACAGAGGTGGTCCTG for 30 cycles at 60°C. PCR products were cloned using ZERO-blunt PCR cloning kit (Invitrogen) and sequenced using M13 universal primers. Sequence analysis was performed using SeqMan software (DNASTAR).
FACS analysis.
Unless otherwise stated, all other antibodies were from BD Biosciences: B220-PercP-Cy5.5, B220-APC-Cy7, B220-FITC, IgM-APC (Jackson ImmunoResearch Laboratories), CD95-PE, CD95-PE-Cy7, GL7-FITC, GL7-APC (APC conjugation was performed using Phycolink from Prozyme), CD43-PE, CD93-biotin, CD138-PE, CD4-PE-Cy5, CD8-PE-Cy5, IgA-Biotin, IgG1-biotin, GR1-Alexa 647 and F4/80-Alexa 647 (eBiosciences), IgD-Biotin, IgD-Alexa 647, Streptavidin-APC, Streptavidin-PE-Cy5, Streptavidin-PercP-Cy5.5, Streptavidin-PE, NP-APC (provided by McHeyzer-Williams), and NP-PE (Biosearch Technologies). Apoptotic cells were gated out using To-Pro-3 (Invitrogen) or DAPI (Sigma-Aldrich). Flow cytometers used were FACSCalibur (Becton Dickinson) and Cyan (DakoCytomation) for acquisition and MoFlo (DakoCytomation) for sorting. FACS data was analyzed with FlowJo software.
Online supplemental material.
Fig. S1 shows expression of AID mRNA by RT-PCR analysis in different tissues of wild-type, AID-GFP, and AID- Cre-YFP transgenic mice. It is available at http://www.jem.org/cgi/content/full/jem.20061952/DC1.
Supplemental Material
Acknowledgments
This study was initiated at the California Institute of Technology in the laboratory of David Baltimore, whom we thank for insight and support. We are also grateful to Michel Nussenzweig for providing the B1-8hi and OcaB−/− mice; Elisabeth Mushinsky for help with immunohistochemistry; Juan Silva and Shirley Pease for transgenic work; Amy Johnson, Marika Orlov, and Sharon Twitty for helpful comments and technical assistance; Michael Litt for assistance with CHIP quantitation; and Art Shaffer and Titus Brown for help with comparative genomic analysis. We thank Dr. McHeyzer-Williams for reagents.
This research was supported in part by the Intramural Research Program of the NIH, NIAMS.
The authors have no conflicting financial interests.
Abbreviations used: Ab-MLV, Abelson murine leukemia virus; AID, activation-induced cytidine deaminase; BAC, bacterial artificial chromosome; CNS, conserved noncoding sequence; GC, germinal center; ILF, isolated lymphoid follicle; NP, nitrophenol; PP, Peyer's patch; QM, quasimonoclonal; SC-RT-PCR, single cell RT-PCR strategy; YFP, yellow fluorescent protein.
E.E. Crouch and Z. Li contributed equally to this work.
References
- 1.Longerich, S., U. Basu, F. Alt, and U. Storb. 2006. AID in somatic hypermutation and class switch recombination. Curr. Opin. Immunol. 18:164–174. [DOI] [PubMed] [Google Scholar]
- 2.Honjo, T., K. Kinoshita, and M. Muramatsu. 2002. Molecular mechanism of class switch recombination: linkage with somatic hypermutation. Annu. Rev. Immunol. 20:165–196. [DOI] [PubMed] [Google Scholar]
- 3.Revy, P., T. Muto, Y. Levy, F. Geissmann, A. Plebani, O. Sanal, N. Catalan, M. Forveille, R. Dufourcq-Labelouse, A. Gennery, et al. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 102:565–575. [DOI] [PubMed] [Google Scholar]
- 4.Fagarasan, S., M. Muramatsu, K. Suzuki, H. Nagaoka, H. Hiai, and T. Honjo. 2002. Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science. 298:1424–1427. [DOI] [PubMed] [Google Scholar]
- 5.McIntosh, R., P. Watson, and A. Weetman. 1998. Somatic hypermutation in autoimmune thyroid disease. Immunol. Rev. 162:219–231. [DOI] [PubMed] [Google Scholar]
- 6.Ramiro, A.R., M. Jankovic, E. Callen, S. Difilippantonio, H.T. Chen, K.M. McBride, T.R. Eisenreich, J. Chen, R.A. Dickins, S.W. Lowe, et al. 2006. Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature. 440:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuppers, R., and R. Dalla-Favera. 2001. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene. 20:5580–5594. [DOI] [PubMed] [Google Scholar]
