

Abstract
Imiquimod (IMQ), a synthetic agonist to Toll-like receptor (TLR) 7, is being successfully used for the treatment of certain skin neoplasms, but the exact mechanisms by which this compound induces tumor regression are not yet understood. While treating basal cell carcinoma (BCC) patients with topical IMQ, we detected, by immunohistochemistry, sizable numbers of both myeloid dendritic cells (mDCs) and plasmacytoid DCs (pDCs) within the inflammatory infiltrate. Surprisingly, peritumoral mDCs stained positive for perforin and granzyme B, whereas infiltrating pDCs expressed tumor necrosis factor–related apoptosis-inducing ligand (TRAIL). The biological relevance of this observation can be deduced from our further findings that peripheral blood–derived CD11c+ mDCs acquired antiperforin and anti–granzyme B reactivity upon TLR7/8 stimulation and could use these molecules to effectively lyse major histocompatibility complex (MHC) class Ilo cancer cell lines. The same activation protocol led pDCs to kill MHC class I–bearing Jurkat cells in a TRAIL-dependent fashion. While suggesting that mDCs and pDCs are directly involved in the IMQ-induced destruction of BCC lesions, our data also add a new facet to the functional spectrum of DCs, ascribing to them a major role not only in the initiation but also in the effector phase of the immune response.
Interest in imiquimod (IMQ) first came from the observation that this imidazoquinoline exerts a profound activity against viral acanthomas that was originally explained by its IFN-inducing capacity (1). When used topically for a prolonged period of time, it can lead to the regression of certain virus-induced (e.g., genital warts [2] and molluscum contagiosum [3]) and other (e.g., basal cell carcinoma [BCC] [4, 5] and lentigo maligna [6]) skin neoplasms. IMQ exerts its biologic activity primarily by ligation of Toll-like receptor (TLR) 7 (7) and, to a lesser extent, TLR8, both of which have been identified as natural receptors for single-stranded RNA (8, 9). Cell stimulation via TLR7 and TLR8 leads to downstream activation of NF-κB and other transcription factors (10, 11). Consequently, several genes encoding mediators and effector molecules of the innate as well as the adaptive immune response are transcribed (12–14). Because of their prominent expression of TLR7 and TLR8, plasmacytoid DCs (pDCs) and myeloid DCs (mDCs) (15, 16), respectively, are therefore likely candidates for the initiation of the IMQ-induced host defense reaction. Other mechanisms explaining the antitumor activity of IMQ are also under discussion. These include (a) the reversal of CD4+ regulatory T cell function (17), (b) a TLR-independent immunostimulatory action of IMQ via adenosine receptor signaling (18), (c) direct (19) and indirect, via IFN-α (20), IMQ-induced proapoptotic effects on tumor cells, and (d) an antiangiogenic activity of IMQ, as shown in a mouse model of angiogenesis (21, 22).
In a recent study, our group investigated IMQ-induced tumor regression in mice and found not only a good clinical response of the tumors to the topically applied compound but also a direct correlation between IMQ-induced tumor regression and the density of DCs in the peritumoral tissue (23). Not infrequently, cancer cells in death were found in close contact with DCs, which was compatible with a tumoricidal property of the latter (24, 25). In this study, we sought to determine whether similar phenomenona also occur during IMQ treatment of human skin cancers and, if so, to unravel the mechanisms responsible for IMQ-induced tumor regression.
RESULTS
Regression of BCC upon IMQ treatment
Seven patients with histopathologically confirmed superficial BCC were treated with IMQ. After a treatment period of 6 wk, we observed a complete clinical (Fig. 1) and histopathological response in all patients. No signs of recurrence were noted in any of the patients followed for at least 10 mo.
Figure 1.
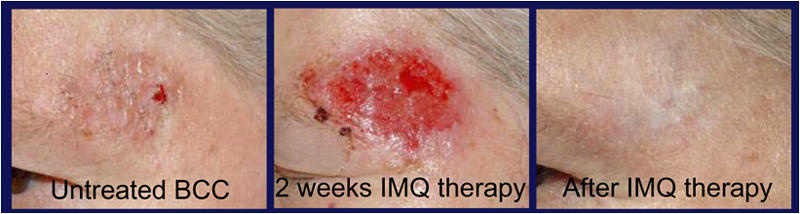
Complete regression of superficial BCC after a 6-wk treatment period with IMQ. IMQ topically applied five times a week for a period of 6 wk led to a local inflammatory response, which resulted in a complete clinical and histopathological tumor clearance in all patients treated. The clinical pictures are representative for all patients (n = 7) treated with IMQ.
In accordance with a previous report (4), all patients developed, before tumor clearance, an inflammatory tissue reaction at the site of IMQ application. It began as erythema after 2–3 d of treatment; became erosive during the second week (Fig. 1); appeared as crusting and, later, scaling plaques after 3–4 wk; and resolved completely after cessation of IMQ treatment. Systemic side effects such as flu-like symptoms, lymphadenopathy, myalgia, or changes in laboratory values never occurred in any of our patients.
Emergence kinetics of leukocytic populations in the peritumoral infiltrate upon IMQ treatment
Biopsies from BCCs were obtained before, during, and after IMQ treatment and subjected to immunofluorescence analysis using a broad panel of antibodies (Table I) to analyze, both quantitatively and qualitatively, the composition and kinetics of the IMQ-induced inflammatory infiltrate. In untreated BCCs we found a sparse infiltrate, mainly consisting of T cells of the helper phenotype (Table II). Upon 2 wk of IMQ treatment, a dramatic increase of CD8+ T cells and, to a much lesser extent, of CD4+ T cells was seen around tumor cell islets (Table II). Fig. 2 (A and B) shows that, after 2 wk of topical IMQ treatment, BCC islets were surrounded and partly infiltrated by dendritically shaped cells exhibiting either the CD11c+/HLA-DR+ mDC (Fig. 2 A) or the CD123+/HLA-DR+ pDC (Fig. 2 B) phenotype. This increase in inflammatory-type mDCs and pDCs occurs at the expense of the resident DC populations of normal human skin (Langerhans cells and CD1c+ dermal DCs; Table II). Other constituents of the peritumoral infiltrate included (a) CD56+/CD94+ NK cells, (b) CD14+ mononuclear phagocytes, and (c) CD15+/HLA-DR− neutrophils (Table II). In contrast, B cell, eosinophil, basophil, and mast cell counts displayed no major alterations upon IMQ treatment. At the end of treatment, when tumor clearance was achieved, the density of the various infiltrating leukocyte subpopulations was comparable to the values before IMQ application (Table II).
Table I.
List of mAbs used in this study
| Antibody specificity | Clone (source of antibodies) | Isotype |
|---|---|---|
| Staining experiments | ||
| BDCA-2 (FITC) | AC144 (Miltenyi Biotec) | mIgG1 |
| CD1a (FITC) | HI149 (BD Biosciences) | mIgG1 |
| CD1c (pur., biot.) | M241 (Ancell) | mIgG1 |
| CD3 (FITC, PerCP) | SK-7 (BD Biosciences) | mIgG1 |
| CD4 (pur.) | SK-3 (BD Biosciences) | mIgG1 |
| CD8 (pur.) | C8/144B (DakoCytomation) | mIgG1 |
| CD8 (allophycocyanin [APC]) | B9.11 (Immunotech Coulter) | mIgG1 |
| CD11c (FITC) | BU15 (Serotec) | mIgG1 |
| CD11c (PE, PE-Cy5) | B-ly6 (BD Biosciences) | mIgG1 |
| CD14 (pur., FITC, PerCP) | MΦP9 (BD Biosciences) | mIgG2b |
| CD15 (FITC) | 80H5 (Immunotech Coulter) | mIgM |
| CD19 (pur., PerCP) | SJ25C1 (BD Biosciences) | mIgG1 |
| CD45RA (pur.) | L48 (BD Biosciences) | mIgG1 |
| CD56 (biot., PE-Cy5) | B159 (BD Biosciences) | mIgG1 |
| CD83 (FITC) | H15a (Immunotech Coulter) | mIgG2b |
| CD94 (pur.) | HP-3D9 (BD Biosciences) | mIgG1 |
| CD117 (c-kit, pur.) | 95C3 (An Der Grub) | mIgG1 |
| CD123 (biot.) | 9F5 (BD Biosciences) | mIgG1 |
| CD203c (PE) | 97A6 (Immunotech Coulter) | mIgG1 |
| CD207 (pur.) | DCGM4 (Immunotech Coulter) | mIgG1 |
| Fas ligand (pur.) | NOK-1 (BD Biosciences) | mIgG1 |
| Granzyme B (pur.) | GrB-7 (Monosan) | mIgG2a |
| Granzyme B (PE) | GB-11 (PeliCluster) | mIgG1 |
| HLA-DR (pur., FITC, APC) | L243 (BD Biosciences) | mIgG2a |
| IFN-α | 225.2C (Chromaprobe) | mIgG2b |
| iNOS (FITC) | 6 (BD Biosciences) | mIgG2a |
| MBP (pur.) | BMK13 (Chemicon) | mIgG1 |
| Pancytokeratin (pur.) | AE1 and AE3 (BioGenex) | mIgG1 |
| Perforin (pur.) | dG9 (Abcam) | mIgG2b |
| Perforin (FITC) | δG9 (BD Biosciences) | mIgG2b |
| TNF-α (FITC) | MAb11 (BD Biosciences) | mIgG1 |
| TRAIL (pur.) | 75402 (R&D Systems) | mIgG1 |
| TRAIL (neutralizing; pur.) | 75411 (R&D Systems) | mIgG1 |
| TRAIL R1 (pur.) | 69036 (R&D Systems) | mIgG1 |
| Cell-sorting experiments | ||
| CD3 (pur.) | UCHT1 (Immunotech Coulter) | mIgG1 |
| CD16 (pur.) | 3G8 (Immunotech Coulter) | mIgG1 |
| CD19 (pur.) | J4.119 (Immunotech Coulter) | mIgG1 |
| CD34 (pur.) | 581 (Immunotech Coulter) | mIgG1 |
| CD41 (pur.) | SZ22 (Immunotech Coulter) | mIgG1 |
| CD56 (pur.) | C218 (Immunotech Coulter) | mIgG1 |
| CD235a (pur.) | 11E4B-7-6 (Immunotech Coulter) | mIgG1 |
biot., biotinylated; MBP, eosinophil major basic protein; pur., purified.
