

Abstract
Altered intestinal O-glycan expression has been observed in patients with ulcerative colitis and colorectal cancer, but the role of this alteration in the etiology of these diseases is unknown. O-glycans in mucin core proteins are the predominant components of the intestinal mucus, which comprises part of the intestinal mucosal barrier. Core 3–derived O-glycans, which are one of the major types of O-glycans, are primarily expressed in the colon. To investigate the biological function of core 3–derived O-glycans, we engineered mice lacking core 3 β1,3-N-acetylglucosaminyltransferase (C3GnT), an enzyme predicted to be important in the synthesis of core 3–derived O-glycans. Disruption of the C3GnT gene eliminated core 3–derived O-glycans. C3GnT-deficient mice displayed a discrete, colon-specific reduction in Muc2 protein and increased permeability of the intestinal barrier. Moreover, these mice were highly susceptible to experimental triggers of colitis and colorectal adenocarcinoma. These data reveal a requirement for core 3–derived O-glycans in resistance to colonic disease.
O-linked oligosaccharides (O-glycans) are the primary components of the intestinal mucus layer that overlies the gastrointestinal epithelium. This layer is a dense, carbohydrate-rich matrix that consists primarily of mucins containing multiple serine and threonine residues, which have been modified by O-glycans and which account for 80% of the mucin mass (1–6). The mucus layer and epithelial cells comprise an intestinal barrier that protects epithelial and intestinal mucosal immune cells from potentially harmful luminal microflora and food components (3–6) and participates in bacterial colonization (7). The role of intestinal epithelial cells in maintaining barrier function and in the pathogenesis of several common intestinal diseases, such as inflammatory bowel disease (IBD) and colorectal cancer, has been well studied (8–15). However, the physiological and pathological significance of the mucus layer has been less explored.
IBD is generally recognized as an immune-mediated disorder resulting from an abnormal interaction between colonic microflora and mucosal immune cells in a genetically susceptible host (14, 16). How this interaction develops is not well understood. A deterioration of the mucus layer of the colon is prominent in patients with ulcerative colitis, which is a common form of IBD (3–6). Altered intestinal O-glycan expression appears early in the pathogenesis of ulcerative colitis (6). Interestingly, similarly altered O-glycans are seen in >90% of colorectal cancers, which have a close association with ulcerative colitis (6, 17, 18). Whether or not this abnormal O-glycan expression contributes to the etiology of these diseases is unknown.
O-glycans containing GalNAc in α-linkage to serine or threonine residues occur on many membrane and secreted proteins, particularly mucins (1, 2). O-glycans have two main core structures, referred to as core 1– and core 3–derived O-glycans (Fig. 1 A). The biosynthesis of these cores is controlled by specific glycosyltransferases. Core 3 β1,3-N-acetylglucosaminyltransferase (C3GnT) activity is enriched in mucin-secreting epithelial tissues, such as gastrointestinal tract, as measured by enzymatic activity assays in tissue lysates (1, 2, 19, 20). The enzyme transfers GlcNAc from UDP-GlcNAc to GalNAcα1-Ser/Thr (Tn antigen) to form the core 3 O-glycan (GlcNAcβ1,3GalNAcα1-Ser/Thr), which can be further modified to form more complex structures, such as core 4 O-glycans (Fig. 1 A). Recently, human core C3GnT (also known as β3Gn-T6 or core 3 synthase) was identified (19, 20). In vitro biochemical analysis suggests that C3GnT is the only enzyme responsible for the biosynthesis of core 3 O-glycans (19, 20).
Figure 1.

Generation of C3GnT−/− mice. (A) The scheme shows the two major O-glycan branching pathways. C3GnT refers to C3GnT. Arrowheads show the possible pathways for further branching, elongation, fucosylation, sialylation, and sulfation. (B and C) Strategy to generate mice lacking core 3–derived O-glycans by targeting the C3GnT gene, and Southern blot genotyping using EcoRV restriction enzyme digestion. (D) RT-PCR confirmed the deletion of the C3GnT gene product. GAPDH was used as an amplification control. (E) C3GnT enzymatic activity in C3GnT +/+ and C3GnT −/− tissues. Error bars represent the mean ± the SD. n = 3. (F) LacZ staining of C3GnT +/+ and C3GnT −/− colonic tissues. Bar, 100 μm.
We hypothesized that core 3–derived O-glycans are a key constituent of the intestinal mucus layer and are important for intestinal barrier function, and that the alteration of core 3–derived O-glycan expression plays a role in the pathogenesis of common intestinal diseases, such as colitis and intestinal tumors. To test these hypotheses, we created mice lacking core 3–derived O-glycans by targeted deletion of the C3GnT gene (C3GnT −/−). We found that deletion of the C3GnT gene eliminated core 3–derived O-glycans and significantly reduced total intestinal glycans. Furthermore, C3GnT −/− mice exhibited an increased susceptibility to experimental colitis and colorectal adenocarcinoma. The results presented in this study indicate that core 3–derived O-glycans are key components of intestinal mucin, and that defects in their expression may be associated with the pathogenesis of colonic disease.
RESULTS
Molecular characterization of the murine C3GnT gene
A Mus musculus chromosome 7 genomic contig (NT_039433) that contains the C3GnT gene and a 2,370-bp, full-length murine cDNA sequence (XM_195661) were identified by BLASTN searches using the published human C3GnT nucleotide sequence (19). The analyses indicated that murine C3GnT is a type II membrane protein with 68% identity to the human C3GnT (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20061929/DC1). Southern blot analysis (unpublished data) confirmed that the murine C3GnT gene has two exons. Exon 1 comprises 36 bp that encode a 5′-untranslated region. Exon 2 comprises 2,318 bp encoding the ATG translational start site, cytoplasmic domain, transmembrane domain, stem region, and the catalytic domain of C3GnT. RT-PCR of RNA extracted from different mouse tissues revealed that murine C3GnT mRNA is predominantly expressed in colonic tissue (Fig. S2 A). The ileum and the salivary glands also expressed low levels of C3GnT transcripts, whereas the rest of the analyzed tissues had no C3GnT expression. To determine if the identified C3GnT gene confers β1,3-N-acetylglucosaminyltransferase activity, we cloned the C3GnT cDNA into a baculovirus expression system (21). Enzymatic analysis indicated that recombinant murine C3GnT exhibited β1,3-N-acetylglucosaminyltransferase activity, which transfers GlcNAc from UDP-GlcNAc to GalNAcβ1α-R, thus confirming the identity of the cloned murine C3GnT gene (Fig. S2, B and C).
Generation of C3GnT −/− mice
To study the biological function of C3GnT, we generated mice lacking the C3GnT gene by targeted homologous recombination in mouse embryonic stem cells (Fig. 1, B and C) using previously described methods (22, 23). RT-PCR and enzymatic assays confirmed that C3GnT mRNA and C3GnT enzyme activity in tissue extracts were eliminated in C3GnT −/− mice (Fig. 1, D and E). To characterize the tissue expression pattern of endogenous C3GnT, we engineered a LacZ reporter gene immediately downstream of the C3GnT ATG translational start site. LacZ staining of different C3GnT −/− tissues confirmed that expression of C3GnT was restricted to colonic tissues (Fig. 1 F and not depicted). Although RT-PCR detected lower levels of endogenous C3GnT mRNA in the small intestine and salivary glands, lacZ staining was not detected in these tissues (unpublished data), which likely reflected sensitivity differences between the two assays.
C3GnT −/− mice developed normally, and both sexes were fertile in a specific pathogen-free barrier facility. Intercrosses between heterozygotes yielded normal-sized litters with Mendelian inheritance. C3GnT −/− mice had normal peripheral blood counts (unpublished data). Gross morphology and histological examinations of 6–20-wk-old mice revealed no observable differences between wild-type littermates (C3GnT +/+) and C3GnT −/− mice in major organs, including heart, liver, salivary glands, stomach, jejunum, ileum, colon, spleen, thymus, and lymph nodes (Fig. S3 and Fig. S4 A, available at http://www.jem.org/cgi/content/full/jem.20061929/DC1; and not depicted). Flow cytometry of C3GnT −/− peripheral blood cells and splenocytes revealed no substantial abnormalities in the absolute number and the ratios of CD3+ T cells (including CD4+, CD8+, and innate-like invariant NKT cells) and of CD19+ B cells (including innate-like marginal zone B cells; Fig. S4 B and not depicted). This suggests that the deficiency of core 3–derived O-glycans does not affect the development of immune cells.