- 8.Okazaki, I.M., H. Hiai, N. Kakazu, S. Yamada, M. Muramatsu, K. Kinoshita, and T. Honjo. 2003. Constitutive expression of AID leads to tumorigenesis. J. Exp. Med. 197:1173–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muramatsu, M., V.S. Sankaranand, S. Anant, M. Sugai, K. Kinoshita, N.O. Davidson, and T. Honjo. 1999. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA- editing deaminase family in germinal center B cells. J. Biol. Chem. 274:18470–18476. [DOI] [PubMed] [Google Scholar]
- 10.Mao, C., L. Jiang, M. Melo-Jorge, M. Puthenveetil, X. Zhang, M.C. Carroll, and T. Imanishi-Kari. 2004. T cell-independent somatic hypermutation in murine B cells with an immature phenotype. Immunity. 20:133–144. [DOI] [PubMed] [Google Scholar]
- 11.William, J., C. Euler, S. Christensen, and M.J. Shlomchik. 2002. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science. 297:2066–2070. [DOI] [PubMed] [Google Scholar]
- 12.Schroder, A.E., A. Greiner, C. Seyfert, and C. Berek. 1996. Differentiation of B cells in the nonlymphoid tissue of the synovial membrane of patients with rheumatoid arthritis. Proc. Natl. Acad. Sci. USA. 93:221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fagarasan, S., K. Kinoshita, M. Muramatsu, K. Ikuta, and T. Honjo. 2001. In situ class switching and differentiation to IgA-producing cells in the gut lamina propria. Nature. 413:639–643. [DOI] [PubMed] [Google Scholar]
- 14.Shikina, T., T. Hiroi, K. Iwatani, M.H. Jang, S. Fukuyama, M. Tamura, T. Kubo, H. Ishikawa, and H. Kiyono. 2004. IgA class switch occurs in the organized nasopharynx- and gut-associated lymphoid tissue, but not in the diffuse lamina propria of airways and gut. J. Immunol. 172:6259–6264. [DOI] [PubMed] [Google Scholar]
- 15.Cattoretti, G., M. Buettner, R. Shaknovich, E. Kremmer, B. Alobeid, and G. Niedobitek. 2006. Nuclear and cytoplasmic AID in extrafollicular and germinal center B cells. Blood. 107:3967–3975. [DOI] [PubMed] [Google Scholar]
- 16.Moldenhauer, G., S.W. Popov, B. Wotschke, S. Bruderlein, P. Riedl, N. Fissolo, R. Schirmbeck, O. Ritz, P. Moller, and F. Leithauser. 2006. AID expression identifies interfollicular large B cells as putative precursors of mature B-cell malignancies. Blood. 107:2470–2473. [DOI] [PubMed] [Google Scholar]
- 17.Gourzi, P., T. Leonova, and F.N. Papavasiliou. 2006. A role for activation-induced cytidine deaminase in the host response against a transforming retrovirus. Immunity. 24:779–786. [DOI] [PubMed] [Google Scholar]
- 18.Heintz, N. 2004. Gene expression nervous system atlas (GENSAT). Nat. Neurosci. 7:483. [DOI] [PubMed] [Google Scholar]
- 19.Copeland, N.G., N.A. Jenkins, and D.L. Court. 2001. Recombineering: a powerful new tool for mouse functional genomics. Nat. Rev. Genet. 2:769–779. [DOI] [PubMed] [Google Scholar]
- 20.Yannoutsos, N., V. Barreto, Z. Misulovin, A. Gazumyan, W. Yu, N. Rajewsky, B.R. Peixoto, T. Eisenreich, and M.C. Nussenzweig. 2004. A cis element in the recombination activating gene locus regulates gene expression by counteracting a distant silencer. Nat. Immunol. 5:443–450. [DOI] [PubMed] [Google Scholar]
- 21.Fields, P.E., G.R. Lee, S.T. Kim, V.V. Bartsevich, and R.A. Flavell. 2004. Th2-specific chromatin remodeling and enhancer activity in the Th2 cytokine locus control region. Immunity. 21:865–876. [DOI] [PubMed] [Google Scholar]
- 22.Litt, M.D., M. Simpson, F. Recillas-Targa, M.N. Prioleau, and G. Felsenfeld. 2001. Transitions in histone acetylation reveal boundaries of three separately regulated neighboring loci. EMBO J. 20:2224–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sayegh, C.E., M.W. Quong, Y. Agata, and C. Murre. 2003. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat. Immunol. 4:586–593. [DOI] [PubMed] [Google Scholar]