Table II.
Number of leukocytes occurring in BCCs before, after 2 wk, and at the end of IMQ treatment
| Cell type | Antigens | Epidermis/dermis | Untreated BCCs | 2 wk IMQ | 6 wk IMQ |
|---|---|---|---|---|---|
| T helper cells | CD4+/CD3+ | epidermis (cells/mm) | 1.8 ± 2 | 3.3 ± 3.3 | 1.4 ± 1.8 |
| dermis (cells/mm2) | 98.2 ± 64.4 | 164 ± 60.6 | 99 ± 56.8 | ||
| Cytotoxic T cells | CD8+/CD3+ | epidermis (cells/mm) | 1.4 ± 0.9 | 5.5 ± 2.6 | 3.7±3.4 |
| dermis (cells/mm2) | 55.7 ± 31.1 | 236.6 ± 61.4 | 63.1±35.3 | ||
| NK cells | CD56+/CD94+ | epidermis (cells/mm) | 0 | 0.7 ± 1.3 | 0 |
| dermis (cells/mm2) | 3 ± 2.2 | 46.8 ± 19 | 10.1 ± 10.7 | ||
| Langerhans cells | CD207+/CD1a+ | epidermis (cells/mm) | 9 ± 4.2 | 3.6 ± 1.7 | 8.3 ± 5.2 |
| dermis (cells/mm2) | 6.2 ± 3.6 | 6.7 ± 4.9 | 5.1 ± 4.7 | ||
| CD11c + mDCs | CD11c+/HLA-DR+ | epidermis (cells/mm) | 0 | 0 | 0 |
| dermis (cells/mm2) | 25.5 ± 4.2 | 58.4 ± 14.6 | 22.9 ± 4.8 | ||
| CD1c + mDCs | CD1c+/HLA-DR+ | epidermis (cells/mm) | 0.7 ± 1.3 | 0.3 ± 0.5 | 0.3 ± 0.4 |
| dermis (cells/mm2) | 55.2 ± 29.7 | 16.3 ± 7.4 | 24.1 ± 12.7 | ||
| Phagocytes | CD14+ | epidermis (cells/mm) | 1.4 ± 3.8 | 2.4 ± 3.6 | 1.3 ± 3 |
| dermis (cells/mm2) | 21.2 ± 15.2 | 47.6 ± 6.1 | 35.1 ± 12.7 | ||
| pDCs | CD123+/ CD45RA+ | epidermis (cells/mm) | 0.2 ± 0.4 | 0 | 0 |
| dermis (cells/mm2) | 9.9 ± 12.1 | 54.7 ± 6.5 | 17.1 ± 8.8 | ||
| Neutrophils | CD15+/HLA-DR− | epidermis (cells/mm) | 0 | 0 | 0 |
| dermis (cells/mm2) | 7.8 ± 8.9 | 62.1 ± 14.3 | 15.4 ± 13 | ||
| Eosinophils | MBP+ | epidermis (cells/mm) | 0 | 0 | 0 |
| dermis (cells/mm2) | 0 | 2.1 ± 3.3 | 0.5 ± 0.9 | ||
| Basophils | CD203c+ | epidermis (cells/mm) | 0 | 0 | 0 |
| dermis (cells/mm2) | 0 | 0 | 0 | ||
| Mast cells | cKit+ | epidermis (cells/mm) | 0 | 0 | 0 |
| dermis (cells/mm2) | 14.3 ± 13.5 | 13.3 ± 2.9 | 10.4 ± 11.7 | ||
| B cells | CD19+ | epidermis (cells/mm) | 0 | 0 | 0 |
| dermis (cells/mm2) | 3.2 ± 4.6 | 2.8 ± 5.6 | 1.1 ± 1.5 |
Cells were visualized using the indicated markers and were evaluated by immunofluorescence analysis. Numbers in bold indicate means ± SE of the mean of cells counted in BCC lesions before, after 2 wk, and after 6 wk of IMQ treatment. MBP, eosinophil major basic protein.
Figure 2.
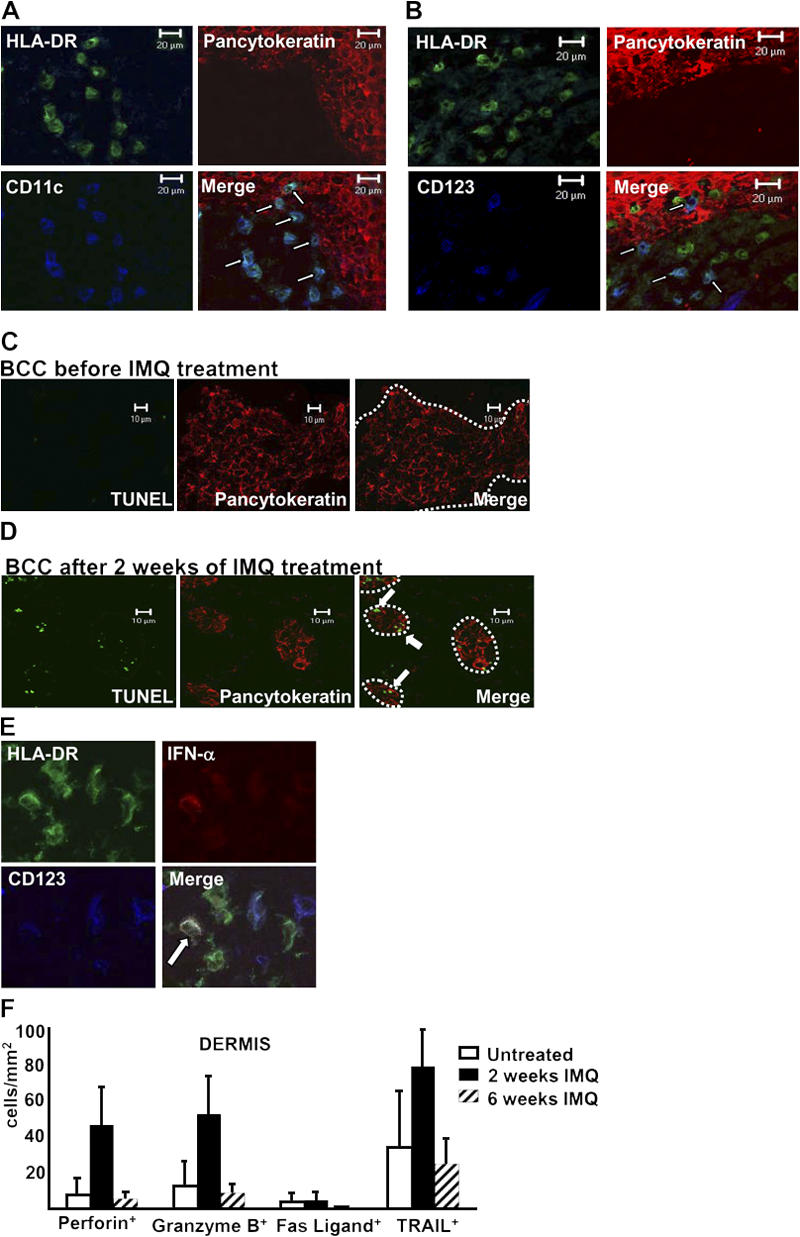
Induction of tumor cell apoptosis is accompanied by peritumoral accumulation of DCs and up-regulation of lytic molecules on inflammatory cells. (A and B) Immunofluorescence triple labeling of cryostat sections of IMQ-treated BCC lesions with antipancytokeratin (TRITC), anti–HLA-DR (FITC), and either (A) anti-CD11c (APC) or (B) anti-CD123 (Cy5) revealed a close proximity of both DC populations with tumor cells. Arrows indicate double-positive DCs. (C) Untreated BCCs and (D) BCCs after 2 wk of IMQ treatment were stained with an antipancytokeratin antibody (TRITC) and TUNEL staining (FITC). Note the double-positive tumor cells occurring after 2 wk of IMQ treatment (arrows). The pictures are representative for all evaluated biopsies. The dotted lines point out the margins of BCCs. (E) Identification of IFN-α–producing pDCs in 2-wk–treated BCCs by triple stainings with anti– IFN-α (TRITC), anti–HLA-DR (FITC), and anti-CD123 (Cy5) mAbs. The arrow indicates a triple-positive cell. (F) Quantitative analysis of lytic molecules expressed by inflammatory cells of the peritumoral infiltrate. Immunofluorescence single stainings were performed using antiperforin, anti–granzyme B, anti-Fas ligand, and anti-TRAIL antibodies (all TRITC). Data are given as absolute numbers of positive cells ± SEM for the indicated markers.
