

Abstract
Tumor suppressor p53-dependent apoptosis is critical in suppressing tumorigenesis. Previously, we reported that DNA double-strand breaks (DSBs) at the V(D)J recombination loci induced genomic instability in the developing lymphocytes of nonhomologous end-joining (NHEJ)–deficient, p53-deficient mice, which led to rapid lymphomagenesis. To test the ability of p53-dependent cell cycle arrest to suppress tumorigenesis in the absence of apoptosis in vivo, we crossbred NHEJ-deficient mice into a mutant p53R172P background; these mice have defects in apoptosis induction, but not cell cycle arrest. These double-mutant mice survived longer than NHEJ/p53 double-null mice and, remarkably, were completely tumor free. We detected accumulation of aberrant V(D)J recombination–related DSBs at the T cell receptor (TCR) locus, and high expression levels of both mutant p53 and cell cycle checkpoint protein p21, but not the apoptotic protein p53-upregulated modulator of apoptosis. In addition, a substantial number of senescent cells were observed among both thymocytes and bone marrow cells. Cytogenetic studies revealed euploidy and limited chromosomal breaks in these lymphoid cells. The results indicate that precursor lymphocytes, which normally possess a high proliferation potential, are able to withdraw from the cell cycle and undergo senescence in response to the persistence of DSBs in a p53–p21–dependent pathway; this is sufficient to inhibit oncogenic chromosomal abnormality and suppress tumorigenesis.
The tumor suppressor p53 is one of the most extensively studied proteins (1, 2). At the cellular level, p53 acts as a “gatekeeper” to eliminate potentially tumorigenic cells by inducing programmed cell death, inhibiting the proliferation of damaged cells through cell cycle arrest (3), or inducing cells to withdraw from the cell cycle, referred to as cellular senescence (4, 5). The ability of p53 to induce apoptosis is considered the primary means by which p53 suppresses tumor growth (6–9). However, experiments have shown that p53 deficiency does not necessarily translate into an interruption in the apoptotic response (10, 11). We hypothesize that the ability of p53 to induce cell cycle arrest or senescence, in some cases, could play a central role in tumor suppression.
At the molecular level, activated p53 functions as a transcription factor by transactivating genes that are important for inducing apoptosis or cell cycle arrest and senescence (12, 13). Therefore, gene expression profiles can be used as markers of apoptosis or senescence. The levels of proapoptotic proteins, such as p53-upregulated modulator of apoptosis (PUMA), are elevated in cells programmed to undergo apoptosis (14, 15), whereas the cyclin-dependent kinase (CDK) inhibitor p21, a well-studied downstream target of p53, is highly expressed in cells undergoing cell cycle arrest and senescence. p21 directly inhibits CDK2 and plays a pivotal role in p53-induced cell cycle arrest (16). It is also an important molecule in one of the mechanisms leading to senescence (17).
The term senescence refers to the finite replicative lifespan of cultured human cells that enter a stable growth-arrested state (18). Currently, senescence is recognized as the final phenotypic state adopted by a cell in response to various stimuli. Senescent cells show a characteristic flattened and enlarged morphology and are positive for acidic β-galactosidase (β-gal) activity (19). Senescent cells also undergo dramatic chromatin remodeling and express proteins that are characteristic of transcriptionally silent heterochromatin. An example of these proteins is the heterochromatin protein 1 (HP1) family member HP1γ, which has been established as a marker for senescence-associated heterochromatin foci (SAHF) (20) in human (21) and murine cells (22). Numerous studies suggest that a major cause of senescence in human cells is telomeric shortening (23), which triggers cellular responses that are identical in many aspects to a DNA damage response (24, 25). Additionally, DNA damage could be considered an extrinsic factor contributing to cellular senescence. Because of its essential property of cell growth arrest, senescence is proposed as a potent tumor suppressor mechanism (26).
DNA damage occurs in the developing lymphocytes when these cells begin to rearrange antigen receptor genes in a process termed V(D)J recombination. This genetically programmed DNA rearrangement process involves the cleavage of DNA double-strands at the recombination signal sequences by the Rag nucleases, which generate two kinds of DNA breaks—signal ends and coding ends (27). Signal ends are blunt and 5′-phosphorylated, whereas coding ends are covalently sealed to form a hairpin structure requiring further processing by the DNA-dependent protein kinase (DNA–PK) and the repair factor Artemis complex before ligation (28). Both signal and coding ends are joined by the nonhomologous end-joining (NHEJ), which is a major DNA repair pathway that is generally responsible for the repair of DNA double-strand breaks (DSBs). The participating factors in this pathway are the DNA–PK complex, which includes the DNA-binding subunits Ku70 and Ku80 and the catalytic subunit of DNA–PK, the repair factor Artemis, and the DNA ligase complex, which is composed of DNA ligase IV, XRCC4, and XLF/Cernunnos (29, 30). All of the NHEJ factors are essential for V(D)J recombination; a severe combined immunodeficient phenotype results from impaired V(D)J recombination in mice deficient in each of the joining factors (31, 32).
In NHEJ-deficient mice, Rag-mediated DNA breaks accumulate because the breaks cannot be joined by other nonspecific end-joining activity. However, the stalled V(D)J recombination breaks induce a p53-dependent apoptosis in developing lymphoid cells. Therefore, NHEJ-deficient mice do not usually develop early lymphomas. In contrast, Rag-mediated DSBs lead to massive genomic instability in the absence of both p53 and NHEJ activity, including chromosomal translocations and oncogenic gene amplifications (33, 34). As a result, double-null mice succumb to early pro-B lymphomas with chromosomal translocation at chromosome 12 at the immunoglobulin locus and chromosome 15 near the protooncogene c-Myc. The involvement of Rag nucleases in tumorigenesis has been confirmed by the lack of lymphoma development in Rag, NHEJ, and p53 triple-deficient mice (35, 36).
A unique p53 mutation, p53R175P (the human equivalent of murine R172P), which was originally identified from human tumor samples (37, 38), provides an excellent model to study the role of p53-dependent cell cycle arrest and senescence in tumor suppression. Although p53R175P lacks the ability to induce apoptosis in response to various stimuli, it retains its partial ability to induce cell cycle arrest through its ability to transactivate key molecules such as the CDK inhibitor p21 (10, 37). In this study, we crossed NHEJ-deficient mice into a p53R172P mutant background to establish an in vivo model to examine the involvement of the p53–p21 pathway in the induction of cell cycle arrest and senescence in response to DNA damage and the consequent suppression of genomic instability and tumorigenesis.