Disruption of the C3GnT gene eliminates core 3–derived O-glycans and exposes the Tn antigen in murine colon
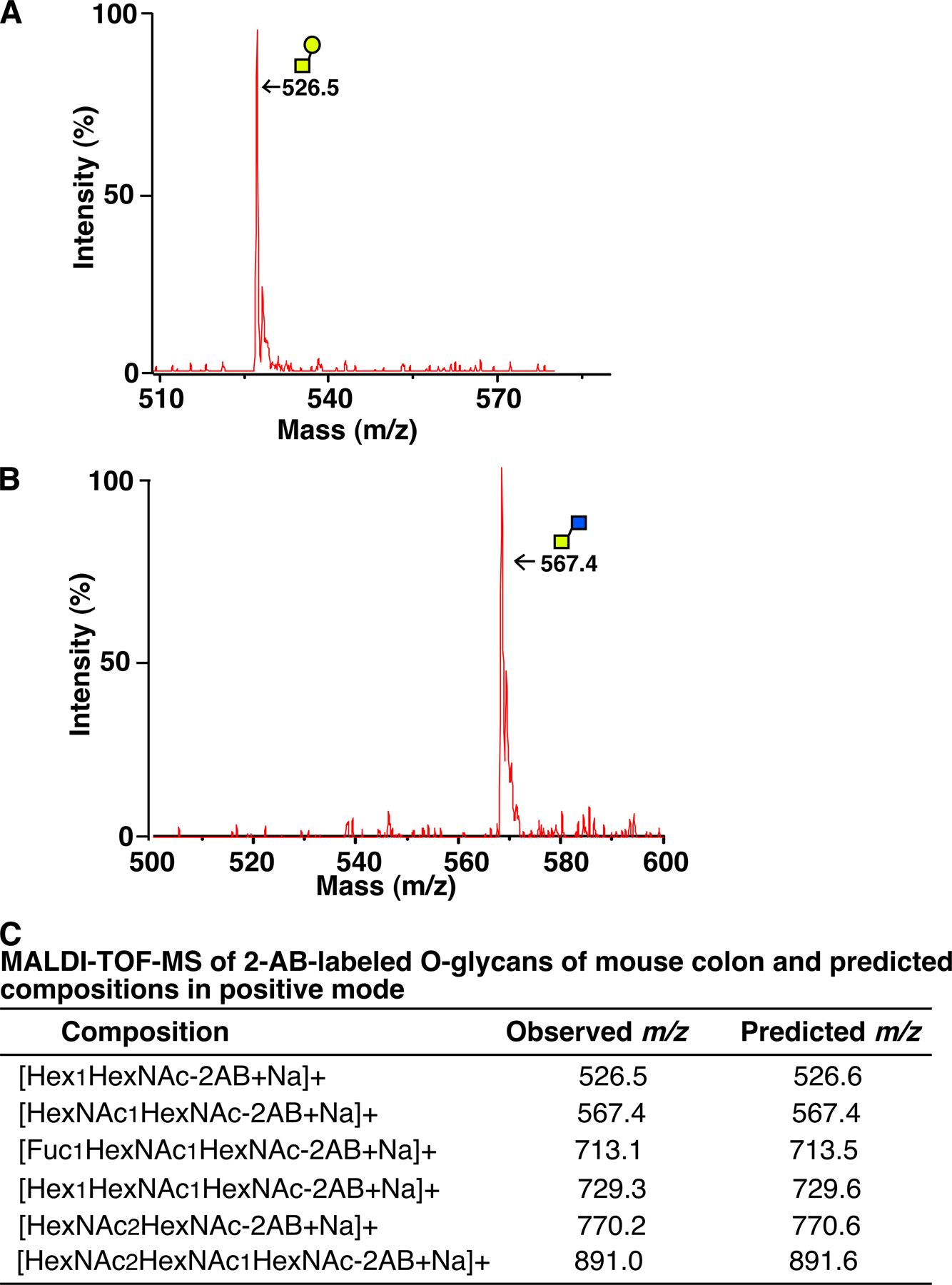
We used matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) to define the O-glycan structures in C3GnT +/+ and C3GnT −/− colon (24). Consistent with previous data, both core 1– and core 3–derived O-glycans (core 3 O-glycans, fucosylated core 3 O-glycans, and core 4 O-glycans) were dominant structures expressed in the C3GnT +/+ colon (Fig. 2 A and Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20061929/DC1) (2, 19, 20, 22). Structural analysis revealed that C3GnT −/− colonic tissue did not express core 3–derived O-glycans (Fig. 2 A and Fig. S5). These results confirmed the deletion of the C3GnT gene and provide definitive evidence that the C3GnT gene encodes the predominant C3GnT activity.
Figure 2.

Disruption of the C3GnT gene eliminates core 3–derived O-glycans and exposes the Tn antigen in murine colons. (A) Annotated spectra of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) analyses of 2-AB–labeled O-glycans of C3GnT +/+ and C3GnT −/− colons in a linear-positive mode. Each annotated molecular mass of O-glycan includes masses of a 2-AB and a Na. Asterisks represent positions of core 3–derived O-glycans that are missing in C3GnT −/− mice. Data are representatives of three experiments. (B) Immunohistochemical staining of mouse colon tissue sections with a mAb to the Tn antigen. The sections were pretreated with or without sialidase. Brown reaction products mark sites of antibody binding. Data are representatives of at least three experiments. (C) C3GnT +/+ and C3GnT −/− colonic tissues stained with PAS (pink color) and Alcian blue (blue color). (D) Pixels of PAS and Alcian blue staining areas were quantified by Photoshop software based on six sections of three independent mice from each group. Error bars represent the mean ± the SD. Bars: (B) 50 μm; (C) 100 μm.
C3GnT catalyzes the formation of core 3–derived O-glycans by adding GlcNAc to its substrate GalNAc-α-Ser/Thr (Tn antigen), which is normally modified with one or more other monosaccharides (Fig. 1 A) (1, 19). We expected that the deletion of the C3GnT gene would expose Tn antigen in C3GnT −/− intestinal tissues. As predicted, immunochemical staining with anti-Tn mAb did not label C3GnT +/+ intestinal tissue. In contrast, anti-Tn mAb labeled C3GnT −/− intestinal epithelial cells, but not other cell types (Fig. 2 B). Enzymatic desialylation of tissue sections did not enhance binding of anti-Tn to C3GnT −/− tissues, suggesting that sialic acid does not appreciably modify the exposed Tn antigen to form the sialyl-Tn antigen (Fig. 1 A) in C3GnT −/− colonic tissue. However, enzymatic desialylation revealed the Tn antigen in C3GnT +/+ colonic tissue, indicating that the sialyl-Tn antigen is normally expressed in wild-type mouse colon (Fig. 2 B).
We then used periodic acid-Schiff's reagent (PAS) and Alcian blue to determine if the deficiency of core 3–derived O-glycans affects the expression of intestinal glycans. PAS stains neutral carbohydrates, whereas Alcian blue recognizes acidic carbohydrates that may represent sialylated, fucosylated, or sulfated sugars. PAS and Alcian blue staining remained unchanged in the jejunum and the ileum of both C3GnT +/+ and C3GnT −/− mice (Fig. S6, available at http://www.jem.org/cgi/content/full/jem.20061929/DC1). However, both PAS staining and Alcian blue staining were significantly reduced in the colons of C3GnT −/− mice compared with those of C3GnT +/+ mice (Fig. 2, C and D). Consistent with the RT-PCR and lacZ staining data, this result demonstrates that core 3–derived O-glycans are predominantly expressed in mouse colon.