Detection of lytic molecules in the peritumoral tissue upon IMQ treatment
Analyzing the mechanisms of IMQ-induced tumor regression, we searched, by Tdt-mediated dUTP-biotin nick-end labeling (TUNEL) staining, for signs of apoptosis in IMQ- treated BCC lesions. Although only a few TUNEL+ tumor cells were detected in untreated BCCs (Fig. 2 C), the decrease in pancytokeratin+ BCC cells after 2 wk of therapy was accompanied by a concomitant increase of TUNEL+ tumor cells (Fig. 2 D). One may therefore conclude that BCC regression upon IMQ treatment is at least partly accomplished by apoptosis of tumor cells.
Based on the observation of IFN-α–induced tumor regression (26) and tumor cell apoptosis (27), we searched for the presence of IFN-α–producing cells in IMQ-treated BCC biopsies and found ∼30% of all pDCs, but no other leukocytes, to exhibit anti–IFN-α staining (Fig. 2 E). It is therefore not unreasonable to assume that IMQ-activated pDCs contribute to BCC apoptosis by IFN-α production.
In addition to IFN-α–induced apoptotic events, perforin/granzyme-based and death receptor ligand–mediated cancer cell lysis are the immune system's major tumoricidal effector mechanisms. When we analyzed BCC biopsies before, during, and after IMQ treatment for the expression of lytic molecules, we could hardly detect any expression of Fas ligand (Fig. 2 F). In contrast, we observed a considerable up-regulation of perforin, granzyme B, and TNF-related apoptosis-inducing ligand (TRAIL) on infiltrating cells after 2 wk of IMQ treatment when compared with the low baseline levels of untreated BCCs (Fig. 2 F).
The majority of anti-TRAIL reactivity was observed on T cells (Fig. 3 A), but CD11c+ mDCs, CD123+ pDCs, and CD14+ mononuclear phagocytes also expressed this molecule (Fig. 3, A and B). The biologic importance of this finding is underscored by the further observation that tumor cells of untreated (Fig. 3 C) and IMQ-treated (Fig. 3 D) BCC lesions were positive for TRAIL receptor 1 (TRAIL R1) and surrounded by a dense rim of CD123+ cells (Fig. 3 D). Anti-TRAIL R2, R3, and R4 stainings yielded negative results (not depicted).
Figure 3.

Upon IMQ treatment, TRAIL R1+ BCC islets are surrounded by TRAIL-expressing pDCs. (A) Detailed quantitative in situ analysis of the expression pattern of TRAIL on leukocytes before, during, and after IMQ treatment was performed by immunofluorescence double stainings with an anti-TRAIL mAb and lineage markers in all patients treated. The data are given as absolute numbers of double-positive cells ± SEM. (B) A representative view of anti-CD123 (FITC), anti–HLA-DR (APC), and anti-TRAIL (TRITC) triple stainings of a BCC lesion after 2 wk of IMQ treatment identifies pDCs as TRAIL-expressing cells. Immunofluorescence triple labeling of (C) untreated and (D) IMQ-treated BCCs with antipancytokeratin (TRITC), anti-TRAIL R1 (A488), and anti-CD123 (Cy5) reveals TRAIL R1+ BCC cells (C and D) surrounded by CD123+ cells (arrows; D).
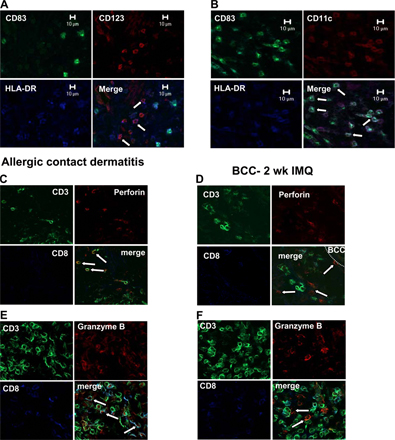
Somewhat to our surprise, we found that only a few T cells and NK cells displayed antiperforin and anti–granzyme B staining and that CD3−CD56−HLA-DR+ cells were the main source of these lytic molecules (Fig. 4, A and C). Additional phenotyping revealed that these cells were CD11c+ (Fig. 4, B and D) and lacked lineage markers such as CD3, CD56, CD14, CD15, or CD19, as well as CD1a and CD207, thus corresponding to mDCs. Similar to what has been observed in untreated psoriatic skin lesions (28), CD11c+/HLA-DR+ DCs in IMQ-treated BCC lesions were also found to react with antibodies against inducible NO synthase (iNOS) and TNF-α (Fig. 4 E). At least phenotypically, these cells therefore corresponded to TNF-α– and iNOS-producing DCs, an mDC population possibly involved in innate immune defense against bacteria (29). In contrast to infiltrating pDCs (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20070021/DC1), the vast majority of CD11c+/HLA-DR+ DCs displayed the maturation marker CD83 (Fig. S1 B). As it was quite unexpected not to find cytotoxic T cells and/or NK cells, but instead to find DCs containing perforin and granzyme B, we compared immunofluorescence triple stainings of IMQ-treated BCCs with those of allergic contact dermatitis, a disease in which perforin- and granzyme B–containing T cells have been previously described (30). In contrast to IMQ-treated BCC lesions in which most perforin- and granzyme B–positive cells were CD3− as well as CD8−, antiperforin and anti–granzyme B reactivity in allergic contact dermatitis was mainly restricted to T cells (Fig. S1, C–F).
Figure 4.

IMQ treatment induces the up-regulation of perforin and granzyme B on mDCs in the peritumoral infiltrate. A detailed quantitative analysis of the expression pattern of (A) perforin and (C) granzyme B on leukocytes before, during, and after IMQ treatment was performed by immunofluorescence double stainings with antibodies against the indicated lytic molecules and lineage markers in all seven treated patients. The data are given as absolute numbers of double-positive cells ± SEM. Representative pictures of immunofluorescence triple stainings with anti-CD11c, anti–HLA-DR, and (B) antiperforin or (D) anti–granzyme B mAbs of the peritumoral infiltrate after 2 wk of treatment with IMQ. (E) Triple immunofluorescence labeling of BCCs upon IMQ treatment with anti-CD11c (PE), anti–HLA-DR (APC), and anti-iNOS (FITC) or anti–TNF-α (FITC) mAbs identifies iNOS and TNF-α to be produced by CD11c+HLA-DR+ cells (arrows).
Detection of perforin and granzyme B in mDCs upon TLR7 and TLR8 stimulation and of TRAIL on pDCs upon TLR7 stimulation
To determine the biological significance of anti-TRAIL and antiperforin/anti–granzyme B staining of DCs in IMQ-treated BCC lesions, we decided, for quantitative reasons, to work with DCs isolated from peripheral blood rather than from these skin lesions. Using the protocol described in Materials and methods, we routinely obtained mDC and pDC populations of 93–99% (mean = 97%) purity. By FACS analysis, these cells were HLA-DR+, expressed either high levels of CD11c or BDCA-2, and lacked the lineage markers CD3, CD56, CD14, and CD19 (Fig. 5 A). None of the lytic molecules searched for were detected on freshly isolated mDCs. In contrast, stimulation of purified peripheral blood–derived mDCs with TLR7 and TLR8 ligands, but not with the vehicle control, led to the intracellular expression of perforin and granzyme B in ∼10–15% of these cells (Fig. 5, B and C). Expression of TRAIL was not detectable on mDCs after stimulation with TLR7/8 ligands (Fig. 5 D).
Figure 5.