RESULTS
Mutant p53R172P rescues embryonic lethality caused by DNA ligase IV deficiency and suppresses tumorigenesis
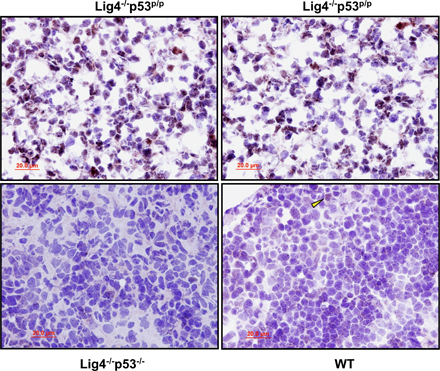
To determine whether the p53R172P mutant was sufficient to suppress tumorigenesis, we crossed heterozygous DNA ligase IV–deficient mice (Lig4+/−) with p53R172P/R172P mice (p53p/p) to generate Lig4+/− p53+/p mice. These animals were subsequently interbred to generate Lig4+/− p53p/p mice, which were viable and fertile. Next, Lig4−/− p53p/p mice were generated from crossing Lig4+/− p53p/p mice. 24% (67 out of 279) of pups from the Lig4+/− p53p/p breeding were genotyped as Lig4−/− p53p/p (Table S1, available at http://www.jem.org/cgi/content/full/jem.20062453/DC1). This number was very close to the expected Mendelian segregation ratio. This indicates that the mutant p53R172P successfully rescued the embryonic lethality caused by the DNA ligase IV deficiency, and the embryonic lethality of Lig4−/− was caused by p53-dependent apoptosis (39, 40). Similar to Lig4−/− p53−/− mice (Fig. S1), Lig4−/− p53p/p mice were smaller than their Lig4+/− or Lig4+/+ littermates (Fig. 1 A). 6 out of 25 Lig4−/− p53p/p mice died shortly after weaning. These mice had difficulty ingesting solid food, and the necropsy of a 4-wk-old Lig4−/− p53p/p mouse revealed the cause of death was starvation and not tumor-related (unpublished data). After the malnourishment problem was corrected by simply providing soft food, all Lig4−/− p53p/p mice survived far beyond the weaning age and significantly longer than the Lig4−/− p53−/− mice (Fig. 1 B; P < 0.001, log rank test). The median survival age of Lig4−/− p53p/p mice was ∼120 d, compared with 65 d for the Lig4−/− p53−/− mice. Of particular note, 2 of the mice survived beyond 6 mo of age (Fig. 1 B), which was never observed in Lig4−/− p53−/− mice. Remarkably, Lig4−/− p53p/p mice were free of lymphomas and any other types of tumor, whereas 14 out of 15 Lig4−/− p53−/− mice died of pro-B lymphomas (Fig. 1, B and C; one Lig4−/− p53−/− mouse died around the weaning age with enlarged incisor teeth). These results clearly demonstrated that the p53R172P mutation successfully rescued the embryonic lethality of Lig4-deficient mice and suppressed the development of lymphomas in Lig4−/− p53p/p mice.
Figure 1.

Phenotype, survival curve, and lymphoma incidence. (A) Lig4−/− p53p/p (top) and Lig4+/− p53p/p (bottom) mice. (B) Survival curve of Lig4−/− p53p/p mice (black) and Lig4−/− p53−/− mice (red). Lig4−/− p53p/p survival is significantly different from Lig4−/− p53−/− mice (P = 0.000204 by log rank test). (C) Lymphoma incidence in Lig4−/− p53p/p and Lig4−/− p53−/− mice.
In the absence of apoptosis, Lig4−/− p53p/p precursor lymphocytes enter senescence
To confirm the defective apoptosis in p53R172P mice, we performed a TUNEL assay and a hematoxylin and eosin (HE) staining of thymic sections from Lig4−/− p53p/p, Lig4−/− p53−/−, and NHEJ-deficient mice (Artemis−/−). As predicted, apoptosis was not observed in thymic sections from Lig4−/− p53−/− or Lig4−/− p53p/p mice, whereas apoptosis was readily detected in thymic sections from Artemis−/− mice (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20062453/DC1). These results confirmed that Rag-mediated DNA DSBs induce a p53-dependent apoptosis in NHEJ-deficient mice, but is completely absent in the p53R172P mutant background. HE staining of thymic sections from Lig4−/− p53p/p mice showed decreased cellular density compared with wild-type (WT) and Lig4−/− p53−/− mice (Fig. S3), but the density was similar to that of Artemis−/− mice. In addition, the cellularity of thymus from a 4-wk-old Lig4−/− p53p/p mouse is only 1.3 million cells, compared with 202 million cells from a Lig4+/+ p53p/p littermate. Analysis by flow cytometry indicated a severe developmental blockage in both thymocytes and bone marrow cells at early stages (Fig. S4). This confirms that the mutant p53R172P is not capable of rescuing V(D)J recombination, but is able to inhibit tumorigenesis in developing lymphoid cells, possibly by responding to the V(D)J recombination– associated breaks and inhibiting genomic instability.
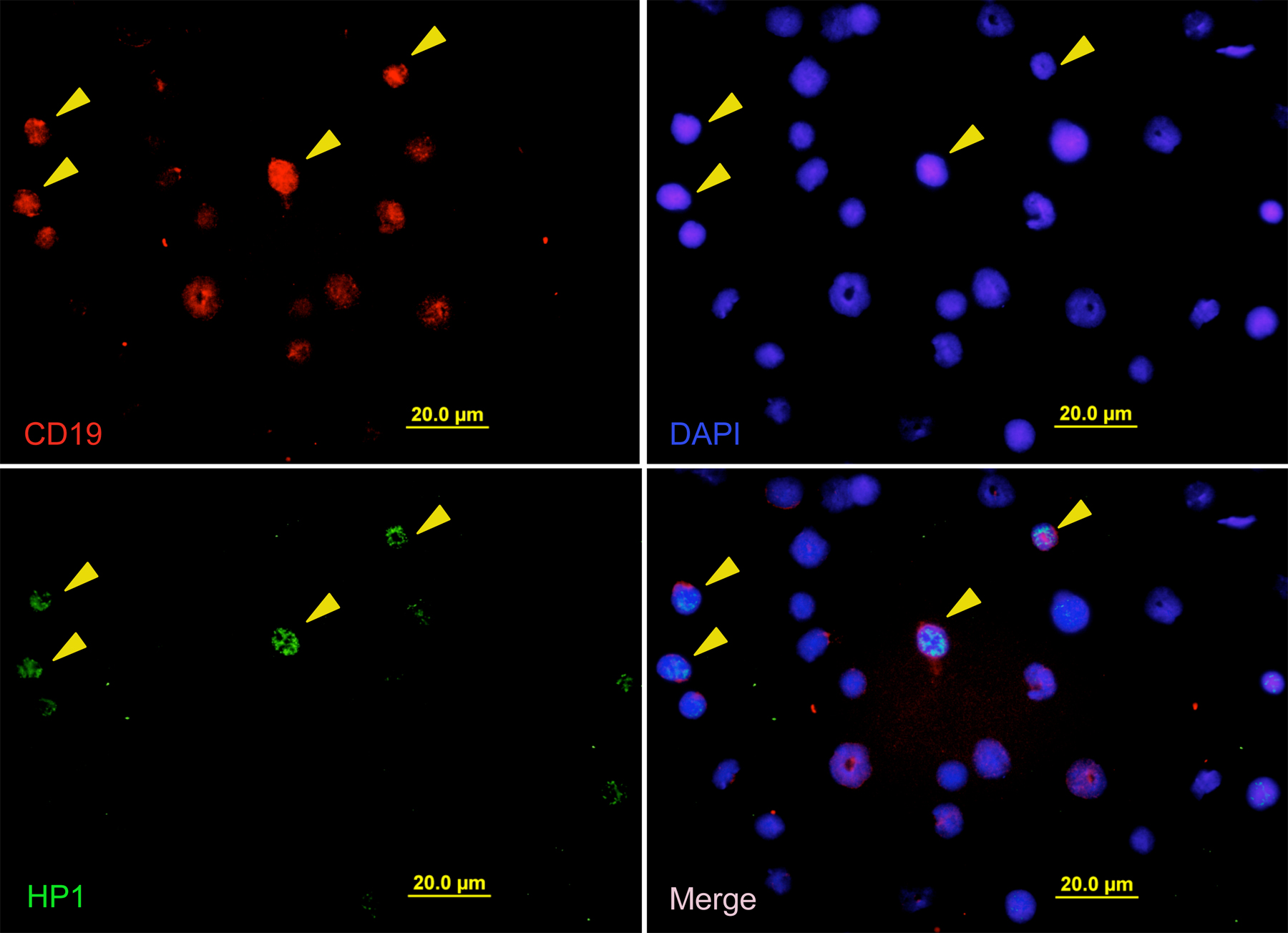
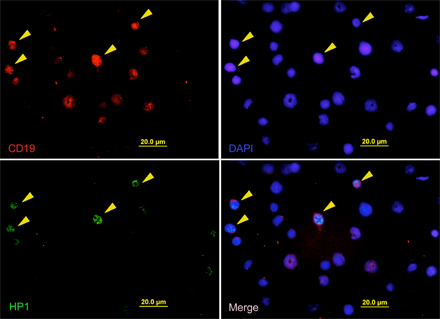