Deficiency of C3GnT results in reduced Muc2 expression in colon and impaired mucosal integrity
To investigate if a deficiency of core 3 O-glycosylation affects the expression of intestinal mucins, we first screened the mRNA levels of the major intestinal mucins by semiquantitative RT-PCR. We observed no significant differences between C3GnT +/+ and C3GnT −/− colons in mRNA levels of Muc2, Muc3, Muc13, and Muc5ac (Fig. S7 A, available at http://www.jem.org/cgi/content/full/jem.20061929/DC1). However, probing with a glycosylation-independent polyclonal antibody revealed a substantial reduction of Muc2 protein, which is the predominant component of the intestinal mucous layer (3–6, 18, 25) in C3GnT −/− colonic tissue (Fig. 3, A and B). We questioned whether the decreased expression of Muc2 was caused by a reduced numbers of goblet cells. However, immunohistochemistry of C3GnT +/+ and C3GnT −/− colon tissues revealed similar expression of intestinal trefoil factor (ITF), which is a nonmucin protein and a product of fully differentiated goblet cells (Fig. 3, A and B) (26). In addition, Alcian blue staining and transmission electron microscopic imaging revealed no considerable morphological difference between C3GnT +/+ and C3GnT −/− goblet cells (Fig. S7, B and C). Thus, goblet cells are preserved in the C3GnT −/− colon, but goblet cell production of Muc2 is reduced. We were unable to examine protein expression of other, less-expressed, intestinal mucins because of the lack of glycosylation-independent antibodies to these murine glycoproteins.
Figure 3.

C3GnT−/− colon has reduced expression of Muc2 and impaired mucosal integrity. (A) Immunohistochemical staining of C3GnT +/+ and C3GnT −/− colonic tissues with an anti-murine Muc2 peptide antibody and with an antibody to ITF, which is a marker of fully differentiated goblet cells. Brown color indicates specific antibody binding. (B) Pixels of positive staining area per crypt were quantified using Photoshop. Six sections of three independent mice from each group were quantified. Error bars represent the mean ± the SD. (C) Serum concentration of the FITC-dextran of C3GnT +/+ and C3GnT −/− mice were measured 4 h after oral administration of FITC-dextran. Error bars represent the mean ± the SD. n = 6. (D) Representative images of fluorescent microscopic analysis of C3GnT +/+ and C3GnT −/− intestinal cryosections 4 h after oral administration of FITC-Dextran (three sections from each mouse, with four mice in each group). (E) Relative amount of 16S rDNA detected by real-time PCR using two independent sets of 16S universal primers. Data are expressed as an n-fold difference between C3GnT +/+ and C3GnT −/− mice. Error bars represent the mean ± the SD. n = 6 mice in each group. The average 16S rDNA value of C3GnT +/+ mice was expressed as 1. Bars: (A) 100 μm; (D) 50 μm.
To investigate if the loss of core 3–derived O-glycans affects mucosal integrity, we fed C3GnT +/+ and C3GnT −/− mice with FITC-dextran by gavage, and 4 h later we measured their serum levels of FITC-dextran to evaluate intestinal permeability. Serum levels of FITC-dextran in C3GnT −/− mice were significantly higher than in C3GnT +/+ mice (Fig. 3 C). Fluorescent microscopic analysis of the C3GnT +/+ and C3GnT −/− intestinal cryosections revealed no fluorescent infiltration of FITC-dextran into both C3GnT +/+ and C3GnT −/− small intestinal villi. In contrast, a higher level of fluorescent intensity was observed in C3GnT −/− colonic tissues compared with that in C3GnT +/+ tissues (Fig. 3 D). Collectively, these data indicate that colonic mucosal integrity is impaired in C3GnT −/− mice.
We reasoned that the deficiency of core 3 O-glycans and the impairment in mucosal integrity might result in an alteration of mucosa-associated bacteria in C3GnT −/− mice. We used 16S bacterial ribosomal DNA (rDNA)–based real-time PCR analysis to address this question. Two sets of universal 16S rDNA primers (16S1 and 16S2) reproducibly detected a significant increase in 16S rDNA in C3GnT −/− colonic tissues in comparison to that in C3GnT +/+ colonic tissues (Fig. 3 E). The increase is not likely caused by variations in luminal bacterial flora because the amount of 16S rDNA in C3GnT +/+ and C3GnT −/− fecal material was similar (Fig. 3 E). Mesenteric nodes had weak, yet detectable, 16S rDNA signal, but there was no difference between C3GnT +/+ and C3GnT −/− groups (unpublished data). Mesenteric bacterial translocation is associated with trafficking of bacteria-bearing dendritic cells from the mucosa, typically after Toll-like receptor sensing or other immune activation (27, 28). Thus, no detectable increase in bacterial translocation in C3GnT −/− mesenteric nodes is consistent with the lack of spontaneous colonic inflammation in C3GnT −/− mice.
C3GnT −/− mice are highly susceptible to dextran sodium sulfate (DSS)–induced colitis
DSS is a chemical commonly used to induce experimental colitis (29). We examined the pathological consequence of the deficiency of core 3 O-glycosylation using this model. We first challenged 6-wk-old C3GnT +/+ and C3GnT −/− males with 2.5% DSS in drinking water for 14 d to determine its lethality rate. We found that 9/10 C3GnT +/+ mice survived the treatment, whereas all C3GnT −/− mice died at the end of the 14-d treatment (Fig. 4 A). We then treated 6-wk-old C3GnT +/+ and C3GnT −/− males with 2% DSS in drinking water for 7 d, followed by 4 d of water without DSS. These experimental conditions induced colitis in both C3GnT +/+ and C3GnT −/− mice. However, the colitis in the C3GnT −/− mice was markedly more severe, with greater weight loss, diarrhea, fecal bleeding, and shortened colon length (Fig. 4, B–D, and Fig. S8, available at http://www.jem.org/cgi/content/full/jem.20061929/DC1). The inflammation was restricted to the colon, especially in the distal colonic region, whereas the small intestine was not substantially affected (Fig. S3). Histological examination showed that both C3GnT +/+ and C3GnT −/− colonic tissues appeared normal before DSS treatment. However, after 2% DSS treatment, C3GnT −/− mice had more severe crypt destruction, larger areas of epithelial ulceration, erosions, and massive inflammatory cell infiltration into the mucosal tissue (Fig. 4, E and F).
Figure 4.

C3GnT−/− mice have more severe DSS-induced colitis. (A) Survival rate of a cohort of 10 age-matched C3GnT +/+ and C3GnT −/− males after a 14-d, 2.5% DSS treatment. (B–D) Percent body weight changes compared with baseline, diarrhea score, and fecal blood score in 6-wk-old C3GnT +/+ and C3GnT −/− males after a 7-d, 2% DSS treatment. Each point represents the mean ± the SEM. n = 10. (E) Hematoxylin and eosin–stained colonic tissues of C3GnT +/+ and C3GnT −/− mice with or without 2% DSS treatment. Bar, 100 μm. (F) Histopathological scores of C3GnT +/+ and C3GnT −/− mice 7 d after 2% DSS treatment based on degrees of inflammatory cell infiltration and epithelial injury. Error bars indicate mean ± the SEM. n = 12.
To characterize the infiltrating inflammatory cells associated with DSS-induced colitis, we probed C3GnT +/+ and C3GnT −/− colonic tissues with anti-CD3 antibody to identify T lymphocytes and anti-F4/80 antibody to identify monocytes/macrophages. Compared with C3GnT +/+ mice, there was a dramatic increase of both T cells and monocytes/macrophages in the lamina propria of C3GnT −/− mice (Fig. 5, A and B).
Figure 5.

C3GnT−/− colon has increased infiltration of T lymphocytes and monocytes/macrophages, and C3GnT−/− CD4 T cells have increased intracellular expression of IL-2, IFN-γ, and TNF-α after 2% DSS treatment. (A) CD3-positive lymphocyte (green) or F4/80-positive monocyte/macrophage (red) infiltrates C3GnT +/+ and C3GnT −/− colon after 2% DSS treatment. Bar, 50 μm. (B) Positive staining areas were quantified as pixels per high-powered microscopic field (20×) using Photoshop based on six sections from three independent mice from each group. Error bars indicate the mean ± the SD. (C) Flow cytometric analysis of intracellular cytokine expression in splenic CD4+ T cells isolated from either C3GnT +/+ or C3GnT −/− mice after 2% DSS treatment. The plots were gated on CD3+ population. The number in each image indicates the percentage of CD4+ T cells. This result is representative of data of three experiments (three C3GnT +/+ and three C3GnT −/− mice in each experiment). (D) Flow cytometric analysis of intracellular cytokine expression in CD3+ IEL and LPL isolated from the colon of C3GnT +/+ or C3GnT −/− mice without DSS treatment. The number in each image indicates the percentages of cytokine-expressing CD3+ cells. This result is representative of data of age-matched males in the 129/SvlmJ background (three C3GnT +/+ and four C3GnT −/− mice).