- 24.Wittschieben, B.O., G. Otero, T. de Bizemont, J. Fellows, H. Erdjument-Bromage, R. Ohba, Y. Li, C.D. Allis, P. Tempst, and J.Q. Svejstrup. 1999. A novel histone acetyltransferase is an integral subunit of elongating RNA polymerase II holoenzyme. Mol. Cell. 4:123–128. [DOI] [PubMed] [Google Scholar]
- 25.Misulovin, Z., X.W. Yang, W. Yu, N. Heintz, and E. Meffre. 2001. A rapid method for targeted modification and screening of recombinant bacterial artificial chromosome. J. Immunol. Methods. 257:99–105. [DOI] [PubMed] [Google Scholar]
- 26.Shapiro-Shelef, M., K.I. Lin, L.J. McHeyzer-Williams, J. Liao, M.G. McHeyzer-Williams, and K. Calame. 2003. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. 19:607–620. [DOI] [PubMed] [Google Scholar]
- 27.Driver, D.J., L.J. McHeyzer-Williams, M. Cool, D.B. Stetson, and M.G. McHeyzer-Williams. 2001. Development and maintenance of a B220− memory B cell compartment. J. Immunol. 167:1393–1405. [DOI] [PubMed] [Google Scholar]
- 28.Blink, E.J., A. Light, A. Kallies, S.L. Nutt, P.D. Hodgkin, and D.M. Tarlinton. 2005. Early appearance of germinal center–derived memory B cells and plasma cells in blood after primary immunization. J. Exp. Med. 201:545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shih, T.A., M. Roederer, and M.C. Nussenzweig. 2002. Role of antigen receptor affinity in T cell-independent antibody responses in vivo. Nat. Immunol. 3:399–406. [DOI] [PubMed] [Google Scholar]
- 30.Shaffer, A.L., K.I. Lin, T.C. Kuo, X. Yu, E.M. Hurt, A. Rosenwald, J.M. Giltnane, L. Yang, H. Zhao, K. Calame, and L.M. Staudt. 2002. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 17:51–62. [DOI] [PubMed] [Google Scholar]
- 31.Cheung, W.C., J.S. Kim, M. Linden, L. Peng, B. Van Ness, R.D. Polakiewicz, and S. Janz. 2004. Novel targeted deregulation of c-Myc cooperates with Bcl-X(L) to cause plasma cell neoplasms in mice. J. Clin. Invest. 113:1763–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srinivas, S., T. Watanabe, C.S. Lin, C.M. William, Y. Tanabe, T.M. Jessell, and F. Costantini. 2001. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim, U., X.F. Qin, S. Gong, S. Stevens, Y. Luo, M. Nussenzweig, and R.G. Roeder. 1996. The B-cell-specific transcription coactivator OCA-B/OBF-1/Bob-1 is essential for normal production of immunoglobulin isotypes. Nature. 383:542–547. [DOI] [PubMed] [Google Scholar]
- 34.Nielsen, P.J., O. Georgiev, B. Lorenz, and W. Schaffner. 1996. B lymphocytes are impaired in mice lacking the transcriptional co-activator Bob1/OCA-B/OBF1. Eur. J. Immunol. 26:3214–3218. [DOI] [PubMed] [Google Scholar]
- 35.Schubart, D.B., A. Rolink, M.H. Kosco-Vilbois, F. Botteri, and P. Matthias. 1996. B-cell-specific coactivator OBF-1/OCA-B/Bob1 required for immune response and germinal centre formation. Nature. 383:538–542. [DOI] [PubMed] [Google Scholar]
- 36.Weill, J.C., and C.A. Reynaud. 2005. Do developing B cells need antigen? J. Exp. Med. 201:7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiu, Y.L., V.B. Soros, J.F. Kreisberg, K. Stopak, W. Yonemoto, and W.C. Greene. 2005. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature. 435:108–114. [DOI] [PubMed] [Google Scholar]
- 38.Newman, E.N., R.K. Holmes, H.M. Craig, K.C. Klein, J.R. Lingappa, M.H. Malim, and A.M. Sheehy. 2005. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 15:166–170. [DOI] [PubMed] [Google Scholar]
- 39.Rosenberg, N. 1994. Abl-mediated transformation, immunoglobulin gene rearrangements and arrest of B lymphocyte differentiation. Semin. Cancer Biol. 5:95–102. [PubMed] [Google Scholar]