Given that cellular senescence is proposed to be an anticancer mechanism (4), we thought the Lig4−/− p53p/p mice served as an excellent in vivo model for investigating this potential antitumor activity. To determine whether the mutant p53R172P protein was able to induce senescence in response to Rag-mediated DNA breaks, lymphoid cells from Lig4−/− p53p/p mice were examined using the senescence-associated acidic β-gal assay. β-gal staining of thymic sections revealed a large number of Lig4−/− p53p/p thymocytes had entered a senescent state, whereas no β-gal–positive thymocytes were observed in Lig4+/+ p53p/p littermates or age-matched Lig4−/− p53−/− mice (Fig. 2 A). Because Lig4−/− p53−/− mice succumb to pro-B lymphomas at an early age (35), we examined the extent to which cellular senescence occurs in developing B lymphocytes. Bone marrow cells were isolated and stained for β-gal activity. As shown in Fig. 2 B, bone marrow cells from Lig4−/− p53p/p, but not cells from Lig4+/+ p53p/p littermates or age-matched Lig4−/− p53−/− mice, were stained positive for β-gal. Furthermore, similar results were obtained when thymic sections (Fig. 2 C) and bone marrow cells (Fig. 2 D) were stained with HP1γ, which is a different marker for senescence. To verify that these β-gal–positive bone marrow cells were precursor B cells, the cells were also stained with the anti-CD19 surface marker. The results revealed that most of the β-gal–positive cells were, indeed, CD19 positive (Fig. 2 E), thereby confirming their B cell lineage. Similarly, HP1γ-positive cells in the bone marrow and thymus of Lig4−/− p53p/p were also stained positive for the surface markers CD19 and CD25, respectively (Fig. 2, F and G; and Figs. S5 and S6, available at http://www.jem.org/cgi/content/full/jem.20062453/DC1) and HP1γ staining was not detected in Lig4−/− p53−/− thymocytes (Fig. S5). These results indicate that both Lig4−/− p53p/p T and B precursor lymphoid cells undergo senescence in a p53-dependent pathway. This strongly suggests that unrepaired Rag-mediated DSBs are able to activate p53R172P and that its residual activity is sufficient to induce senescence, which successfully blocks tumorigenesis in the lymphoid precursor cells.
Figure 2.

Developing lymphoid cells undergo senescence. β-gal staining for thymic sections (A) and bone marrow cells (B) of Lig4−/− p53p/p, Lig4−/− p53−/−, and Lig4+/+ p53p/p mice showing positive staining only in Lig4−/− p53p/p mice. Anti-HP1γ staining of thymic sections (C) and bone marrow cells (D) of Lig4−/− p53p/p, Lig4−/− p53−/−, and Lig4+/+ p53p/p mice; positive staining was observed only in Lig4−/− p53p/p mice. (E) Bone marrow cells from Lig4−/− p53p/p mice, positive for β-gal staining, were costained with the B cell marker CD19 (dark brown cells); arrows show double-positive cells and an example of bone marrow cells from Lig4−/− p53p/p mice that are positively stained for CD19 and HP1γ (F), showing single channel of green fluorescence (HP1), red (CD19) and blue (DAPI), and merged image; (G) thymocytes staining positive for HP1γ (green), CD25 (red), and DAPI (blue). Images with wider views of both stainings are shown in Figs. S5 and S6, available at http://www.jem.org/cgi/content/full/jem.20062453/DC1.
Accumulation of aberrant V(D)J recombination breaks in Lig4−/− p53p/p thymocytes
Cleavage of DNA by the Rag nucleases generates blunt signal ends and hairpin coding ends. Signal ends are protected in the postcleavage complex and are persistent in WT lymphoid precursor cells (41), whereas hairpin coding ends are usually resolved rapidly. As a result, coding ends never accumulate in WT mice. However, hairpin coding ends were previously detected in the thymocytes of DNA–PKcs–deficient, Artemis-deficient, and Ku80-deficient mice (42–44) because of a defect in processing and opening the hairpins. Because DNA ligase IV functions in the last step of V(D)J recombination end joining, we tested whether coding ends, either hairpin or open-end forms, were present in the Lig4−/− p53p/p lymphoid cells. Coding ends have been previously detected by the ligation-mediated PCR (LMPCR) assay (45, 46), which is the same methodology used in this study. Our LMPCR assay revealed that signal ends were clearly present at the TCR Dδ2 site in WT and Lig4−/− p53p/p samples (Fig. 3, A and B). ApaLI digestion cut the perfect signal ends and reduced the fragment size from 123 bp to 98 bp (Fig. 3 B, left). However, in Lig4−/− p53p/p, but not in WT thymocytes, DNA breaks in addition to the signal ends were clearly detected. These were potential coding ends. This was further demonstrated by the increased intensity of bands in those DNA samples treated with DNA polymerase or mung bean nuclease. Because LMPCR assays can only detect DNA breaks with open blunt termini, treatment with DNA polymerase (which does not open hairpin coding ends) converts those ends with single-strand overhangs to blunt ends. On the other hand, mung bean nuclease opens the hairpin ends, thus allowing the detection of these sealed ends. Our results revealed that both treatments enhanced the extent to which potential coding ends were detected, suggesting the presence of coding ends with a single-strand overhang and hairpin ends. This is the first report of the direct detection of processed (open) coding ends in the mouse, mechanistically implicating a stepwise processing of hairpin coding ends by the DNA–PKcs–Artemis complex.
Figure 3.