Peripheral blood–derived DCs express lytic molecules after TLR7/8 stimulation. (A) Characterization of isolated peripheral blood–derived pDCs and mDCs as HLA-DR+BDCA-2+ and HLA-DR+CD11c+, respectively, and lineage-negative cell populations by flow cytometry. (B and C) Analysis of intracellular expression levels of perforin and granzyme B of isolated mDCs before and after culture with TLR7/8 agonists by flow cytometry. (B) Representative dot plots of mDCs show double-positive cells for perforin (FITC) and granzyme B (PE) after stimulation with TLR7, TLR8, and TLR7/8 agonists. (C) Quantitative analysis of granzyme B+ and perforin+ mDCs. Data are given as mean percentages of perforin- or granzyme B–positive mDCs ± SEM from three independent experiments. (D) Freshly isolated and TLR7/8-stimulated mDCs do not display membrane-bound TRAIL (A488). Representative histogram plots from three independent experiments show the anti-TRAIL reactivity (bold line) and the matching isotype control (dashed line). (E and F) Determination of intracellular expression levels of perforin and granzyme B on isolated pDCs before and after stimulation with TLR agonists. (E) Representative dot plots of pDCs show a decrease of granzyme B–positive cells after stimulation with TLR7 and TLR7/8 agonists. (F) Quantitative analysis of pDCs expressing perforin and granzyme B. Data are given as mean percentages of perforin- and granzyme B–positive cells ± SEM from three independent experiments. (G) TLR7/8-stimulated, but not freshly isolated, pDCs display membrane-bound TRAIL (A488). Representative histogram plots from three different experiments show the anti-TRAIL reactivity (bold line) and the matching isotype control (dashed line). Dead cells were excluded by gating out propidium iodide–positive cells in all experiments.
Unstimulated pDCs abundantly contained granzyme B within their cytoplasm (Fig. 5 E). After 12 h of stimulation with ligands to TLR7 and TLR7/8, but not with the vehicle control or the TLR8 ligand alone, the intracellular expression of granzyme B in pDCs had decreased. (Fig. 5, E and F). This is consistent with the previous finding of pDCs expressing TLR7 but not TLR8 (15). In contrast to mDCs, TLR7/8 agonists induced up-regulation of TRAIL on the surface of pDCs (Fig. 5 G). Similar to the pretreatment situation, the cell populations stimulated with TLR7/8 agonists for 12 h failed to express CD3 and CD56, strongly arguing against the possibility of the involvement of T cells and/or NK cells. These findings indicate that, upon stimulation with TLR7/8 agonists, mDCs and pDCs from peripheral blood acquire the phenotype of mDCs and pDCs found in the peritumoral tissue of IMQ-treated BCC lesions.
mDCs release perforin and granzyme B upon TLR7 and TLR8 stimulation
The cytotoxic effect of the perforin–granzyme B pathway is only functional if the cytotoxic proteins are degranulated and come into contact with target cells. To reveal possible cytotoxic functions of perforin- and granzyme B–expressing TLR7/8-stimulated DCs, we analyzed perforin and granzyme B levels in the supernatants of sorted DCs after TLR7 and TLR8 ligation by ELISA. The viability of DCs was controlled by annexin V (flow cytometry) and trypan blue stainings (counting chamber) and exceeded 90% before and after cultures. Supernatants of PMA/ionomycin-stimulated, negatively selected T cells served as positive controls. We observed a substantial release of perforin and granzyme B after stimulation of mDCs with TLR7 and TLR8 ligands, in contrast to the vehicle control (Fig. 6, A and B). We detected granzyme B in the supernatants of pDCs after TLR7 and TLR7/8 ligation but not after TLR8 ligation alone or vehicle control (Fig. 6 B), which was consistent with our FACS data described in the previous section. Perforin could not be detected at any setting in the supernatant of pDCs (Fig. 6 A). These data demonstrate that mDCs and pDCs can be activated to release lytic molecules in a biologically relevant fashion.
Figure 6.

Secretion of perforin and granzyme B by mDCs upon TLR7 and TLR8 stimulation. (A) Perforin and (B) granzyme B protein levels were determined by ELISA in media conditioned by purified blood–derived mDCs and pDCs after 12 h of stimulation with TLR7 and TLR8 agonists. Media conditioned by 12-h PMA/ionomycin-stimulated T cells were used as positive controls. Data are given as means ± SEM from two independent experiments with 2 × 105 cells in each setting.
TLR7/8-stimulated mDCs and pDCs become killer cells: involvement of different cytotoxic pathways
To evaluate the functional significance of lytic molecules expressed by DCs after TLR7 and TLR8 ligation, we performed cytotoxicity assays with purified peripheral blood–derived mDCs and pDCs as effector cells, and perforin-sensitive K562 (Fig. S2 A, available at http://www.jem.org/cgi/content/full/jem.20070021/DC1) and TRAIL-sensitive Jurkat cell lines (31) as target cells. Although freshly isolated mDCs hardly displayed any cytotoxic activity against K562 and Jurkat cells, mDCs stimulated with TLR7/8 agonists effectively lysed K562 but not Jurkat cells (Fig. 7 A). We observed the opposite effect in pDCs, in that they exhibited substantial cytotoxic activity against Jurkat and not K562 cells after TLR7/8 stimulation, but not when unstimulated (Fig. 7 B). This diverse killing behavior indicated distinct mechanisms operative in DC-mediated cytotoxicity after stimulation with TLR7/8 ligands. To investigate the different cytotoxic pathways in greater detail, we performed inhibition experiments with (a) concanamycin A, a specific inhibitor of the perforin-based cytotoxic pathway (32), (b) a neutralizing anti-TRAIL antibody to block TRAIL-mediated killing, and (c) a neutralizing anti-Fas ligand antibody to inhibit Fas-induced killing. As shown in Fig. 7 C, coincubation of TLR7/8-stimulated mDCs and K562 in the presence of concanamycin A led to a complete disappearance of the specific lysis, whereas anti-TRAIL and anti-Fas ligand antibodies had no effect on the mDC-mediated cytotoxicity. On the other hand, preincubation of TLR7/8-stimulated pDCs with an inhibitory anti-TRAIL antibody essentially abolished the pDC cytotoxicity against Jurkat cells (Fig. 7 D); the specificity of this inhibition is also documented by the failure of concanamycin A and anti-Fas ligand antibodies to block the killing of Jurkat cells by TLR7/8-activated pDCs. To exclude the theoretical possibility of contaminating NK/NKT cells exhibiting cytotoxic activity within isolated DC populations, we performed cytotoxicity assays with effector cells consisting of irrelevant filler cells (i.e., A431) containing titrated numbers of NK cells. In a setting of 7% NK cells titrated into A431 as filler cells, we observed a 10% cytotoxic activity against K562 and Jurkat (Fig. S2 B), which represented less than half of the mDC-mediated killing and less than one third of pDC-mediated cytotoxicity. In addition, NK cells killed K562 and Jurkat cells with a comparable intensity and did not show selective killing like mDCs and pDCs. As NK cells and T cells never accounted for >1% within purified DC populations (Fig. 5 A), these experiments strongly argue against contaminating cells being even partly responsible for the cytotoxic activity of the DC populations.
Figure 7.
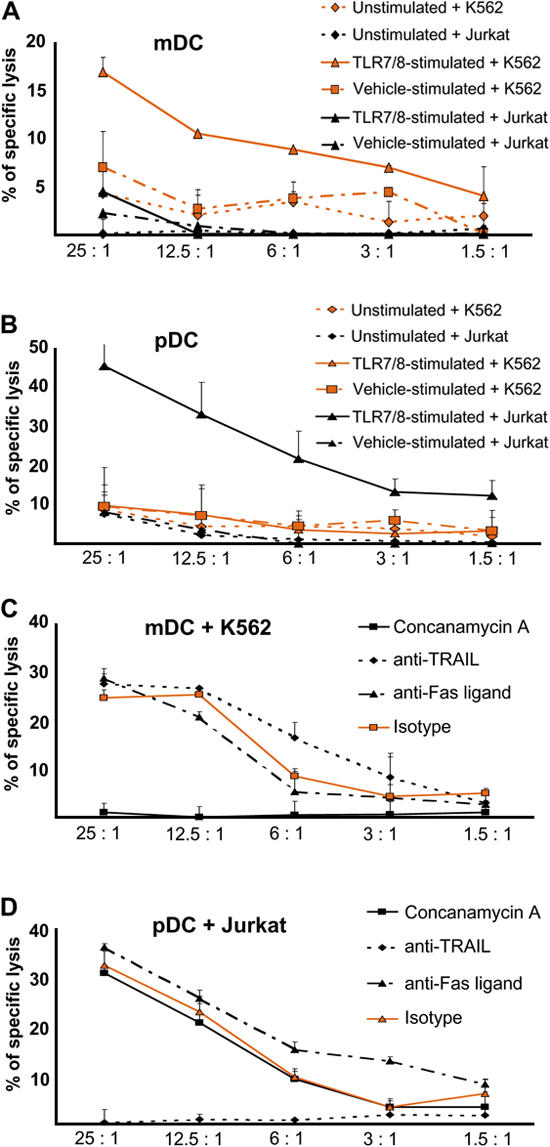
Tumoricidal activity of blood-derived mDCs and pDCs after stimulation with TLR7/8 agonists. Unstimulated, TLR7/8- stimulated, or vehicle-stimulated (A) mDCs and (B) pDCs were cultured with K562 or Jurkat as target cells for a conventional 2-h europium-TDA release assay at the indicated effector/target cell ratios. Data represent the means of triplicate wells ± SEM from two independent experiments. (C and D) For inhibition of different cytotoxicity-inducing pathways, 12-h TLR7/8-stimulated mDCs and pDCs as effector cells were preincubated with 100 nM concanamycin A for 120 min, 5 μg/ml anti-TRAIL for 30 min, 5 μg/ml anti-Fas ligand for 30 min, or 5 μg/ml of an IgG1 isotype for 30 min. Target cells were (C) K562 for TLR7/8-stimulated mDCs and (D) Jurkat for TLR7/8-stimulated pDCs. Cytotoxicity was determined by a 2-h europium-TDA release assay at the indicated effector/target cell ratios. Data represent the means of triplicate wells ± SEM from two independent experiments.