- 40.Nobrega, M.A., I. Ovcharenko, V. Afzal, and E.M. Rubin. 2003. Scanning human gene deserts for long-range enhancers. Science. 302:413. [DOI] [PubMed] [Google Scholar]
- 41.Kim, T.H., L.O. Barrera, M. Zheng, C. Qu, M.A. Singer, T.A. Richmond, Y. Wu, R.D. Green, and B. Ren. 2005. A high-resolution map of active promoters in the human genome. Nature. 436:876–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pokholok, D.K., C.T. Harbison, S. Levine, M. Cole, N.M. Hannett, T.I. Lee, G.W. Bell, K. Walker, P.A. Rolfe, E. Herbolsheimer, et al. 2005. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 122:517–527. [DOI] [PubMed] [Google Scholar]
- 43.Bayly, R., L. Chuen, R.A. Currie, B.D. Hyndman, R. Casselman, G.A. Blobel, and D.P. LeBrun. 2004. E2A-PBX1 interacts directly with the KIX domain of CBP/p300 in the induction of proliferation in primary hematopoietic cells. J. Biol. Chem. 279:55362–55371. [DOI] [PubMed] [Google Scholar]
- 44.Bradney, C., M. Hjelmeland, Y. Komatsu, M. Yoshida, T.P. Yao, and Y. Zhuang. 2003. Regulation of E2A activities by histone acetyltransferases in B lymphocyte development. J. Biol. Chem. 278:2370–2376. [DOI] [PubMed] [Google Scholar]
- 45.Sciammas, R., A.L. Shaffer, J.H. Schatz, H. Zhao, L.M. Staudt, and H. Singh. 2006. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity. 25:225–236. [DOI] [PubMed] [Google Scholar]
- 46.Neumann, B., A. Luz, K. Pfeffer, and B. Holzmann. 1996. Defective Peyer's patch organogenesis in mice lacking the 55-kD receptor for tumor necrosis factor. J. Exp. Med. 184:259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vajdy, M., M.H. Kosco-Vilbois, M. Kopf, G. Kohler, and N. Lycke. 1995. Impaired mucosal immune responses in interleukin 4–targeted mice. J. Exp. Med. 181:41–53. [DOI] [PubMed] [Google Scholar]
- 48.Kitamura, D., J. Roes, R. Kuhn, and K. Rajewsky. 1991. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 350:423–426. [DOI] [PubMed] [Google Scholar]
- 49.Macpherson, A.J., A. Lamarre, K. McCoy, G.R. Harriman, B. Odermatt, G. Dougan, H. Hengartner, and R.M. Zinkernagel. 2001. IgA production without mu or delta chain expression in developing B cells. Nat. Immunol. 2:625–631. [DOI] [PubMed] [Google Scholar]
- 50.Machida, K., K.T. Cheng, V.M. Sung, S. Shimodaira, K.L. Lindsay, A.M. Levine, M.Y. Lai, and M.M. Lai. 2004. Hepatitis C virus induces a mutator phenotype: enhanced mutations of immunoglobulin and protooncogenes. Proc. Natl. Acad. Sci. USA. 101:4262–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tobollik, S., L. Meyer, M. Buettner, S. Klemmer, B. Kempkes, E. Kremmer, G. Niedobitek, and B. Jungnickel. 2006. Epstein-Barr virus nuclear antigen 2 inhibits AID expression during EBV-driven B-cell growth. Blood. 108:3859–3864. [DOI] [PubMed] [Google Scholar]
- 52.Uchida, J., T. Yasui, Y. Takaoka-Shichijo, M. Muraoka, W. Kulwichit, N. Raab-Traub, and H. Kikutani. 1999. Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science. 286:300–303. [DOI] [PubMed] [Google Scholar]
- 53.Gourzi, P., T. Leonova, and F.N. Papavasiliou. 2007. Viral induction of AID is independent of the interferon and the Toll-like receptor signaling pathways but requires NF-κB. J. Exp. Med. 204:259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kallies, A., J. Hasbold, D.M. Tarlinton, W. Dietrich, L.M. Corcoran, P.D. Hodgkin, and S.L. Nutt. 2004. Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. J. Exp. Med. 200:967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Casola, S., G. Cattoretti, N. Uyttersprot, S.B. Koralov, J. Segal, Z. Hao, A. Waisman, A. Egert, D. Ghitza, and K. Rajewsky. 2006. Tracking germinal center B cells expressing germ-line immunoglobulin gamma1 transcripts by conditional gene targeting. Proc. Natl. Acad. Sci. USA. 103:7396–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}