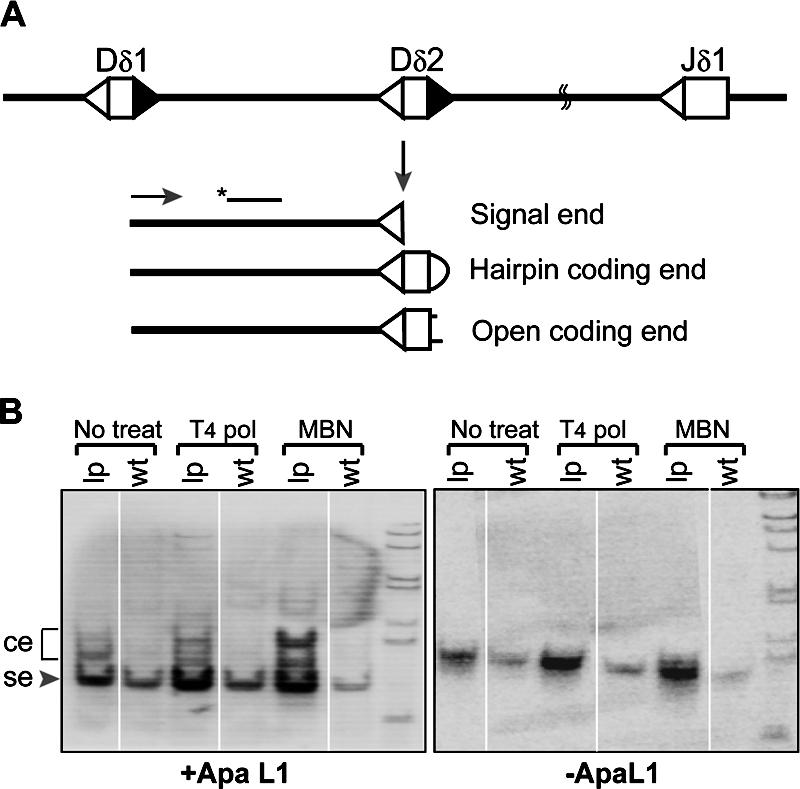
Accumulation of open coding ends in Lig4−/− p53p/p thymocytes. (A) Diagram of V(D)J recombination activity at the TCRδ locus. Cleavage by Rag nucleases generates a blunt signal end (se) and hairpin coding end (ce), which can be further processed by the DNA–PK–Artemis complex. (B) LMPCR assay of WT and Lig4−/− p53p/p (LP) thymocytes, signal ends can be digested by ApaLI to reduce the PCR products from 123 to 98 bp. The residual species are coding ends, which was confirmed by cloning and sequencing (Fig. S6). MBN, mung bean nuclease; T4 Pol, T4 DNA polymerase. Fig. S6 is available at http://www.jem.org/cgi/content/full/jem.20062453/DC1.
To confirm that these ends were coding ends, we cloned and sequenced the PCR products that were resistant to ApaLI digestion. Three individual PCRs of the DNA polymerase-treated samples were performed, and the products were cloned into Escherichia coli and screened for ApaLI-resistant colonies. Sequencing analyses revealed that these were indeed coding ends. We found perfect coding ends containing the entire Dδ2 coding sequences, as well as coding ends with 3–6 nucleotide deletions (Fig. S7, available at http://www.jem.org/cgi/content/full/jem.20062453/DC1). Our results indicate that in Lig4 deficiency, Rag-liberated hairpin coding ends cannot be joined, but they can still be processed by the DNA–PK and Artemis complex. However, whereas aberrant ends normally induce a p53-dependent DNA damage response that leads to apoptosis, these ends instead induce mutant p53 response in Lig4−/− p53p/p mice that drives cells with these aberrant ends to undergo cell cycle arrest and become senescent.
Euploid profile of Lig4−/− p53p/p cells with limited chromosomal breaks
Our previous study conducted in NHEJ/p53−/− mice indicated that Rag-mediated DSBs formed during V(D)J recombination and, if not properly controlled during the cell cycle, lead to genomic instability and tumorigenesis (35). Because Lig4−/− p53p/p mice do not succumb to lymphomas, we compared the extent of genomic instability in precursor lymphoid cells from these two genotypic backgrounds. To ensure a fair comparison, liver cells from newborn mice of both genotypes before the onset of tumors were examined. 50 metaphases were analyzed in cells of each genotype; typical metaphase spreads with telomeric probes are shown in Fig. 4 (A and B). Each chromosomal spread was examined using confocal microscopy to identify telomeric signals. A chromosomal arm without a telomeric signal was scored as a chromatid break (47). Most of the Lig4−/− p53p/p metaphases contained one or two chromatid breaks, and the majority of metaphases were euploid (46 out of 50 metaphases had the normal 40 chromosomes). In contrast, Lig4−/− p53−/− metaphase spreads predominantly had one or two chromosomal breaks, and only 7 out of 50 metaphases (14%) had a normal number of chromosomes (Fig. 4, C and D). These results suggest that aberrant V(D)J recombination breaks result in chromatid breaks in cells of each genotype. However, the effects of these breaks differ between the two different genotypes; in the absence of the tumor suppressor p53, these breaks lead to genomic instability and the activation of oncogenes, possibly via abnormal chromosomal replication related to aneuploidy. In contrast, the mutant p53R172P induces cell cycle arrest and senescence, which is sufficient to block genomic instability and inhibit tumorigenesis. These results underscore the important role of abnormal chromosomal/centromere duplication in tumorigenesis by inducing chromosomal translocation and amplification under the effect of p53 deficiency.
Figure 4.

Analysis of genomic instability in mouse fetal liver cells. Typical chromosomal spreads from Lig4−/− p53p/p (A) and Lig4−/− p53−/− (B) newborn liver cells. (C) Abnormal chromosomal count was observed in a total of 50 metaphases from 3 different Lig4−/− p53−/− (red column) and 9 different Lig4−/− p53p/p mice (blue column); and (D) chromosomal breaks in Lig4−/− p53−/− cells (red column) and Lig4−/− p53p/p mice (blue column). Numbers in A and B represent chromosome counts only. The inset in A shows a typical normal chromosome with 4 telomeric signals and a chromosome with a missing telomeric signal.
DNA damage response in Lig4−/− p53p/p thymocytes: stabilized mutant p53 and activation of p21 protein leads to cell cycle arrest and senescence
To understand the p53 activation occurring in Lig4−/− p53p/p mice, thymocytes were harvested and examined for the expression of the mutant p53 by Western blot analysis. A significant increase in the p53R172P protein level was observed in Lig4−/− p53p/p thymocytes (Fig. 5, lane 2). This p53 level was comparable to that observed in Lig4+/+ p53p/p and WT thymocytes after irradiation (Fig. 5, lanes 3 and 5), indicating that p53R172P could be activated and stabilized in the same way as WT p53 in response to DNA damage. The expression of p53 was not increased in nonirradiated Lig4+/+ p53p/p thymocytes (Fig. 5, lane 4), suggesting that V(D)J recombination intermediates, which persist only in thymocytes of Lig4−/− p53p/p (Fig. 3 B), induced an elevation in mutant p53 levels. The elevated p53 protein level in the Lig4−/− p53p/p thymocytes was also confirmed by immunohistochemistry (Fig. S8, available at http://www.jem.org/cgi/content/full/jem.20062453/DC1). We conclude from our findings that the p53R172P is able to respond to DNA damage, including V(D)J recombination-associated DSBs, and become activated similarly to the WT p53, despite the mutation at R172P.