To further understand the immunological features of DSS-induced colitis in C3GnT −/− mice, we evaluated intracellular production of the cytokines IL-2, IL-17, IFN-γ, and TNF-α in splenocytes (30). Compared with DSS-treated C3GnT +/+ mice, splenic CD4 T cells from DSS-treated C3GnT −/− mice exhibited modestly elevated levels of IL-2, IFN-γ, and TNF-α, suggesting that effector activation of CD4+ T cells associated with DSS-induced mucosal inflammation is greater in C3GnT −/− mice (Fig. 5 C).
C3GnT −/− mice are highly susceptible to DSS-induced colitis. We speculated that C3GnT −/− intestinal mucosal lymphocytes might be activated because of an impaired mucosal barrier function, even in the absence of DSS challenge. We thus examined TNF-α, IFN-γ, IL-17, and IL-6 expression in intraepithelial lymphocytes (IELs) and lamina propria lymphocytes (LPLs) from colons of C3GnT +/+ and C3GnT −/− mice without DSS treatment. Notably, the expression of the proinflammatory cytokines TNF-α, IFN-γ, and IL-17 by CD3+ IELs and LPLs was significantly increased in C3GnT −/− mice as compared with C3GnT +/+ mice (Fig. 5 C). This suggests an inflammation-prone status in intestines of C3GnT −/− mice. Interestingly, IL-6 expression was increased in the IELs of C3GnT −/− mice (Fig. 5 D), which may contribute to the increased susceptibility to colorectal tumor in the mice (31).
Accelerated colorectal tumorigenesis in C3GnT −/− mice after azoxymethane (AOM) and DSS treatment
Patients with ulcerative colitis exhibit increased risk of colorectal cancer (32, 33). Abnormal O-glycans such as the Tn antigen are expressed in >90% of colorectal cancers, but are absent in normal colonic mucosa (6, 17). Thus, we explored whether mice lacking core 3 O-glycans are susceptible to colorectal tumorigenesis. We used the well-established AOM/DSS model (34). 12 or 20 wk after AOM/DSS treatment, C3GnT −/− colon and rectum exhibited a fivefold increase in tumor volume (Fig. 6, A and B). Occasionally, small adenomas were found in the proximal colon, but no tumors were observed in the cecum, ileum, and jejunum of C3GnT +/+ or C3GnT −/− mice (unpublished data). Histologically, most lesional tissues were composed of proliferating adenomatous columnar epithelium with prominent architectural and cytological atypia. The changes were consistent with tubular adenoma featuring extensive high-grade dysplasia (intramucosal carcinoma). In C3GnT −/− mice, 20 wk after treatment, some of the adenomas progressed to adenocarcinomas that invaded muscularis mucosae and submucosa (Fig. 6 C).
Figure 6.

Accelerated colorectal tumorigenesis in C3GnT−/− mice after AOM/DSS treatment. (A) Representative luminal views of C3GnT +/+ and C3GnT −/− colons. Arrows indicate tumors. (B) Quantification of colonic tumor volume in C3GnT +/+ and C3GnT −/− mice. Error bars represent the mean ± the SD. n = 7. (C) Representative histopathological features of colorectal adenocarcinomas in C3GnT −/− mice. Arrowhead points to invasive neoplastic glands. Dashed lines depict the muscularis mucosae. (D) Quantification of BrdU-labeled proliferating colonic epithelial cells of C3GnT +/+ and C3GnT −/− mice with or without AOM/DSS treatments. Error bars indicate the mean ± the SD. n = 5. (E) Expression of β-catenin, TCF-4, c-myc, and cyclin-D1 in C3GnT +/+ and C3GnT −/− colonic tissues measured by Western blots. β-Actin was used as a loading control. The data represent at least three experiments. (F) Immunohistochemical staining illustrates nuclear localization of β-catenin in C3GnT −/− colonic neoplastic tissues (dark brown, arrows), whereas the nuclei of control normal epithelial cells had no β-catenin staining (white arrowheads). The data are representative of at least three experiments. Bars: (A) 1 cm; (C) 100 μm; (F) 50 μm.
To investigate the cellular mechanisms by which a deficiency of core 3–derived O-glycans affects colorectal tumorigenesis, we used bromodeoxyuridine (BrdU) metabolic labeling to examine the rate of colonic epithelial proliferation (25). 3 h after BrdU intraperitoneal injection, the number of BrdU-positive colonic epithelial cells per crypt in C3GnT −/− mice was significantly greater than those in C3GnT +/+ mice with or without AOM/DSS treatment (Fig. 6 D). These results indicate that the absence of core 3–derived O-glycans increases colonic epithelial cell proliferation in C3GnT −/− mice.
Wnt signaling is one of the key signaling pathways in colorectal cancer development in humans (35). Western blot analysis showed that the expression of β-catenin and TCF4, which are components of the Wnt pathway, was significantly greater in tumor and nontumor colon tissue extracts from C3GnT −/− mice (Fig. 6 E). C3GnT −/− tissues also had much more cyclin D1 and c-myc, which are downstream targets of β-catenin and TCF-4. Immunohistochemistry revealed strong nuclear translocation of β-catenin in C3GnT −/− tumor tissues (Fig. 6 F). These data suggest that the molecular mechanism of tumorigenesis in C3GnT −/− mice is similar to that in humans.
DISCUSSION
To investigate the function of core 3–derived O-glycans, we generated mice with a targeted deletion of the C3GnT gene. Disruption of the gene eliminated core 3–derived O-glycans in the mouse colon, resulting in reduced expression of Muc2 and increased intestinal barrier permeability. Furthermore, C3GnT −/− mice exhibited increased susceptibility to DSS-induced colitis and AOM/DSS-induced colorectal adenocarcinomas. These data demonstrate the importance of core 3–derived O-glycans in intestinal function.
Factors controlling O-glycan structure in the colon
Unlike core 1-derived O-glycans that are present in most tissues (1, 22), core 3–derived O-glycans are expressed predominantly in colonic epithelial cells (demonstrated here by lacZ reporter staining in C3GnT −/− mice), implying an important function in colon. Our MALDI-TOF-MS analyses indicated that disruption of the C3GnT gene eliminated all core 3–derived O-glycans, including core 4, and other core 3–derived O-glycan variants. These data provide definitive evidence that C3GnT gene encodes all measurable C3GnT activity. This distinguishes C3GnT from many glycosyltransferases that usually exist as multigene families with related structures and functions (1, 2). Interestingly, the common core 1 O-glycan Galβ1,3GalNAcα1-Ser/Thr is generated by the activity of the core 1 β1,3-galactosyltransferase (T-synthase), which, like C3GnT, is also encoded by a single gene (36).
Tn antigen (GalNAcα-O-Ser/Thr), which is the only structure common to all mucin-type O-glycans, is normally hidden because it is a substrate for different types of additional glycosylation. These include sialylation to form the sialyl-Tn antigen or extensions to form core 1– or core 3–derived O-glycans (Fig. 1 A). As expected, we found that deficiency of C3GnT resulted in the exposure of Tn antigen. Desialylation did not enhance Tn expression, indicating that sialic acid did not cap most of the exposed Tn antigens to form the sialyl-Tn antigen. Furthermore, HPLC and MALDI-TOF-MS analyses demonstrated that the absence of core 3–derived O-glycans did not cause compensatory increase in the biosynthesis of core 1-derived O-glycans (Fig. 2 A and not depicted). These data are consistent with our previous observation that the Tn antigen, which was exposed by global targeted deletion of core 1–derived O-glycans, was not modified by sialylation during mouse embryonic development (22).