DISCUSSION
IMQ is now an established therapeutic option in the treatment of certain skin neoplasms such as genital warts (2), actinic keratoses (33), and BCCs (4, 5). In BCCs, cure rates are similar to those achieved with other therapeutic procedures (34, 35). Our study confirms these observations.
After binding to and signaling through TLR7 (7), IMQ leads to up-regulation of cytokines with growth-inhibitory (e.g., IFN-α) and/or proinflammatory (e.g., TNF-α, IL-1, and IL-6) properties (1, 36). TLR-independent mechanisms, operating directly by induction of apoptosis (19) and/or indirectly by adenosine receptor signaling (18), could also contribute to the clinically observed tumor regression. Even though the IMQ-induced regression pattern of skin cancers shows no signs of T cell memory, evidence exists that CD8+ T cells are abundantly present in the inflammatory infiltrate of IMQ-treated skin cancers (12, 14). A role for NK cells has also been claimed (37). To address the involvement of cellular cytotoxicity in IMQ-induced tumor regression, in this study we searched for the expression of lytic molecules on/in different cell populations forming this infiltrate.
As opposed to the pretreatment situation, we detected sizable numbers of perforin/granzyme B–positive cells, as well as TRAIL-expressing cells, after 2 wk of IMQ treatment. Interestingly, we observed that the majority of CD8+ T cells and CD56+ NK cells, which together formed a major portion of the IMQ-induced infiltrate, were largely devoid of antiperforin and anti–granzyme B reactivity. The reasons for this finding are not entirely clear. One could argue that CD8+ T cells and NK cells are simply bystander cells within the inflammatory infiltrate and are not concerned with cytotoxic effector functions. It is also conceivable that CD8+ T cells discharge their cytotoxic granules directly after migration into the peritumoral tissue; after 2 wk of IMQ treatment, the time point we had chosen for taking the biopsies, these granules would therefore no longer be detectable within their cytoplasm. We cannot definitively exclude this possibility. The occurrence of staining artifacts is unlikely, as we detected perforin- and granzyme B–positive T cells in skin biopsies of allergic contact eczema (Fig. S1, C and E). To our surprise, double and triple labeling of IMQ treated BCC sections revealed perforin and granzyme B to be expressed predominantly by lineage-negative CD11c+/HLA-DR+ mDCs. On the other hand, TRAIL, a molecule directly involved in tumor cell killing by binding to its corresponding death receptors (38–40), was expressed on several cell types, including pDCs. We sought to better characterize the mDC subset expressing cytotoxic molecules in situ and found that they coexpress TNF-α and iNOS, two proinflammatory mediators with antitumor activity (41, 42). Thus, these cells corresponded to TNF-α– and iNOS-producing DCs, which were shown to originate from blood-derived monocytes (43) and exhibit effector functions in bacterial infections in mice (29) and to correlate with disease activity in psoriasis in humans (28).
We were not able to isolate DCs from IMQ-treated BCC lesions in numbers that would have sufficed to conduct meaningful functional experiments. We therefore decided to test the functional relevance of our immunostaining results in studies with peripheral blood–derived mDCs and pDCs stimulated with TLR7 and TLR8 agonists. When activated in vitro with such compounds, peripheral blood–derived mDCs not only expressed and released perforin and granzyme B but also exhibited substantial cytotoxicity against MHC class I–negative (K562) but not MHC class I–positive (Jurkat) tumor cell lines. This lytic activity was abolished by pretreatment of effector cells with concanamycin A, an inhibitor of perforin-mediated cytotoxicity. To rule out the possibility that T cells and/or NK/NKT cells would contaminate the mDC effector cell population, we analyzed purified DCs of the peripheral blood by FACS stainings and by real-time PCR. Results obtained showed that these cells lacked all lineage markers at the protein level (Fig. 5 A) and were devoid of CD3ɛ transcripts (unpublished data). We also titrated CD56+ cells into a population of nonfunctional filler cells and found that the lytic capacity of this cell mixture was considerably lower than that of purified TLR7/8-activated DCs, even when numbers of CD56+ NK cells were greater than the peak levels of all lineage−CD11c− cells ever encountered in our mDC preparations (Fig. S2 B). Evidence for a direct cytotoxic potential of mDCs has also been presented by other investigators, but their findings differ from ours with regard to the activation status of the DCs and the type of lytic molecules involved (24, 44–48).
The emergence of pDCs in the peritumoral tissue of IMQ-treated skin cancers has been demonstrated in mice (23) and in humans (49). We confirmed the occurrence of IFN-α–producing pDCs in the peritumoral infiltrate during IMQ treatment. As IFN-α induces cytotoxic molecules on NK cells (50) and T cells (51), pDCs may indirectly contribute to the elimination of tumor cells. In this study, we asked whether pDCs could directly acquire effector functions via up-regulation of cytotoxic molecules and, thus, play an active role in tumor clearance. In contrast to mDCs, pDCs occurring within the peritumoral infiltrate expressed neither perforin nor granzyme B. At first glance, this was rather surprising, because unstimulated pDCs from peripheral blood clearly displayed intracellular anti–granzyme B (Fig. 5 E) (52, 53). Our further observation that pDC expression levels of granzyme B are continuously decreasing upon TLR7 ligation may be an explanation for the results of our in situ stainings. In any event, granzyme B does not seem to contribute to the antitumor activity of pDCs, as inhibition of the perforin/granzyme B–mediated cytotoxic pathway of TLR7/8-stimulated pDCs had no effect on the cytotoxic activity of these cells. The physiological role of granzyme B in pDCs therefore remains to be established.
On the basis of our findings that pDCs occurring in the IMQ-induced infiltrate expressed TRAIL and that TLR7-stimulated peripheral blood–derived pDCs displayed TRAIL on their surface and used this molecule for cytotoxic purposes, we would like to propose that pDCs have the potential to act as anticancer effector cells. Our results confirm and extend observations by others demonstrating the TRAIL- mediated cytotoxicity of pDCs after stimulation by influenza virus or TLR agonists (25).
The biological importance of our findings was underlined by the observation that IMQ-treated BCC cells, surrounded by CD123+ pDCs, are TRAIL 1 (DR4)–positive (Fig. 3 D) and express only negligible amounts of MHC class I molecules in situ (unpublished data). Even though we are aware of the fact that TLR7/8-stimulated DCs from peripheral blood are not necessarily equivalent to DCs that accumulate in the skin in response to TLR activation, our data suggest that perforin/granzyme B–bearing cytotoxic mDCs and TRAIL-expressing pDCs participate in IMQ-induced tumor regression by MHC class I–independent and death receptor–dependent killing of BCC cells, respectively. This does not exclude the possibility that other effector molecules (e.g., TNF-α) are also involved in this process.
Many questions remain to be resolved. To begin with, it has to be determined whether all DCs or only subpopulations thereof can be transformed into effector cells upon appropriate stimulation. Although we did not find NKG2D, CD94, CD16, or CD56 to be expressed on cytotoxic mDCs or pDCs before or after TLR7/8 stimulation (unpublished data), the relationship of human DCs described in our study to the newly described IFN-producing killer DCs in mice (54, 55) has to be defined. It also needs to be clarified whether natural TLR7/8 ligands (viral single-stranded RNA) or stimuli other than TLR7/8 agonists can endow DCs with lytic properties, which signaling cascades are involved in this process, and whether tumor cells, by an as of yet unknown recognition process, are triggering the release of cytotoxic granules by activated DCs. All of this information is required to devise strategies for the use of cytotoxic DCs as tools in anticancer immunotherapy.
Finally, it will be of great interest to determine whether DC cytotoxicity ever occurs in the course of pathophysiological DC-driven immune responses. If so, the attractive possibility exists that DCs may use this effector mechanism to down-regulate the immune reactions that they initiate themselves.
MATERIALS AND METHODS
Patients and tissue material.
Seven patients with histologically confirmed superficial BCC (three females and four males; mean age = 78.3 ± 8.2) were enrolled in the study after giving their written informed consent. BCCs were located on the head (n = 2), the neck (n = 2), and the back (n = 3). None of the patients were immunosuppressed or had any dermatological disease that could exacerbate upon IMQ (Aldara; 3M Pharmaceuticals) treatment. BCCs included in this study were therapy naive and did not receive any BCC-specific therapy (e.g., photodynamic therapy, 5-fluorouracil, or IFN-α) before this study.