Figure 5.

Induction of p53, p21, and PUMA proteins in response to DSBs. p53p/p and WT thymocytes exposed to IR (5 Gy), and nonirradiated Lig4−/− p53−/− and Lig4−/− p53p/p thymocytes were analyzed by Western blotting, using antibodies against p53 and β-actin and p21, PUMA, and β-actin.
Next, we assessed whether stabilization of the mutant p53 could activate the normal p53 downstream effectors. First, we examined the level of p21 expression, which is a key factor in p53-dependent cell cycle arrest and senescence. Western blot analysis revealed that p21 was drastically elevated in Lig4−/− p53p/p thymocytes (Fig. 5, lane 2); this was also clearly observed in the results of immunohistochemistry (Fig. S9, available at http://www.jem.org/cgi/content/full/jem.20062453/DC1). This was surprising because a previous study has shown that the p53R172P mutant has a partial defect in its ability to activate p21 (10). We ruled out the possibility of p53-independent activation of p21 because no increase was observed in Lig4−/− p53−/− cells (Fig. 5, lane 1). The higher level of p21 observed in Lig4−/− p53p/p mice may result from the persistence of DSBs that maintain a high level of mutant p53, which still possesses a residual ability to transactivate p21. On the other hand, the level of the proapoptotic protein PUMA was not increased in Lig4−/− p53p/p or Lig4−/− p53−/− mice (Fig. 5 B, lanes 2 and 1), which contrasts with the increased level of this protein in WT thymocytes (Fig. 5 B, lane 5) undergoing apoptosis after irradiation (48). This result was consistent with the absence of apoptosis in Lig4−/− p53p/p thymocytes. Together, our results indicate that the V(D)J recombination– related DSBs induce an elevation in the level of p53R172P, which lacks the ability to induce the apoptotic pathway, possibly caused by a defect in transactivating proapoptotic genes such as PUMA and BAX (38, 49). Instead, the mutant p53 transactivates a high level of p21, which subsequently causes cells to enter cell cycle arrest and become senescent.
Although it is well established that p53 is a key factor in inducing senescence in response to DNA damage, other pathways may also lead to cell senescence independent of p53 (50, 51). One key molecule in this mechanism is p16INK4a (50), which is a positive regulator of the tumor suppressor retinoblastoma protein (pRB) and causes cells to senesce in response to certain stimuli. To determine whether this pathway might mediate cellular senescence induced by DNA DSBs in this in vivo system, we examined the expression levels of p16INK4a and pRB in Lig4−/− p53p/p thymocytes. Western blot analysis showed a very low level of the p16INK4a in Lig4−/− p53p/p, Lig4+/+ p53p/p, and WT thymocytes, eliminating p16INK4a as the factor inducing senescence in our system (Fig. S10, available at http://www.jem.org/cgi/content/full/jem.20062453/DC1). To further rule out any involvement of this pathway, Western blot analysis did not show any change in the expression of the pRB protein in Lig4−/− p53p/p thymocytes compared with WT and Lig4+/+ p53p/p thymocytes (Fig. S11). Thus, although the p16–pRB pathway may induce senescence in some instances, V(D)J recombination breaks mainly induce a p53–p21 pathway in Lig4−/− p53p/p thymocytes that in turn, cause the cells to enter senescence.
DISCUSSION
In this study, we have successfully established and characterized an in vivo model showing that the mutant p53R172P induces cell cycle arrest and senescence, rather than apoptosis, most likely through a p21-dependent pathway in response to persistent DNA damage. This response is sufficient to inhibit genomic instability and suppress tumorigenesis. We report for the first time the detection of aberrant V(D)J recombination intermediates (open coding ends) in thymocytes of Lig4−/− p53p/p mice. We also detected stabilized mutant p53 and high level of p21 in thymocytes, which coincides with large numbers of senescent cells in CD25-positive (DN) thymocytes and CD19-positive bone marrow cells. Consistent with the finding of senescence in precursor lymphoid cells, we found a very low level of the proapoptotic protein PUMA in thymocytes. In addition, cytogenetic analysis of the lymphoid cells of Lig4−/− p53p/p mice showed normal chromosomal counts with few chromosomal breaks, suggesting that the p53–p21 pathway was able to suppress genomic instability and inhibit tumorigenesis. This was unexpected, taking into consideration a previous study in which DNA ligase IV–p53 double-deficient mice succumbed to aggressive pro-B cell lymphomas in a Rag-dependent manner, with chromosomal translocation and oncogenic amplification (35).
Based on the findings of this study in Lig4−/− p53p/p mice, we propose the model shown in Fig. 6. When V(D)J recombination–induced DSBs remain unrepaired as a result of a deficiency in the NHEJ pathway, p53 is activated, leading to apoptosis. In the absence of p53 activity, these DSBs induce genomic instability and lymphomagenesis. However, when the p53 gene contains the R172P mutation, this same DNA damage signal produces a very different outcome. Cells with persistent DSBs induce activation of mutant p53 and, subsequently, increase the level of the CDK inhibitor p21 that causes cells to undergo cell cycle arrest and, ultimately, senescence; this is sufficient to inhibit tumorigenesis. In addition, a very low level of proapoptotic protein PUMA was found in the Lig4−/− p53p/p thymocytes, which is consistent with previous studies of the inability of the mutant p53 protein to transactivate another crucial proapoptotic protein, BAX (38, 49). We suggest the inability of p53R172P to activate apoptosis-related genes explains at least partially why the p53R172P mice are deficient in apoptosis, confirming the essential function of the transactivation activity of p53 in inducing apoptosis. Our results also implicate the critical role of p53 in determining cell cycle arrest versus apoptosis, in addition to the upstream signaling. It is intriguing, however, that the mutant p53 is able to transactivate p21 to a very high level. Previous studies have indicated that p53R175P was able to activate p21 in irradiated cells, but at a reduced rate (10, 37, 38). Although we cannot discard the possibility that p53R172P may function independently of p21 in preventing genomic instability, it is improbable because we observed increased levels of both p53 and p21 in only Lig4−/− p53p/p lymphoid cells, and because recent evidence showed that p21 deficiency accelerates the tumor onset and chromosomal aberrations in p53p/p mice (52). We interpret a high p21 level found in Lig4−/− p53p/p thymocytes as likely caused by a combination of persistent unrepaired DNA breaks resulting from the deficiency of DNA ligase IV and the failure of p53 to transactivate other effector genes key to inducing apoptosis, such as PUMA and BAX (38). Perhaps this activation of p21 serves as a “backup” pathway in the event that the primary goal, apoptosis, cannot be achieved. Another possibility is the accumulation of senescent cells with high levels of p21 to maintain their senescent status in thymus and bone marrow.
Figure 6.