Mouse colon expresses enzymes to generate both core 1– and core 3–derived O-glycans, and expresses α2,6-sialyltransferase that modifies the Tn antigen (1, 2, 19, 22). Therefore, our data argue against the idea that the formation of core 1 and core 3 O-glycans may compete because both types of O-glycans share Tn antigen as a substrate, and that exposed Tn antigens are always modified by sialylation. Such notions have been suggested as potential mechanisms for the aberrant expression of some cancer cell-related O-glycan antigens (20). Our data suggest that substrate specificities of the glycosyltransferases for core 1 or core 3 O-glycans may be determined not only by the Tn antigen, but also by other factors, such as rate-limiting enzymatic activity, sequence of the acceptor peptide, or neighboring glycosylation, as exhibited by some of the UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases (37).
Core 3 O-glycans and colitis
Ulcerative colitis is limited to the colon, and typically presents in the rectum, where the mucus layer is the most elaborate (3–6, 38, 39). Although the factors responsible for this regional variation are unknown, the deterioration of the mucus layer of the colon is prominent in ulcerative colitis. Indeed, the impaired expression of mucins has long been documented in patients with ulcerative colitis (3–5, 18, 38, 39). The expression of Muc2, which is the primary component of colonic mucus layer, is lower in patients with active ulcerative colitis (6, 40). Truncated intestinal O-glycans, such as Tn antigen, are also detected in patients with ulcerative colitis and are associated with abnormal mucin expression (6, 17, 18). In such clinical studies, it is difficult to ascertain whether mucin deficiency or altered O-glycan expression is causal or is secondary to inflammation. The relationship between impaired mucin expression and altered O-glycans has also been unclear. Recently, the possibility that impaired mucin production is a causal factor in colitis was supported by targeted deletion of the gene encoding Muc2 in mice, which developed spontaneous colitis and more severe DSS-induced colitis (41).
Our study adds a new dimension to this theme by demonstrating that a qualitative change of O-glycan in colonic mucin is sufficient to increase susceptibility to colitis, as well as to colorectal tumorigenesis. Unlike the recent report on Muc2 −/− mice (41), C3GnT −/− mice did not undergo spontaneous colitis, and their inflammatory response to DSS was manifested more by mononuclear inflammation than by crypt abscesses. These differences may reflect distinct effects of quantitative vs. qualitative Muc2 abnormalities on the colonic mucosal response to injury. Quantitatively, Muc2 protein levels in the absence of core 3 O-glycans were reduced, but still reached ∼40% of native levels. The function of this residual Muc2 protein may be sufficient to protect mice from spontaneous colitis. However, it should also be noted that genetic background and environmental factors, such as resident microbiota, contribute to the colitis phenotype in genetically susceptible mice (42). Indeed, spontaneous colitis in Muc2 −/− mice appeared to depend on the 129SV genetic background, as it was not observed in the initial description of the Muc2 −/− mice on the C57BL6/129SV mixed background (25, 41). Most C3GnT −/− mice used in this study were in a C57/BL6-129/SvlmJ mixed background, which may contribute to the variations in the colitis phenotype.
The increased DSS susceptibility of C3GnT −/− mice suggests that altered O-glycosylation is a causal factor for abnormal mucin expression in colitis. Abnormal expression of O-glycans may change mucin intracellular trafficking and secretion, or may increase access to proteolytic degradation. Alterations in the structure or expression of glycosyltransferases have been implicated in the etiology of several human diseases (1, 2, 43). Dysregulated expression or genetic mutations of the C3GnT gene might contribute to the pathogenesis of some patients with ulcerative colitis. Genome-wide scans have identified some loci that are associated with IBD susceptibility. No genes regulating O-glycosylation have yet been identified in the known IBD loci (44). However, glycosylation pathways are complex, and they are sometimes promoted by enzymes or factors that function indirectly (1, 21). For example, the activity of T-synthase, which is a key enzyme for core 1–derived O-glycans, is dependent on a unique molecular chaperone called Cosmc (21). Thus, strategies focusing on the variety of genes controlling O-glycosylation may test whether they represent genetic risk factors for ulcerative colitis and colorectal cancer.
It is not yet known whether patients with ulcerative colitis or colorectal cancer have impaired mucin formation as a primary trait or as a secondary response to inflammation (3–5, 17, 18, 38, 39). Our findings indicate that, in either context, such an impairment undermines mucosal resistance to inflammation and to carcinogenesis. A recent study suggested that dysfunction of the intestinal epithelial barrier is the primary cause of SAMP ileitis susceptibility (45). Our results underscore an etiologic role of intestinal barrier function in the pathogenesis of IBD.
Enhanced Escherichia coli adherence and invasion has been found in Crohn's disease and colon cancer, presumably because of altered mucosal glycosylation (46). We found that bacterial 16S rDNA was also increased in C3GnT −/− colonic issues, suggesting that lack of core 3–derived O-glycans causes either an enhanced adherence of bacteria to colonic mucosa or an increase in translocation of bacteria into colonic mucosa. Notably, the expression of several proinflammatory cytokines was increased in C3GnT −/− mucosal lymphocytes from mice that did not receive DSS. This result indicates that deficiency of core 3–derived O-glycans may result in abnormal interactions of intestinal microorganisms and/or their products with mucosal immune cells, may perturb mucosal immune homeostasis, and may thus render C3GnT −/− mice more susceptible to DSS challenge. Moreover, the inflammatory products of these cells have been mechanistically linked to increased epithelial permeability through activation of epithelial myosin light chain kinase (47, 48). Accordingly, conditions that reduce barrier integrity and increase inflammation may promote susceptibility to colitis and inflammation-dependent epithelial neoplasia.
The immunologic response to DSS injury in C3GnT −/− mice included modest TH1 skewing (IL-2, IFN-γ, and TNF-α), but progressed to neither TH17 nor TH2 immune colitis. This distinguishes C3GnT −/− mice from models of TH1 or TH17 colitis (42, 49, 50). In this respect, Muc2 −/− and C3GnT −/− mice may represent different facets of the inflammation that is associated with human ulcerative colitis. The relatively modest immune inflammatory components in Muc2 −/− and C3GnT −/− mice suggest that deficiencies in mucus layer integrity alone are insufficient to drive immune colitis, without the contribution of additional traits affecting innate immune function or immunoregulation (42).
Glycosylation regulates lymphocyte trafficking, and mice transgenic for human α1,2-fucosyltransferase exhibit colitis as a result of defective T cell development resulting from altered lymphocyte glycosylation (51). However, it is unlikely that the C3GnT −/− phenotypes are caused by abnormal glycosylation of lymphocytes because core 3–derived O-glycans are not expressed in hematopoietic cells (19). In addition, the immune organs of thymus and spleen of C3GnT −/− mice were normal, and C3GnT −/− peripheral blood had a normal distribution of various lymphocyte subsets. These data provide no evidence that core 3–derived O-glycans cause abnormal systemic immune development.
Core 3 O-glycans and carcinogenesis
The biologic mechanisms linking mucin abnormalities with colitis and carcinogenesis are not well understood. Altered core 3 O-glycosylation may impair intestinal mucus integrity, allowing abnormal interaction of intestinal luminal contents and commensal microflora with mucosal immune cells. Increased intestinal permeability observed in C3GnT −/− mice supports this possibility. In addition, loss of core 3–derived O-glycans may cause an alteration in the variety and/or the density of intestinal commensal microflora. Thus, compromised intestinal homeostasis may result in chronic activation of mucosal immune cells in C3GnT −/− mice, even without challenge. Elevated expression of TNF-α, IFN-γ, and IL-6 in the C3GnT −/− IELs and LPLs supports this hypothesis. In addition, the increases in lamina propria CD4+ T cells and macrophages and in splenocyte cytokine production suggest that the immune-mediated response to intestinal injury is increased after DSS challenge in C3GnT −/− mice. Products of these and other immune cell types can induce abnormal epithelial cell growth and survival that contribute to carcinogenesis (9, 52).
Greater epithelial cell proliferation in C3GnT −/− mice was observed even in the absence of AOM/DSS treatment. Thus, growth and survival characteristics of C3GnT −/− epithelial cells may respond to increased constitutive exposure to luminal contents, altered commensal microbial composition, or the products that they induce in the local resident mucosal cells. This is comparable to mice impaired in epithelial barrier function because of deficiency of the multiple drug resistance type 1 gene, which also display spontaneous colitis and colonic dysplasia, particularly in the presence of intestinal injury or certain bacterial colonists (33, 53–55). Also, down-regulation of C3GnT expression has been observed in colon carcinoma (20), which may indicate that this trait could be a feature of some epithelium at risk for carcinogenesis. The altered expression of β-catenin/TCF-4, c-myc, and cyclin D1 observed during carcinogenesis in C3GnT −/− mice relates its molecular pathogenesis to that commonly observed in human colorectal cancer (35).