Patients received a once daily, five-times-a-week dosing regimen of IMQ 5% cream for 6 wk, according to the official treatment guidelines approved for BCC. The cream was applied as a thin layer without occlusion overnight exactly on the BCC sites. Safety and efficacy were clinically monitored weekly and by blood examinations at weeks 3 and 6. After 6 wk of treatment, response rates were determined by clinical examination and histological analysis of a punch biopsy taken from formerly affected BCC sites.
For immunofluorescence analysis, 4-mm punch biopsies were taken under local anesthesia (1% lidocaine) from untreated BCCs after 2 wk of IMQ treatment, when the clinical signs of inflammation were strongest, and after 6 wk of IMQ therapy at the treatment endpoint. The biopsies were embedded in optimum cutting tissue compound (Tissue-Tek; Sakura Finetek), snap-frozen in liquid nitrogen, and stored at –80°C until further processing.
Studies involving patient material were performed according to the Declaration of Helsinki. Biopsies were routinely taken from every patient for diagnostic purposes, followed by an informed consent about a possible scientific use of the data obtained as recommended by the local ethics committee (Federal Academic Hospital Feldkirch).
Immunofluorescence staining.
Single and multicolor immunofluorescence staining procedures were performed as previously described (56). The mAbs used in this study and their sources are shown in Table I.
Apoptotic cells were detected using a kit from Roche Molecular Biochemicals to visualize DNA breaks as TUNEL+, according to the manufacturer's guidelines. After blocking with normal mouse serum for 20 min to diminish background staining, sections were counterstained with a purified antipancytokeratin mAb overnight at 4°C. Purified antibody binding was visualized by incubating the sections with rhodamine (TRITC)-conjugated AffiniPure F(ab′)2 fragment goat anti–mouse IgG (H+L). The evaluation of immunofluorescence results was performed as previously described (56). In brief, biopsy specimens were read in a blinded fashion by two independent investigators with a mean observer coefficient <10%. Labeled cells were enumerated per visual field and expressed as the number of cells per millimeter/basement membrane epidermis and millimeter2 dermis ± SEM.
Cell preparations from peripheral blood.
Peripheral blood samples were purchased as buffy coats from the Vienna Red Cross Center. DCs were isolated as follows: PBMCs were obtained by Ficoll-Plaque density gradient centrifugation (Histopaque-1077; Sigma-Aldrich) and depleted of T cells, B cells, NK cells, hemopoietic stem cells, monocytes, platelets, and erythrocytes by anti-CD3/CD19/CD56/CD34/CD14/CD16/CD41/CD235a (3 μg/ml each) immunolabeling and anti–mouse IgG immunomagnetic depletion (MACS; Miltenyi Biotec). The remaining cell fraction was then divided into mDCs and pDCs by positive selection using anti-CD1c and, thereafter, anti-CD304 (BDCA-4) microbeads (MACS). The purity of isolated DC populations was 93–99%, as determined by flow cytometric analysis (FACScan; BD Biosciences) with either anti-CD11c/HLA-DR or anti-CD303 (BDCA-2)/HLA-DR stainings. None of the other leukocyte subpopulations tested individually (T cells, B cells, NK cells, and monocytes) accounted for > 1% of the sorted cell population. T cells were prepared as a positive control for the analysis of the perforin and granzyme B release by negative selection from PBMCs using anti-CD11c/CD14/CD16/CD19/CD34/CD41/CD56/CD123/CD235a (3 μg/ml each) immunolabeling and anti–mouse IgG immunomagnetic depletion (MACS). NK cells used as positive controls for cytotoxicity assays were separated by anti-CD56 (3 μg/ml each) immunolabeling and anti–mouse IgG immunomagnetic selection after T cell depletion (MACS). The purity of T cells and NK cells was >99%, as determined by FACS analysis.
DC stimulation with TLR agonists.
Synthetic agonists to TLR7 (3M-001), TLR8 (3M-002), TLR7/8 (3M-003), and an inactive small molecule TLR7/8 analogue that served as a negative control (3M-006) were provided by R.L. Miller (3M Pharmaceuticals, St. Paul, MN). Isolated mDCs and pDCs were washed and resuspended in RPMI 1640 medium (Invitrogen) supplemented with heat-inactivated 10% FCS (Invitrogen) and 1% penicillin/streptomycin (Invitrogen) and cultured with 4 μM 3M-001, 3M-002, 3M-003, or 3M-006 for 12 h at 2 × 106 cells/ml in 0.25 ml in 96-well plates or 0.5 ml in 48-well plates. Viability of the cells was measured with trypan blue and annexin V (BD Biosciences) before and after cultures, according to the manufacturer's instructions. Cells were analyzed by FACS stainings for detection of lytic molecules or used as effector cells in cytotoxicity assays. Media conditioned by DC subsets were collected and stored at −20°C for further analysis.
Flow cytometry.
To analyze lytic molecules on blood-derived DCs, we performed intra- and extracellular FACS stainings of isolated DC populations before and after culture with TLR agonists. After staining of DC populations with anti–BDCA-2 (Miltenyi Biotec) or anti-CD11c (BD Biosciences) mAbs, cells were fixed and permeabilized with a cell permeabilization kit (An Der Grub), according to the manufacturer's instructions for intracellular protein detection. Cells were incubated with antiperforin FITC (BD Biosciences) and anti–granzyme B PE (PeliCluster; Sanquin) antibodies. Surface TRAIL expression was visualized by staining with an anti-TRAIL mAb (R&D Systems) after labeling the purified antibody with a protein labeling kit (FluoReporter Oregon Green A488; Invitrogen), according to the manufacturer's instructions. Purity controls of DC populations before and after culture were performed by triple stainings with antibodies against HLA-DR FITC (BD Biosciences), BDCA-2 PE (Miltenyi Biotec), or CD11c PE (BD Biosciences) and CD3 PerCp, CD14 PerCp, CD19 PerCp, or CD56 PECy5.
After incubation with these mAbs, DCs were washed twice with ice-cold PBS and subjected to flow cytometric analysis using a flow cytometer (FACScan).
Measurement of perforin and granzyme B in the supernatants.
Supernatants were harvested after stimulation of isolated mDCs and pDCs with TLR7, TLR8, or TLR7/8 agonists or vehicle control for 12 h and stored at −20°C. Perforin and granzyme B levels were quantified by ELISA kits (Diaclone Research), according to the manufacturer's instructions. Supernatants of T cells stimulated with 50 ng/ml PMA (Sigma-Aldrich) and 1 μg/ml ionomycin (Sigma-Aldrich) for 12 h served as positive controls.
Cytotoxicity assays.
The ability of DCs to exert cytotoxicity was assessed in a conventional 2-h europium-TDA release assay (PerkinElmer), as previously described (57), according to the manufacture's instructions. Data were expressed as the percentage of cytotoxicity calculated by the following formula: cytotoxicity (%) = (experimental release – spontaneous release)/(maximum release – spontaneous release) × 100.
We used the chronic myelogenous leukemia cell line K562 (provided by C. Wagner, Medical University of Vienna, Vienna, Austria) and the acute T cell leukemia cell line Jurkat (provided by P. Meraner, Medical University of Vienna, Vienna, Austria) as target cells. In a 96-well plate, 5 × 103 target cells per well were incubated with mDCs and pDCs in different effector/target ratios (starting at 25:1) in triplicates. For inhibition of TRAIL- and Fas ligand–dependent lysis, 5 μg/ml of azide-free neutralizing anti-TRAIL (clone 75411; R&D Systems) and 5 μg/ml of anti–Fas ligand (clone NOK-1; BD Biosciences), respectively, were added to effector cells 30 min before the addition of target cells. A 30-min preculture of effector cells with 5 μg/ml of an IgG1 isotype (Sigma-Aldrich) served as negative control. For inhibition of the perforin-based cytotoxicity, effector cells were treated for 120 min with 100 nM concanamycin A (Sigma-Aldrich) before target cells were added.
Online supplemental material.
Fig. S1 shows that mDCs, but not pDCs, display CD83 in IMQ-treated BCC and that perforin/granzyme B–containing T cells occur in lesions of allergic contact dermatitis, but not IMQ-treated BCC. Fig. S2 depicts NK cell–mediated cytotoxicity. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20070021/DC1.
Supplemental Material
Acknowledgments
We thank Richard L. Miller (3M Pharmaceuticals) for providing agonists to TLR7 and TLR8 and Drs. Dieter Maurer and Ethan Shevach for critically reading the manuscript. We would also like to thank Sabine Altrichter for technical advice and Andreas Ebner for preparing the figures.
The authors have no conflicting financial interests.
Abbreviations used: APC, allophycocyanin; BCC, basal cell carcinoma; IMQ, imiquimod; iNOS, inducible NO synthase; mDC, myeloid DC; pDC, plasmacytoid DC; TLR, Toll-like receptor; TRAIL, TNF-related apoptosis-inducing ligand; TUNEL, Tdt-mediated dUTP-biotin nick-end labeling.