Proposed model depicting the outcomes of WT or mutant p53 activation in response to DNA damage in NHEJ deficiency.
Our previous study of mice with the Lig4−/− p53−/− genotype convincingly demonstrated that the tumorigenic process requires chromosomal translocation and rounds of replication to amplify the oncogene c-Myc by means of breakage–fusion–bridge cycles (35). In this study, our data suggest that in the absence of an apoptosis pathway, persistent DNA breaks activate the p53–p21 pathway, which blocks DNA replication and cell cycle, thereby inhibiting further genomic instability, which is a key event for tumorigenesis. Our study further reveals a strong correlation between aneuploidy and oncogene activation, implying that p53 has an essential tumor suppressor role in preventing chromosomal abnormality. Aneuploidy is characteristic of p53-deficient cells and can result from a failure of controlling centrosome duplication, which is normally coordinated with other cell cycle events, such as DNA replication. Indeed, centromere dysregulation is commonly found in p53-deficient cells, but normal centrosome duplication can be restored through the reintroduction of p53 (53). Furthermore, the main control mechanism of centromere duplication occurs through the CDK2, where the kinase can be inhibited by p21, which is a major p53 effector expressed at high levels in Lig4−/− p53p/p thymocytes. These observations lead us to further hypothesize that the V(D)J recombination–mediated DNA DSBs signal for the activation of p53 and, subsequently, p21. This is sufficient to inhibit abnormal centromere duplication and maintain the euploidy status of Lig4−/− p53p/p lymphoid cells, thereby preventing further genomic instability required for tumorigenesis.
Cellular senescence is a state in which cells withdraw from the cell cycle, and it has been proposed to be a tumor suppression mechanism (26). In human cells, telomere shortening is the primary event that causes cells to undergo senescence. Telomere shortening is a result of excessive cellular proliferation and DNA replication, culminating in a critical state in which the chromosome ends lack the usual protection. It has been proposed that a shortened telomere can be recognized as a broken chromosome, which sends a cellular signal evoking the activation of p53. This makes our model useful for studying the pathway that leads to cellular senescence and its role in suppressing tumorigenesis, as telomere shortening does not frequently occur in murine cells because of the extra length of their telomeres. Our study further indicates a critical role of p21 in inducing cellular senescence to block further genomic instability and tumorigenesis in response to persistent DNA DSBs. Other senescent pathways, such as p16–pRB, are important in response to different stimuli.
In addition, cellular senescence, particularly premature senescence, has been linked to aging. These Lig4−/− p53p/p mice could also be an excellent model for studying the connection between senescence and aging. A compelling finding in this regard was that although tumors do not develop in Lig4−/− p53p/p mice, they die early of an unknown cause; further pathological analyses are required to explore the cause of death. One possible cause of death in these animals could be premature aging. In support of this, it is well established that nonspecific DNA DSBs occur as a result of normal metabolism and environmental exposure and that DNA ligase IV plays an important role in the repair of DSBs in general. We hypothesize that these nonspecific breaks could accumulate in the Lig4−/− background and that the mutant p53 inhibits tumorigenesis by inducing senescence, which might lead to premature aging and some aging-related diseases.
MATERIALS AND METHODS
Mouse colony and maintenance.
Mutant mouse strains (mixed genetic background of C57BL/6 and 129SV) and their colony maintenance have been previously described (10, 35, 54). Protocols used in this study were approved by the Institutional Animal Care and Use Committee at the University of Texas M.D. Anderson Cancer Center.
Cell senescence assay and immunohistochemistry studies.
Acidic β-gal activity, which indicates cell senescence, was detected as previously described (19). In brief, 6-μm-thick frozen thymic sections from newborn mice of different genotypes were processed for the β-gal assay (staining kit was purchased from Cell Signaling Technology). Bone marrow cell suspension was obtained from 4-mo-old mice, cytospun, and fixed with 1:1 acetone/methanol (100% vol/vol). The cells were stained with β-gal and observed under a brightfield microscope.
The cells were also stained for the CD19 B cell marker using a standard protocol. In brief, after the β-gal assay, the cells were processed for antigen retrieval using the proteinase K method, blocked using the streptavidin- biotin system (Vector Laboratories), and incubated with rat anti-CD19 antibody (BD Biosciences) with its corresponding isotype control. The CD19- positive cells were detected using an anti–rat biotinylated antibody and streptavidin–horseradish peroxidase (HRP) (BD).
Thymocytes, thymic sections, and bone marrow cells were stained with anti-HP1γ monoclonal antibody (clone 42s2; Millipore), followed by HRP/DAB detection system. Thymocytes or thymic sections were also costained with anti-HP1γ and CD25 (BD Biosciences), followed by fluorescent detection.
LMPCR assays.
To detect the signal and coding ends in thymocyte DNA, LMPCR assays were performed as previously described (45).
TUNEL assay.
A TUNEL assay (DeadEnd Fluorometric TUNEL System; Promega) was performed to identify apoptotic cells in thymic sections of newborn mice, including a DNase-treated positive control. In brief, frozen sections were pretreated with 10 μg/ml proteinase K and equilibrated; DNA breaks were labeled with fluorescein-12-dUTP. The sections were subsequently stained with DAPI (Sigma-Aldrich). The green fluorescence of apoptotic cells against a blue background (DAPI) was detected by fluorescence microscopy.
Western blot.
Thymocytes were cultured in Dulbecco's modified Eagle medium supplemented with 10% fetal calf serum and 10 mM Hepes at a density of 106 cells/ml. The cells were subsequently exposed to 5 Gy of γ-radiation and cultured for 4 h. Whole-cell protein extracts were prepared (11) and quantified by the Bradford method (Bio-Rad Laboratories). Protein (50–150 μg/lane) was electrophoretically separated on SDS-PAGE and transferred to a PVDF membrane. The membranes were blotted with antibodies specific for p53, which recognizes p53R172P (CM5; Vector Laboratories), p21 (C19; Santa Cruz Biotechnology), PUMA (Cell Signaling Technology), and β-actin (Sigma-Aldrich). An anti–rabbit or –mouse antibody conjugated to HRP was used, and the signal was detected using a chemiluminescence kit (PerkinElmer).
Fluorescence in situ hybridization (FISH).
For FISH, fetal liver cells were obtained from newborn mice of different genotypes. Metaphase preparations were prepared as previously described (35). Telomeric FISH was used to detect chromosomal breaks using a Cy3-labeled protein nucleic acid probe (Applied Biosystems).
Online supplemental material.
Fig. S1 shows that Lig4−/−p53−/− mice are smaller. Fig. S2 shows a TUNEL assay on thymic sections of newborn mice. Fig. S3 shows HE staining of thymic sections of newborn mice. Fig. S4 shows the SCID phenotype of Lig4−/− p53p/p mice. Fig. S5 shows that HP1γ-positive cells are CD25+ thymocytes. Fig. S6 shows that HP1γ-positive cells are CD19+ bone marrow cells. Fig. S7 shows sequencing of coding ends at the TCR Dδ2 locus. Fig. S8 shows immunohistochemical detection of p53 protein on thymic sections from WT and Lig4−/−p53p/p mice. Fig. S9 shows immunohistochemical detection of p21 protein on thymic sections from WT and Lig4−/− p53p/p mice. Fig. S10 shows p16INK4a and β-actin protein expression in WT, Lig4+/+p53p/p, and Lig4−/−p53p/p thymocytes. Fig. S11 shows phosphorylated retinoblastoma protein expression in WT, Lig4+/+p53p/p, and Lig4−/−p53p/p thymocytes. Table S1 shows that p53R172P rescues embryonic lethality of Lig4−/− mice. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20062453/DC1.