In summary, we have discovered that C3GnT-deficient mice lack core 3–derived O-glycans, exhibit impaired Muc2 expression primarily in colonic tissues, and are highly susceptible to chemical-induced colitis and colorectal tumorigenesis. These data underscore an important in vivo role of core 3–derived O-glycans in intestinal function. The C3GnT −/− mice should be useful to dissect how mucus layer integrity can be manipulated to promote or prevent chronic colitis and colorectal carcinogenesis. Because core 1–derived O-glycans are also a major component of intestinal mucus, generation of mice specifically lacking intestinal core 1–derived O-glycans and mice lacking both core 1– and core 3–derived O-glycans will be valuable to evaluate the overall contribution of O-glycans to intestinal function and to pathogenesis of common intestinal diseases.
MATERIALS AND METHODS
Generation of C3GnT −/− mice.
Based on published human C3GnT DNA and amino acid sequences, National Center for Biotechnology Information database searches were used to identify the corresponding murine C3GnT gene. We generated conventional C3GnT gene-deficient mice (C3GnT −/−), as illustrated in Fig. 1 B, by targeted homologous recombination in murine CJ7 embryonic stem cells (129/SvlmJ origin) using previously described methods (22, 23). In the gene-targeting construct, a neomycin selection marker was used to replace the major part of the coding exon 2 of C3GnT gene (Fig. 1 B). Because there is no available antibody or other molecular probe to C3GnT or core 3 O-glycans, a lacZ reporter was genetically integrated immediately after the endogenous C3GnT promoter region to characterize the expression pattern of C3GnT (Fig. 1 B). Genotypes of mice were initially verified by Southern blotting and then routinely determined by PCR of genomic DNA from tail biopsies. RT-PCR was used to measure C3GnT mRNA transcripts.
Unless specified, 6-wk-old C3GnT −/− male mice in the 129/SvlmJ and C57BL/6J mixed background were used for experiments. Wild-type littermates (C3GnT +/+) were used as controls. All mouse experiments were performed in compliance with protocols approved by the Institutional Animal Care and Use Committees of the Oklahoma Medical Research Foundation and the University of California.
LacZ staining.
5-μm-thick cryosections of different tissues from C3GnT −/− mice were stained for lacZ based on the previously published method (56). In brief, sections on slides were treated with cold 0.2% glutaraldehyde in PBS for 10 min and rinsed three times with lacZ wash buffer (2 mM MgCl2, 0.01% sodium deoxycholate, and 2.5% Nonidet-P40 in PBS). The slides were stained overnight in 0.5 mg/ml X-gal, 5 mM potassium ferrocyanide, and 5 mM potassium ferricyanide in lacZ wash buffer at 37°C. After staining, the slides were rinsed with PBS and counterstained with nuclear fast red.
C3GnT activity assay.
C3GnT activity was measured using GalNAcα1-O-p-Nitrophenyl (GalNAcα-pNP; Sigma-Aldrich) as an acceptor following the established methods, with minor modifications (22). In brief, the assay was performed in a 50-μl reaction containing 50 mM Tris-HCl, pH 7.0, 2 mM GalNAcα-pNP, 200 μM UDP-[3H]GlcNAc (60,000–90,000 cpm; PerkinElmer Life Science), 20 mM MnCl2, 0.1% Triton X-100, and 100 μg protein from tissue extracts from C3GnT +/+ and C3GnT −/− mice. The reactions were incubated at 37°C for 1 h and stopped by adding 950 μl cold H2O. The products were separated from free UDP-[3H]GlcNAc by Sep-Pak (C18) column chromatography and quantified.
Expression of soluble recombinant epitope-tagged C3GnT.
The putative catalytic domain of mouse C3GnT was amplified by PCR using mouse colon cDNA as a template and cloned into a baculovirus expression vector that contains DNA sequence encoding a signal peptide and a human protein C epitope, which is recognized by a Ca2+-dependent mAb (HPC4), upstream of the inserted C3GnT sequence (21). Sf9 insect cells transfected with the vector and an empty vector were used to produce high-titer viral stocks. High Five insect cells grown in serum-free medium at 27°C were infected by the recombinant viruses for 72 h. The soluble recombinant C3GnT was purified from the conditioned medium using a HPC4-UltraLink resin column. 100 ng of purified, epitope-tagged C3GnT or 20 μl of transfected or mock-transfected media were electrophoresed on a SDS-PAGE (4–20%), transferred to a nitrocellulose membrane, and detected with the HPC4 antibody.
Histology.
For histological analysis, organs from C3GnT +/+ and C3GnT −/− mice were fixed in 10% formalin, processed, and embedded in paraffin. 5-μm-thick tissue sections were stained with hematoxylin and eosin. In another experiment, PAS (Sigma-Aldrich) and Alcian blue (Newcomer Supply) were used to stain general intestinal carbohydrate moieties. Transmission electron microscopy was performed as previously described (22).
Intestinal glycan structure analysis.
The analysis was performed following our published protocol (24). In brief, intestinal mucus, including the epithelial cell layer, was collected by gently scraping the luminal surface of intestines from C3GnT +/+ and C3GnT −/− mice. Samples were dried and glycans were released by ammonia-based β elimination. The glycans were labeled with 2-aminobenzamide (2-AB) and separated by a phase column (4.6 × 250 mm; Zorbax NH2; Agilent Technologies) using HPLC equipped with a fluorescence detector (Ex 330 nm and Em 420 nm). O-glycans labeled with 2-AB were collected from the HPLC and analyzed for structure composition using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS; Applied Biosystem) in a linear positive mode. All acquired spectra were smoothed by applying a 19-point Savitzky-Golay smoothing routine. The matrix used for the positive mode was 10 mg/ml 2,5-dihydroxybenzoic acid prepared in 50% acetonitrile and 0.1% trifluoroacetic acid.
In vivo intestinal permeability.
We used an established method to determine if the loss of O-glycans increases intestinal barrier permeability (57). In brief, C3GnT +/+ and C3GnT −/− mice were administered 200 μl of FITC-dextran (600 mg/kg body weight; 4 kD; Sigma-Aldrich) by gavage. Blood was collected 4 h later by retroorbital bleeding. The serum concentration of the FITC-dextran was determined using a fluorimeter (PerkinElmer Life Sciences) with an excitation wavelength at 490 nm and an emission wavelength of 530 nm. Serial-diluted FITC-dextran was used to generate a standard curve. In another experiment, mice were treated with FITC-dextran as described above, and cryosections of the small intestines and colons were prepared for fluorescence microscopy.
Real-time PCR.
Two sets of universal primers specific for the conserved regions of the bacterial 16S rDNA gene were used (58–60). These included 16S1 (forward, 5′-CCATGAAGTCGGAATCGCTAG-3′; reverse, 5′-ACTCCCATGGTGTGACGG-3′) and 16S2 (forward, 5′-TCCTACGGGAGGCAGCAGT-3′; reverse, 5′-GGACTACCAGGGTATCTAATCCTGTT-3′). DNA extracted from colonic tissues and colonic fecal materials were used as a template. Colonic tissues were washed carefully before extraction of DNA to remove residual feces. For the quantification of fecal bacterial 16S rDNA, DNA from each mouse fecal sample was quantified, and 2.5 pg DNA was used as a template in each PCR reaction. For the quantification of 16S rDNA in colonic tissues, a pair of primers specific to the mouse P-selectin gene (forward, 5′-ATGATTAGCAAATTCTAGCTCCTGTTT-3′; reverse, 5′-TAGGTCTCTTAGGATCTCCCTTCAAT-3′) was used in a separate reaction as the endogenous control to normalize the DNA loading between samples. Real-time PCR was performed on an ABI Prism 7000 spectrofluorometric thermal cycler (Applied Biosystems) using SYBR-green as a double-stranded DNA–specific binding dye. The relative amount of 16S rDNA in each sample was estimated using the ΔΔCT method following the manufacturer's protocol. Each sample was assayed in duplicate.