T. Kopp and G. Stingl contributed equally to this study.
References
- 1.Weeks, C.E., and S.J. Gibson. 1994. Induction of interferon and other cytokines by imiquimod and its hydroxylated metabolite R-842 in human blood cells in vitro. J. Interferon Res. 14:81–85. [DOI] [PubMed] [Google Scholar]
- 2.Garland, S.M., R. Waddell, A. Mindel, I.M. Denham, and J.C. McCloskey. 2006. An open-label phase II pilot study investigating the optimal duration of imiquimod 5% cream for the treatment of external genital warts in women. Int. J. STD AIDS. 17:448–452. [DOI] [PubMed] [Google Scholar]
- 3.Theos, A.U., R. Cummins, N.B. Silverberg, and A.S. Paller. 2004. Effectiveness of imiquimod cream 5% for treating childhood molluscum contagiosum in a double-blind, randomized pilot trial. Cutis. 74:134–138, 141–142. [PubMed] [Google Scholar]
- 4.Gollnick, H., C.G. Barona, R.G. Frank, T. Ruzicka, M. Megahed, V. Tebbs, M. Owens, and P. Stampone. 2005. Recurrence rate of superficial basal cell carcinoma following successful treatment with imiquimod 5% cream: interim 2-year results from an ongoing 5-year follow-up study in Europe. Eur. J. Dermatol. 15:374–381. [PubMed] [Google Scholar]
- 5.Peris, K., E. Campione, T. Micantonio, G.C. Marulli, M.C. Fargnoli, and S. Chimenti. 2005. Imiquimod treatment of superficial and nodular basal cell carcinoma: 12-week open-label trial. Dermatol. Surg. 31:318–323. [DOI] [PubMed] [Google Scholar]
- 6.Fleming, C.J., A.M. Bryden, A. Evans, R.S. Dawe, and S.H. Ibbotson. 2004. A pilot study of treatment of lentigo maligna with 5% imiquimod cream. Br. J. Dermatol. 151:485–488. [DOI] [PubMed] [Google Scholar]
- 7.Akira, S., and H. Hemmi. 2003. Recognition of pathogen-associated molecular patterns by TLR family. Immunol. Lett. 85:85–95. [DOI] [PubMed] [Google Scholar]
- 8.Diebold, S.S., T. Kaisho, H. Hemmi, S. Akira, and C. Reis e Sousa. 2004. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 303:1529–1531. [DOI] [PubMed] [Google Scholar]
- 9.Heil, F., H. Hemmi, H. Hochrein, F. Ampenberger, C. Kirschning, S. Akira, G. Lipford, H. Wagner, and S. Bauer. 2004. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 303:1526–1529. [DOI] [PubMed] [Google Scholar]
- 10.Medzhitov, R., P. Preston-Hurlburt, and C.A. Janeway Jr. 1997. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 388:394–397. [DOI] [PubMed] [Google Scholar]
- 11.Akira, S., and K. Takeda. 2004. Toll-like receptor signalling. Nat. Rev. Immunol. 4:499–511. [DOI] [PubMed] [Google Scholar]
- 12.Michalopoulos, P., N. Yawalkar, M. Bronnimann, A. Kappeler, and L.R. Braathen. 2004. Characterization of the cellular infiltrate during successful topical treatment of lentigo maligna with imiquimod. Br. J. Dermatol. 151:903–906. [DOI] [PubMed] [Google Scholar]
- 13.Barnetson, R.S., A. Satchell, L. Zhuang, H.B. Slade, and G.M. Halliday. 2004. Imiquimod induced regression of clinically diagnosed superficial basal cell carcinoma is associated with early infiltration by CD4 T cells and dendritic cells. Clin. Exp. Dermatol. 29:639–643. [DOI] [PubMed] [Google Scholar]
- 14.Smith, K.J., S. Hamza, and H. Skelton. 2004. Topical imidazoquinoline therapy of cutaneous squamous cell carcinoma polarizes lymphoid and monocyte/macrophage populations to a Th1 and M1 cytokine pattern. Clin. Exp. Dermatol. 29:505–512. [DOI] [PubMed] [Google Scholar]
- 15.Kadowaki, N., S. Ho, S. Antonenko, R.W. Malefyt, R.A. Kastelein, F. Bazan, and Y.J. Liu. 2001. Subsets of human dendritic cell precursors express different Toll-like receptors and respond to different microbial antigens. J. Exp. Med. 194:863–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jarrossay, D., G. Napolitani, M. Colonna, F. Sallusto, and A. Lanzavecchia. 2001. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur. J. Immunol. 31:3388–3393. [DOI] [PubMed] [Google Scholar]
- 17.Peng, G., Z. Guo, Y. Kiniwa, K.S. Voo, W. Peng, T. Fu, D.Y. Wang, Y. Li, H.Y. Wang, and R.F. Wang. 2005. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 309:1380–1384. [DOI] [PubMed] [Google Scholar]
- 18.Schön, M.P., M. Schön, and K.N. Klotz. 2006. The small antitumoral immune response modifier imiquimod interacts with adenosine receptor signaling in a TLR7- and TLR8-independent fashion. J. Invest. Dermatol. 126:1338–1347. [DOI] [PubMed] [Google Scholar]
- 19.Schön, M., A.B. Bong, C. Drewniok, J. Herz, C.C. Geilen, J. Reifenberger, B. Benninghoff, H.B. Slade, H. Gollnick, and M.P. Schön. 2003. Tumor-selective induction of apoptosis and the small-molecule immune response modifier imiquimod. J. Natl. Cancer Inst. 95:1138–1149. [DOI] [PubMed] [Google Scholar]
- 20.Grebenova, D., K. Kuzelova, O. Fuchs, P. Halada, V. Havlicek, I. Marinov, and Z. Hrkal. 2004. Interferon-alpha suppresses proliferation of chronic myelogenous leukemia cells K562 by extending cell cycle S-phase without inducing apoptosis. Blood Cells Mol. Dis. 32:262–269. [DOI] [PubMed] [Google Scholar]
- 21.Majewski, S., M. Marczak, B. Mlynarczyk, B. Benninghoff, and S. Jablonska. 2005. Imiquimod is a strong inhibitor of tumor cell-induced angiogenesis. Int. J. Dermatol. 44:14–19. [DOI] [PubMed] [Google Scholar]
- 22.Li, V.W., W.W. Li, K.E. Talcott, and A.W. Zhai. 2005. Imiquimod as an antiangiogenic agent. J. Drugs Dermatol. 4:708–717. [PubMed] [Google Scholar]
- 23.Palamara, F., S. Meindl, M. Holcmann, P. Lührs, G. Stingl, and M. Sibilia. 2004. Identification and characterization of pDC-like cells in normal mouse skin and melanomas treated with imiquimod. J. Immunol. 173:3051–3061. [DOI] [PubMed] [Google Scholar]
- 24.Fanger, N.A., C.R. Maliszewski, K. Schooley, and T.S. Griffith. 1999. Human dendritic cells mediate cellular apoptosis via tumor necrosis factor–related apoptosis-inducing ligand (TRAIL). J. Exp. Med. 190:1155–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chaperot, L., A. Blum, O. Manches, G. Lui, J. Angel, J.P. Molens, and J. Plumas. 2006. Virus or TLR agonists induce TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. J. Immunol. 176:248–255. [DOI] [PubMed] [Google Scholar]
- 26.Tucker, S.B., J.W. Polasek, A.J. Perri, and E.A. Goldsmith. 2006. Long-term follow-up of basal cell carcinomas treated with perilesional interferon alfa 2b as monotherapy. J. Am. Acad. Dermatol. 54:1033–1038. [DOI] [PubMed] [Google Scholar]
- 27.Thyrell, L., S. Erickson, B. Zhivotovsky, K. Pokrovskaja, O. Sangfelt, J. Castro, S. Einhorn, and D. Grander. 2002. Mechanisms of Interferon-alpha induced apoptosis in malignant cells. Oncogene. 21:1251–1262. [DOI] [PubMed] [Google Scholar]
- 28.Lowes, M.A., F. Chamian, M.V. Abello, J. Fuentes-Duculan, S.L. Lin, R. Nussbaum, I. Novitskaya, H. Carbonaro, I. Cardinale, T. Kikuchi, et al. 2005. Increase in TNF-alpha and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a). Proc. Natl. Acad. Sci. USA. 102:19057–19062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Serbina, N.V., T.P. Salazar-Mather, C.A. Biron, W.A. Kuziel, and E.G.P. Am. 2003. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 19:59–70. [DOI] [PubMed] [Google Scholar]