Supplemental Material
Acknowledgments
We are grateful to Drs. Frederick Alt and Guillermina Lozano for providing mutant mice to make this study possible, and to Dr. Cara Benjamin for helping with our experiments and reviewing the manuscript. We would also like to thank Drs. Lawrence Donehower, John Manis, and Honnavara N. Ananthaswamy for critical review, and Betty Notzon for editing the manuscript.
This study was financially supported by a University of Texas M.D. Anderson Institution grant, the Leukemia Research Foundation, the American Cancer Society–Bonnie Kies Lymphatic System Research Scholar grant (to C. Zhu), and Vietnam Education Foundation (to T. Van Nguyen).
The authors declare no conflicting financial interests.
Abbreviations used: CDK, cyclin-dependent kinase; DSB, double-strand break; FISH, fluorescence in situ hybridization; HE, hematoxylin and eosin; HP, heterochromatin protein; HRP, horseradish peroxidase; LMPCR, ligation-mediated PCR; NHEJ, nonhomologous end-joining; PUMA, p53-upregulated modulator of apoptosis; SAHF, senescence-associated heterochromatin foci.
T. Van Nguyen and N. Puebla-Osorio contributed equally to this paper.
References
- 1.Donehower, L.A., M. Harvey, B.L. Slagle, M.J. McArthur, C.A. Montgomery Jr., J.S. Butel, and A. Bradley. 1992. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 356:215–221. [DOI] [PubMed] [Google Scholar]
- 2.Jacks, T., L. Remington, B.O. Williams, E.M. Schmitt, S. Halachmi, R.T. Bronson, and R.A. Weinberg. 1994. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 4:1–7. [DOI] [PubMed] [Google Scholar]
- 3.Levine, A.J. 1997. p53, the cellular gatekeeper for growth and division. Cell. 88:323–331. [DOI] [PubMed] [Google Scholar]
- 4.Campisi, J. 2005. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 120:513–522. [DOI] [PubMed] [Google Scholar]
- 5.Ko, L.J., and C. Prives. 1996. p53: puzzle and paradigm. Genes Dev. 10:1054–1072. [DOI] [PubMed] [Google Scholar]
- 6.Wang, X.W. 1999. Role of p53 and apoptosis in carcinogenesis. Anticancer Res. 19:4759–4771. [PubMed] [Google Scholar]
- 7.Fridman, J.S., and S.W. Lowe. 2003. Control of apoptosis by p53. Oncogene. 22:9030–9040. [DOI] [PubMed] [Google Scholar]
- 8.Attardi, L.D. 2005. The role of p53-mediated apoptosis as a crucial anti-tumor response to genomic instability: lessons from mouse models. Mutat. Res. 569:145–157. [DOI] [PubMed] [Google Scholar]
- 9.Symonds, H., L. Krall, L. Remington, M. Saenz-Robles, S. Lowe, T. Jacks, and T. Van Dyke. 1994. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell. 78:703–711. [DOI] [PubMed] [Google Scholar]
- 10.Liu, G., J.M. Parant, G. Lang, P. Chau, A. Chavez-Reyes, A.K. El-Naggar, A. Multani, S. Chang, and G. Lozano. 2004. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat. Genet. 36:63–68. [DOI] [PubMed] [Google Scholar]
- 11.Chao, C., D. Herr, J. Chun, and Y. Xu. 2006. Ser18 and 23 phosphorylation is required for p53-dependent apoptosis and tumor suppression. EMBO J. 25:2615–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laptenko, O., and C. Prives. 2006. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ. 13:951–961. [DOI] [PubMed] [Google Scholar]
- 13.Vogelstein, B., D. Lane, and A.J. Levine. 2000. Surfing the p53 network. Nature. 408:307–310. [DOI] [PubMed] [Google Scholar]
- 14.Nakano, K., and K.H. Vousden. 2001. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell. 7:683–694. [DOI] [PubMed] [Google Scholar]
- 15.Yu, J., and L. Zhang. 2003. No PUMA, no death: implications for p53-dependent apoptosis. Cancer Cell. 4:248–249. [DOI] [PubMed] [Google Scholar]
- 16.Harper, J.W., G.R. Adami, N. Wei, K. Keyomarsi, and S.J. Elledge. 1993. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 75:805–816. [DOI] [PubMed] [Google Scholar]
- 17.Brown, J.P., W. Wei, and J.M. Sedivy. 1997. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science. 277:831–834. [DOI] [PubMed] [Google Scholar]
- 18.Hayflick, L., and P.S. Moorhead. 1961. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25:585–621. [DOI] [PubMed] [Google Scholar]
- 19.Dimri, G.P., X. Lee, G. Basile, M. Acosta, G. Scott, C. Roskelley, E.E. Medrano, M. Linskens, I. Rubelj, O. Pereira-Smith, et al. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA. 92:9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narita, M., S. Nunez, E. Heard, M. Narita, A.W. Lin, S.A. Hearn, D.L. Spector, G.J. Hannon, and S.W. Lowe. 2003. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 113:703–716. [DOI] [PubMed] [Google Scholar]
- 21.Collado, M., J. Gil, A. Efeyan, C. Guerra, A.J. Schuhmacher, M. Barradas, A. Benguria, A. Zaballos, J.M. Flores, M. Barbacid, et al. 2005. Tumour biology: senescence in premalignant tumours. Nature. 436:642. [DOI] [PubMed] [Google Scholar]
- 22.Braig, M., S. Lee, C. Loddenkemper, C. Rudolph, A.H. Peters, B. Schlegelberger, H. Stein, B. Dorken, T. Jenuwein, and C.A. Schmitt. 2005. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 436:660–665. [DOI] [PubMed] [Google Scholar]
- 23.Rodier, F., S.H. Kim, T. Nijjar, P. Yaswen, and J. Campisi. 2005. Cancer and aging: the importance of telomeres in genome maintenance. Int. J. Biochem. Cell Biol. 37:977–990. [DOI] [PubMed] [Google Scholar]
- 24.von Zglinicki, T., G. Saretzki, J. Ladhoff, F. d'Adda di Fagagna, and S.P. Jackson. 2005. Human cell senescence as a DNA damage response. Mech. Ageing Dev. 126:111–117. [DOI] [PubMed] [Google Scholar]
- 25.Lou, Z., and J. Chen. 2006. Cellular senescence and DNA repair. Exp. Cell Res. 312:2641–2646. [DOI] [PubMed] [Google Scholar]
- 26.Campisi, J. 2001. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 11:S27–S31. [DOI] [PubMed] [Google Scholar]
- 27.Gellert, M. 2002. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu. Rev. Biochem. 71:101–132. [DOI] [PubMed] [Google Scholar]