Immunohistochemistry.
To characterize intestinal inflammatory cell infiltrates, cryosections of colons from C3GnT −/− and C3GnT +/+ mice were first incubated with a hamster polyclonal antibody to the lymphocyte marker, CD3 (1:50 dilution; BD Biosciences), or a rat mAb to the macrophage marker, F4/80 (1:200 dilution; Accurate Chemical and Scientific Co.) for 1 h at room temperature. The sections were then incubated for 30 min with biotin-conjugated anti–hamster or rat IgG, and subsequently incubated for 30 min with fluorescein isothiocyanate–conjugated avidin D or Texas red avidin D (Vector Laboratories), respectively. The sections were mounted with Vectashield mounting medium (Vector Laboratories) and analyzed by a fluorescent microscope (ECLIPSE E600; Nikon).
For immunohistochemical detection of Muc2, ITF, and β-catenin, deparaffinized sections were boiled for 20 min in 0.01 M citrate buffer, pH 6.0, for epitope retrieval. The sections were then blocked for 20 min using a protein blocking kit (DakoCytomation) for nonspecific antibody binding. Endogenous avidin-binding proteins were blocked using avidin/biotin blocking reagents (Vector Laboratories). Sections were incubated overnight at 4°C with rabbit anti-Muc2 antibody (1:200; Santa Cruz Biotechnologies), with goat anti-ITF antibody (1:200; Santa Cruz Biotechnologies), with mouse anti–β-catenin mAb (1:500; Transduction Laboratories), or with isotype-matched control IgG, respectively. The sections were subsequently incubated with biotinylated anti–rabbit, –goat, or –mouse IgG antibodies (Vector Laboratories) for 45 min, followed by a 20-min incubation with 0.6% H2O2 in methanol to inhibit endogenous peroxidase activity. The sections were finally incubated with horseradish peroxidase (HRP)-streptavidin (Vector Laboratories) for 30 min, and then developed with a diaminobenzidine substrate and counterstained with hemotoxylin.
For Tn antigen staining, deparaffinized sections were incubated with or without 0.5 U/ml sialidase from Arthrobacter ureafaciens (Roche) at 37°C for 3 h. Sections were incubated for 30 min with biotinylated mAb against the Tn antigen (mouse IgG) or with isotype-matched control mouse IgG. Bound antibodies were detected with HRP-conjugated streptavidin (Vector Laboratories).
Immunoblots.
Colon extracts (40 μg of total protein) from C3GnT −/−and C3GnT +/+ mice were resolved by SDS-PAGE under reducing conditions and transferred to a nitrocellulose membrane (Bio-Rad Laboratories). The membrane was blocked with 5% nonfat dry milk overnight and incubated with a rabbit polyclonal antibody to β-catenin (Lab Vision Corp.), a mouse mAb to TCF-4 (Millipore), a rabbit polyclonal antibody to c-myc (Santa Cruz Biotechnologies), a mouse mAb to cyclin D1 (BD Biosciences), or an anti–β-actin antibody (Affinity BioReagents) for loading control. Binding was detected with HRP-conjugated anti–rabbit or –mouse IgG (Vector Laboratories) using enhanced chemiluminescence (GE Healthcare).
DSS-induced colitis model.
To determine the lethality of DSS, mice were fed 2.5% DSS (40 kD; MP Biomedicals, Inc.) in drinking water for 14 d. To induce colitis, C3GnT +/+ and C3GnT −/− males were treated with 2% DSS dissolved in drinking water for 7 d, followed by 4 d of regular water. Mice were then killed for histological analysis.
Body weight, the presence of occult or gross blood per rectum, stool consistency, and mortality were monitored daily during the course of treatments. The clinical score was assessed based on these measurements and determined according to a published method (61). Histological scoring was performed in a blinded fashion as a combined score of inflammatory cell infiltration and tissue damage (61).
Intracellular cytokine staining.
Intracellular cytokine staining was performed to assess the level of cytokines produced by the lymphocytes from the spleens of DSS-treated C3GnT +/+ and C3GnT −/− mice, as well as intestinal mucosal lymphocytes from C3GnT +/+ and C3GnT −/− mice without DSS treatment.
In brief, splenocytes were isolated from DSS-treated C3GnT +/+ and C3GnT −/− mice after lysis of erythrocytes with ACK Lysis Buffer (Cambrex Bio Science). Intestinal IELs and LPLs were isolated from the large intestines of C3GnT +/+ and C3GnT −/− males (129/SvlmJ background), as previously described with minor modifications (62). For intracellular cytokine staining, isolated lymphocytes were activated with Leukocyte Activation Cocktail containing BD GolgiPlug (BD Biosciences) at 5 × 105 cells/well in 96-well plates and cultured for 4–5 h in 5% CO2 at 37°C. Activated cells were harvested for surface and intracellular cytokine staining. Surface staining of the cells was performed by incubating with FITC-, APC-, and PerCP-conjugated mAbs specific to CD3, CD4, and CD19 for 30 min on ice. After surface staining, the cells were fixed and permeabilized for 20 min at room temperature using BD Cytofix/Cytoperm buffer and washed twice with the BD Perm/Wash buffer (both BD Biosciences). Intracellular cytokine staining was performed by incubating with the PE-conjugated antibodies against murine IL-2, -6, -10, -12, -17, TNF-α, and IFN-γ on ice for 15 min. The cells were washed three times with BD Perm/Wash buffer and analyzed with a FACSCalibur flow cytometer (BD Biosciences).
AOM/DSS-induced mouse colorectal tumorogenesis model.
6-wk-old C3GnT +/+ and C3GnT −/− males were given a single intraperitoneal injection of AOM (10 mg/kg body weight; Sigma-Aldrich) (34). Starting 1 wk after injection, mice were fed with 1.75% DSS in drinking water for 7 d and then received no further treatment. Mice were killed for histopathological analysis 12 or 20 wk after the 7-d DSS treatment. The colon was cut open longitudinally and examined for the presence of tumors. The number and size of tumors were recorded. The tumor volumes were determined by measuring the length (l) and the width (w) and calculated volume (V = lw 2/2), as previously described (63). Tissues were fixed in 10% formalin and embedded in paraffin for histopathological analysis. Fresh colonic tissue lysates from C3GnT +/+ and C3GnT −/− mice with or without AOM/DSS treatments were prepared for Western blot analysis.
In vivo BrdU labeling assay.
Mice were injected intraperitoneally with BrdU (20 mg/kg of body weight; Sigma-Aldrich) (25). 3 h after BrdU injection, mice were killed, and colonic tissues were processed for immunochemical analysis. Incorporation of BrdU in proliferating intestinal epithelial cells was detected with a rat mAb to BrdU (1:100 dilution; Accurate Chemical and Scientific Co.). The number of BrdU-positive cells per crypt column was quantified.
Statistical analysis.
Data were analyzed by the Student's t test. The Wilcoxon signed-rank test was used to compare data that did not satisfy the Student's t test. P < 0.05 was considered significant.
Online supplemental material.
Fig. S1 shows murine C3GnT genomic structure and deduced protein sequence. Fig. S2 shows characterization of murine C3GnT. Fig. S3 presents histology (hematoxylin and eosin stain) of C3GnT +/+ and C3GnT −/− small intestines from 6-wk-old mice. Fig. S4 demonstrates that the immune organs and immune cells develop normally in C3GnT −/− mice. Fig. S5 shows MADI-TOF-MS standards of 2-AB–labeled core 1 and core 3 O-glycans, as well as MALDI-TOF-MS of 2-AB–labeled O-glycans of mouse colon and predicted compositions in a linear positive mode. Fig. S6 shows glycan expression profiles in C3GnT +/+ and C3GnT −/− small intestines. Fig. S7 is an analysis of mRNA transcripts of different intestinal mucins from C3GnT +/+ and C3GnT −/− colons, as well as morphological comparisons of C3GnT +/+ and C3GnT −/− goblet cells. Fig. S8 compares colon length of C3GnT +/+ and C3GnT −/− mice after DSS-induced colitis. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20061929/DC1.