- 30.Yawalkar, N., R.E. Hunger, C. Buri, S. Schmid, F. Egli, C.U. Brand, C. Mueller, W.J. Pichler, and L.R. Braathen. 2001. A comparative study of the expression of cytotoxic proteins in allergic contact dermatitis and psoriasis: spongiotic skin lesions in allergic contact dermatitis are highly infiltrated by T cells expressing perforin and granzyme B. Am. J. Pathol. 158:803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zauli, G., S. Sancilio, A. Cataldi, N. Sabatini, D. Bosco, and R. Di Pietro. 2005. PI-3K/Akt and NF-kappaB/IkappaBalpha pathways are activated in Jurkat T cells in response to TRAIL treatment. J. Cell. Physiol. 202:900–911. [DOI] [PubMed] [Google Scholar]
- 32.Kataoka, T., N. Shinohara, H. Takayama, K. Takaku, S. Kondo, S. Yonehara, and K. Nagai. 1996. Concanamycin A, a powerful tool for characterization and estimation of contribution of perforin- and Fas-based lytic pathways in cell-mediated cytotoxicity. J. Immunol. 156:3678–3686. [PubMed] [Google Scholar]
- 33.Korman, N., R. Moy, M. Ling, R. Matheson, S. Smith, S. McKane, and J.H. Lee. 2005. Dosing with 5% imiquimod cream 3 times per week for the treatment of actinic keratosis: results of two phase 3, randomized, double-blind, parallel-group, vehicle-controlled trials. Arch. Dermatol. 141:467–473. [DOI] [PubMed] [Google Scholar]
- 34.Nikkels, A.F., C. Pierard-Franchimont, N. Nikkels-Tassoudji, R. Bourguignon, and G.E. Pierard. 2005. Photodynamic therapy and imiquimod immunotherapy for basal cell carcinomas. Acta Clin. Belg. 60:227–234. [DOI] [PubMed] [Google Scholar]
- 35.Dixon, A.J. 2005. Multiple superficial basal cell carcinomata–topical imiquimod versus curette and cryotherapy. Aust. Fam. Physician. 34:49–52. [PubMed] [Google Scholar]
- 36.Sidky, Y.A., E.C. Borden, C.E. Weeks, M.J. Reiter, J.F. Hatcher, and G.T. Bryan. 1992. Inhibition of murine tumor growth by an interferon-inducing imidazoquinolinamine. Cancer Res. 52:3528–3533. [PubMed] [Google Scholar]
- 37.Sullivan, T.P., T. Dearaujo, V. Vincek, and B. Berman. 2003. Evaluation of superficial basal cell carcinomas after treatment with imiquimod 5% cream or vehicle for apoptosis and lymphocyte phenotyping. Dermatol. Surg. 29:1181–1186. [DOI] [PubMed] [Google Scholar]
- 38.Wiley, S.R., K. Schooley, P.J. Smolak, W.S. Din, C.P. Huang, J.K. Nicholl, G.R. Sutherland, T.D. Smith, C. Rauch, C.A. Smith, et al. 1995. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 3:673–682. [DOI] [PubMed] [Google Scholar]
- 39.Schneider, P., M. Thome, K. Burns, J.L. Bodmer, K. Hofmann, T. Kataoka, N. Holler, and J. Tschopp. 1997. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity. 7:831–836. [DOI] [PubMed] [Google Scholar]
- 40.Wang, S., and W.S. El-Deiry. 2003. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 22:8628–8633. [DOI] [PubMed] [Google Scholar]
- 41.Xu, W., L. Liu, and I.G. Charles. 2002. Microencapsulated iNOS-expressing cells cause tumor suppression in mice. FASEB J. 16:213–215. [DOI] [PubMed] [Google Scholar]
- 42.Mocellin, S., M. Provenzano, M. Lise, D. Nitti, and C.R. Rossi. 2003. Increased TIA-1 gene expression in the tumor microenvironment after locoregional administration of tumor necrosis factor-alpha to patients with soft tissue limb sarcoma. Int. J. Cancer. 107:317–322. [DOI] [PubMed] [Google Scholar]
- 43.Serbina, N.V., and E.G.P. Am. 2006. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 7:311–317. [DOI] [PubMed] [Google Scholar]
- 44.Janjic, B.M., G. Lu, A. Pimenov, T.L. Whiteside, W.J. Storkus, and N.L. Vujanovic. 2002. Innate direct anticancer effector function of human immature dendritic cells. I. Involvement of an apoptosis-inducing pathway. J. Immunol. 168:1823–1830. [DOI] [PubMed] [Google Scholar]
- 45.Schmitz, M., S. Zhao, Y. Deuse, K. Schakel, R. Wehner, H. Wohner, K. Holig, F. Wienforth, A. Kiessling, M. Bornhauser, et al. 2005. Tumoricidal potential of native blood dendritic cells: direct tumor cell killing and activation of NK cell-mediated cytotoxicity. J. Immunol. 174:4127–4134. [DOI] [PubMed] [Google Scholar]
- 46.Lu, G., B.M. Janjic, J. Janjic, T.L. Whiteside, W.J. Storkus, and N.L. Vujanovic. 2002. Innate direct anticancer effector function of human immature dendritic cells. II. Role of TNF, lymphotoxin-alpha(1)beta(2), Fas ligand, and TNF-related apoptosis-inducing ligand. J. Immunol. 168:1831–1839. [DOI] [PubMed] [Google Scholar]
- 47.Vanderheyde, N., E. Aksoy, Z. Amraoui, P. Vandenabeele, M. Goldman, and F. Willems. 2001. Tumoricidal activity of monocyte- derived dendritic cells: evidence for a caspase-8-dependent, Fas-associated death domain-independent mechanism. J. Immunol. 167:3565–3569. [DOI] [PubMed] [Google Scholar]
- 48.Trinite, B., C. Chauvin, H. Peche, C. Voisine, M. Heslan, and R. Josien. 2005. Immature CD4- CD103+ rat dendritic cells induce rapid caspase-independent apoptosis-like cell death in various tumor and nontumor cells and phagocytose their victims. J. Immunol. 175:2408–2417. [DOI] [PubMed] [Google Scholar]
- 49.Urosevic, M., R. Dummer, C. Conrad, M. Beyeler, E. Laine, G. Burg, and M. Gilliet. 2005. Disease-independent skin recruitment and activation of plasmacytoid predendritic cells following imiquimod treatment. J. Natl. Cancer Inst. 97:1143–1153. [DOI] [PubMed] [Google Scholar]
- 50.Sato, K., S. Hida, H. Takayanagi, T. Yokochi, N. Kayagaki, K. Takeda, H. Yagita, K. Okumura, N. Tanaka, T. Taniguchi, and K. Ogasawara. 2001. Antiviral response by natural killer cells through TRAIL gene induction by IFN-alpha/beta. Eur. J. Immunol. 31:3138–3146. [DOI] [PubMed] [Google Scholar]
- 51.Kayagaki, N., N. Yamaguchi, M. Nakayama, H. Eto, K. Okumura, and H. Yagita. 1999. Type I interferons (IFNs) regulate tumor necrosis factor– related apoptosis-inducing ligand (TRAIL) expression on human T cells: a novel mechanism for the antitumor effects of type I IFNs. J. Exp. Med. 189:1451–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rissoan, M.C., T. Duhen, J.M. Bridon, N. Bendriss-Vermare, C. Peronne, B. de Saint Vis, F. Briere, and E.E. Bates. 2002. Subtractive hybridization reveals the expression of immunoglobulin-like transcript 7, Eph-B1, granzyme B, and 3 novel transcripts in human plasmacytoid dendritic cells. Blood. 100:3295–3303. [DOI] [PubMed] [Google Scholar]
- 53.Santoro, A., A. Majorana, L. Roversi, F. Gentili, S. Marrelli, W. Vermi, E. Bardellini, P. Sapelli, and F. Facchetti. 2005. Recruitment of dendritic cells in oral lichen planus. J. Pathol. 205:426–434. [DOI] [PubMed] [Google Scholar]
- 54.Taieb, J., N. Chaput, C. Menard, L. Apetoh, E. Ullrich, M. Bonmort, M. Pequignot, N. Casares, M. Terme, C. Flament, et al. 2006. A novel dendritic cell subset involved in tumor immunosurveillance. Nat. Med. 12:214–219. [DOI] [PubMed] [Google Scholar]
- 55.Chan, C.W., E. Crafton, H.N. Fan, J. Flook, K. Yoshimura, M. Skarica, D. Brockstedt, T.W. Dubensky, M.F. Stins, L.L. Lanier, et al. 2006. Interferon-producing killer dendritic cells provide a link between innate and adaptive immunity. Nat. Med. 12:207–213. [DOI] [PubMed] [Google Scholar]
- 56.Stary, G., C. Bangert, G. Stingl, and T. Kopp. 2005. Dendritic cells in atopic dermatitis: expression of FcepsilonRI on two distinct inflammation-associated subsets. Int. Arch. Allergy Immunol. 138:278–290. [DOI] [PubMed] [Google Scholar]
- 57.Blomberg, K., R. Hautala, J. Lovgren, V.M. Mukkala, C. Lindqvist, and K. Akerman. 1996. Time-resolved fluorometric assay for natural killer activity using target cells labelled with a fluorescence enhancing ligand. J. Immunol. Methods. 193:199–206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}