- 28.Ma, Y., U. Pannicke, K. Schwarz, and M.R. Lieber. 2002. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 108:781–794. [DOI] [PubMed] [Google Scholar]
- 29.Burma, S., B.P. Chen, and D.J. Chen. 2006. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst.). 5:1042–1048. [DOI] [PubMed] [Google Scholar]
- 30.Sekiguchi, J.M., and D.O. Ferguson. 2006. DNA double-strand break repair: a relentless hunt uncovers new prey. Cell. 124:260–262. [DOI] [PubMed] [Google Scholar]
- 31.Dudley, D.D., J. Chaudhuri, C.H. Bassing, and F.W. Alt. 2005. Mechanism and control of V(D)J recombination versus class switch recombination: similarities and differences. Adv. Immunol. 86:43–112. [DOI] [PubMed] [Google Scholar]
- 32.Bassing, C.H., and F.W. Alt. 2004. The cellular response to general and programmed DNA double strand breaks. DNA Repair (Amst.). 3:781–796. [DOI] [PubMed] [Google Scholar]
- 33.Mills, K.D., D.O. Ferguson, and F.W. Alt. 2003. The role of DNA breaks in genomic instability and tumorigenesis. Immunol. Rev. 194:77–95. [DOI] [PubMed] [Google Scholar]
- 34.Ferguson, D.O., and F.W. Alt. 2001. DNA double strand break repair and chromosomal translocation: lessons from animal models. Oncogene. 20:5572–5579. [DOI] [PubMed] [Google Scholar]
- 35.Zhu, C., K.D. Mills, D.O. Ferguson, C. Lee, J. Manis, J. Fleming, Y. Gao, C.C. Morton, and F.W. Alt. 2002. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell. 109:811–821. [DOI] [PubMed] [Google Scholar]
- 36.Difilippantonio, M.J., S. Petersen, H.T. Chen, R. Johnson, M. Jasin, R. Kanaar, T. Ried, and A. Nussenzweig. 2002. Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. J. Exp. Med. 196:469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rowan, S., R.L. Ludwig, Y. Haupt, S. Bates, X. Lu, M. Oren, and K.H. Vousden. 1996. Specific loss of apoptotic but not cell-cycle arrest function in a human tumor derived p53 mutant. EMBO J. 15:827–838. [PMC free article] [PubMed] [Google Scholar]
- 38.Ludwig, R.L., S. Bates, and K.H. Vousden. 1996. Differential activation of target cellular promoters by p53 mutants with impaired apoptotic function. Mol. Cell. Biol. 16:4952–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao, Y., Y. Sun, K.M. Frank, P. Dikkes, Y. Fujiwara, K.J. Seidl, J.M. Sekiguchi, G.A. Rathbun, W. Swat, J. Wang, et al. 1998. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 95:891–902. [DOI] [PubMed] [Google Scholar]
- 40.Gao, Y., D.O. Ferguson, W. Xie, J.P. Manis, J. Sekiguchi, K.M. Frank, J. Chaudhuri, J. Horner, R.A. DePinho, and F.W. Alt. 2000. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature. 404:897–900. [DOI] [PubMed] [Google Scholar]
- 41.Roth, D.B., C. Zhu, and M. Gellert. 1993. Characterization of broken DNA molecules associated with V(D)J recombination. Proc. Natl. Acad. Sci. USA. 90:10788–10792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu, C., M.A. Bogue, D.S. Lim, P. Hasty, and D.B. Roth. 1996. Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell. 86:379–389. [DOI] [PubMed] [Google Scholar]
- 43.Gao, Y., J. Chaudhuri, C. Zhu, L. Davidson, D.T. Weaver, and F.W. Alt. 1998. A targeted DNA-PKcs-null mutation reveals DNA-PK-independent functions for KU in V(D)J recombination. Immunity. 9:367–376. [DOI] [PubMed] [Google Scholar]
- 44.Rooney, S., J. Sekiguchi, C. Zhu, H.L. Cheng, J. Manis, S. Whitlow, J. DeVido, D. Foy, J. Chaudhuri, D. Lombard, and F.W. Alt. 2002. Leaky Scid phenotype associated with defective V(D)J coding end processing in Artemis-deficient mice. Mol. Cell. 10:1379–1390. [DOI] [PubMed] [Google Scholar]
- 45.Zhu, C., and D.B. Roth. 1995. Characterization of coding ends in thymocytes of scid mice: implications for the mechanism of V(D)J recombination. Immunity. 2:101–112. [DOI] [PubMed] [Google Scholar]
- 46.Schlissel, M., A. Constantinescu, T. Morrow, M. Baxter, and A. Peng. 1993. Double-strand signal sequence breaks in V(D)J recombination are blunt, 5′-phosphorylated, RAG-dependent, and cell cycle regulated. Genes Dev. 7:2520–2532. [DOI] [PubMed] [Google Scholar]
- 47.Franco, S., M. Gostissa, S. Zha, D.B. Lombard, M.M. Murphy, A.A. Zarrin, C. Yan, S. Tepsuporn, J.C. Morales, M.M. Adams, et al. 2006. H2AX Prevents DNA Breaks from Progressing to Chromosome Breaks and Translocations. Mol. Cell. 21:201–214. [DOI] [PubMed] [Google Scholar]
- 48.Jeffers, J.R., E. Parganas, Y. Lee, C. Yang, J. Wang, J. Brennan, K.H. MacLean, J. Han, T. Chittenden, J.N. Ihle, et al. 2003. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 4:321–328. [DOI] [PubMed] [Google Scholar]
- 49.Crook, T., N.J. Marston, E.A. Sara, and K.H. Vousden. 1994. Transcriptional activation by p53 correlates with suppression of growth but not transformation. Cell. 79:817–827. [DOI] [PubMed] [Google Scholar]
- 50.Lowe, S.W., and C.J. Sherr. 2003. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr. Opin. Genet. Dev. 13:77–83. [DOI] [PubMed] [Google Scholar]
- 51.Drayton, S., J. Rowe, R. Jones, R. Vatcheva, D. Cuthbert-Heavens, J. Marshall, M. Fried, and G. Peters. 2003. Tumor suppressor p16INK4a determines sensitivity of human cells to transformation by cooperating cellular oncogenes. Cancer Cell. 4:301–310. [DOI] [PubMed] [Google Scholar]
- 52.Barboza, J.A., G. Liu, Z. Ju, A.K. El-Naggar, and G. Lozano. 2006. p21 delays tumor onset by preservation of chromosomal stability. Proc. Natl. Acad. Sci. USA. 103:19842–19847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tarapore, P., H.F. Horn, Y. Tokuyama, and K. Fukasawa. 2001. Direct regulation of the centrosome duplication cycle by the p53-p21Waf1/Cip1 pathway. Oncogene. 20:3173–3184. [DOI] [PubMed] [Google Scholar]
- 54.Frank, K.M., J.M. Sekiguchi, K.J. Seidl, W. Swat, G.A. Rathbun, H.L. Cheng, L. Davidson, L. Kangaloo, and F.W. Alt. 1998. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature. 396:173–177. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}