Supplemental Material
Acknowledgments
We thank Rodger P. McEver for insightful suggestions and critical reading of the manuscript. We thank Zoltan Laszik for pathology consultation and Samuel McGee, Zhenghui Liu, Ouyang Hui, and Todd Walker for technical support. Microinjection of blastocysts was performed by Kristy Edwards at the Oklahoma Medical Research Foundation microinjection facility, which is directed by Ute Hochgeschwender. Tissue processing and transmission electron microscopy were performed in the imaging core facility of the Oklahoma Medical Research Foundation.
This work was supported by National Institutes of Health grants RR018758 (L. Xia and R.D. Cummings) and DK069434 (J. Braun), and by Senior Research (L. Xia) and Career Development (B. Wei) Awards from the Crohn's and Colitis Foundation of America.
The authors have no conflicting financial interests.
Abbreviations used: AOM, azoxymethane; C3GnT, core 3 β1,3-N-acetylglucosaminyltransferase; DSS, dextran sodium sulfate; HRP, horseradish peroxidase; IBD, inflammatory bowel disease; IEL, intraepithelial lymphocyte; ITF, intestinal trefoil factor; LPL, lamina propria lymphocyte; O-glycans, O-linked oligosaccharides; PAS, periodic acid-Schiff's reagent.
References
- 1.Varki, A., R. Cummings, J. Esko, H. Freeze, G. Hart, and J. Marth. 1999. Essentials of Glycobiology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York. 653 pp. [PubMed]
- 2.Brockhausen, I., and W. Kuhns. 1997. Glycoproteins and Human Disease. R.G. Landes Company, Austin, Texas. 204 pp.
- 3.Rhodes, J.M. 1996. Unifying hypothesis for inflammatory bowel disease and associated colon cancer: sticking the pieces together with sugar. Lancet. 347:40–44. [DOI] [PubMed] [Google Scholar]
- 4.Rhodes, J.M. 1997. Colonic mucus and ulcerative colitis. Gut. 40:807–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Podolsky, D.K., and K.J. Isselbacher. 1984. Glycoprotein composition of colonic mucosa. Specific alterations in ulcerative colitis. Gastroenterology. 87:991–998. [PubMed] [Google Scholar]
- 6.Corfield, A.P., D. Carroll, N. Myerscough, and C.S. Probert. 2001. Mucins in the gastrointestinal tract in health and disease. Front. Biosci. 6:D1321–D1357. [DOI] [PubMed] [Google Scholar]
- 7.Macfarlane, S., E.J. Woodmansey, and G.T. Macfarlane. 2005. Colonization of mucin by human intestinal bacteria and establishment of biofilm communities in a two-stage continuous culture system. Appl. Environ. Microbiol. 71:7483–7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ponda, P.P., and L. Mayer. 2005. Mucosal epithelium in health and disease. Curr. Mol. Med. 5:549–556. [DOI] [PubMed] [Google Scholar]
- 9.Macdonald, T.T., and G. Monteleone. 2005. Immunity, inflammation, and allergy in the gut. Science. 307:1920–1925. [DOI] [PubMed] [Google Scholar]
- 10.Kabashima, K., T. Saji, T. Murata, M. Nagamachi, T. Matsuoka, E. Segi, K. Tsuboi, Y. Sugimoto, T. Kobayashi, Y. Miyachi, et al. 2002. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J. Clin. Invest. 109:883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clayburgh, D.R., T.A. Barrett, Y. Tang, J.B. Meddings, L.J. Van Eldik, D.M. Watterson, L.L. Clarke, R.J. Mrsny, and J.R. Turner. 2005. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J. Clin. Invest. 115:2702–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Resta-Lenert, S., J. Smitham, and K.E. Barrett. 2005. Epithelial dysfunction associated with the development of colitis in conventionally housed mdr1a−/− mice. Am. J. Physiol. Gastrointest. Liver Physiol. 289:G153–G162. [DOI] [PubMed] [Google Scholar]
- 13.Rakoff-Nahoum, S., J. Paglino, F. Eslami-Varzaneh, S. Edberg, and R. Medzhitov. 2004. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 118:229–241. [DOI] [PubMed] [Google Scholar]
- 14.Elson, C.O., R.B. Sartor, S.R. Targan, and W.J. Sandborn. 2003. Challenges in IBD Research: updating the scientific agendas. Inflamm. Bowel Dis. 9:137–153. [DOI] [PubMed] [Google Scholar]
- 15.Morteau, O., S.G. Morham, R. Sellon, L.A. Dieleman, R. Langenbach, O. Smithies, and R.B. Sartor. 2000. Impaired mucosal defense to acute colonic injury in mice lacking cyclooxygenase-1 or cyclooxygenase-2. J. Clin. Invest. 105:469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elson, C.O., Y. Cong, V.J. McCracken, R.A. Dimmitt, R.G. Lorenz, and C.T. Weaver. 2005. Experimental models of inflammatory bowel disease reveal innate, adaptive, and regulatory mechanisms of host dialogue with the microbiota. Immunol. Rev. 206:260–276. [DOI] [PubMed] [Google Scholar]
- 17.Byrd, J.C., and R.S. Bresalier. 2004. Mucins and mucin binding proteins in colorectal cancer. Cancer Metastasis Rev. 23:77–99. [DOI] [PubMed] [Google Scholar]
- 18.Jass, J.R., and M.D. Walsh. 2001. Altered mucin expression in the gastrointestinal tract: a review. J. Cell. Mol. Med. 5:327–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwai, T., N. Inaba, A. Naundorf, Y. Zhang, M. Gotoh, H. Iwasaki, T. Kudo, A. Togayachi, Y. Ishizuka, H. Nakanishi, and H. Narimatsu. 2002. Molecular cloning and characterization of a novel UDP-GlcNAc:GalNAc-peptide beta1,3-N-acetylglucosaminyltransferase (beta 3Gn-T6), an enzyme synthesizing the core 3 structure of O-glycans. J. Biol. Chem. 277:12802–12809. [DOI] [PubMed] [Google Scholar]
- 20.Iwai, T., T. Kudo, R. Kawamoto, T. Kubota, A. Togayachi, T. Hiruma, T. Okada, T. Kawamoto, K. Morozumi, and H. Narimatsu. 2005. Core 3 synthase is down-regulated in colon carcinoma and profoundly suppresses the metastatic potential of carcinoma cells. Proc. Natl. Acad. Sci. USA. 102:4572–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ju, T., and R.D. Cummings. 2002. A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc. Natl. Acad. Sci. USA. 99:16613–16618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia, L., T. Ju, A. Westmuckett, G. An, L. Ivanciu, J.M. McDaniel, F. Lupu, R.D. Cummings, and R.P. McEver. 2004. Defective angiogenesis and fatal embryonic hemorrhage in mice lacking core 1–derived O-glycans. J. Cell Biol. 164:451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia, L., M. Sperandio, T. Yago, J.M. McDaniel, R.D. Cummings, S. Pearson-White, K. Ley, and R.P. McEver. 2002. P-selectin glycoprotein ligand-1-deficient mice have impaired leukocyte tethering to E-selectin under flow. J. Clin. Invest. 109:939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia, B., J.A. Royall, G. Damera, G.P. Sachdev, and R.D. Cummings. 2005. Altered O-glycosylation and sulfation of airway mucins associated with cystic fibrosis. Glycobiology. 15:747–775. [DOI] [PubMed] [Google Scholar]
- 25.Velcich, A., W. Yang, J. Heyer, A. Fragale, C. Nicholas, S. Viani, R. Kucherlapati, M. Lipkin, K. Yang, and L. Augenlicht. 2002. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science. 295:1726–1729. [DOI] [PubMed] [Google Scholar]
- 26.Itoh, H., P.L. Beck, N. Inoue, R. Xavier, and D.K. Podolsky. 1999. A paradoxical reduction in susceptibility to colonic injury upon targeted transgenic ablation of goblet cells. J. Clin. Invest. 104:1539–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macpherson, A.J., and K. Smith. 2006. Mesenteric lymph nodes at the center of immune anatomy. J. Exp. Med. 203:497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turnbull, E.L., U. Yrlid, C.D. Jenkins, and G.G. Macpherson. 2005. Intestinal dendritic cell subsets: differential effects of systemic TLR4 stimulation on migratory fate and activation in vivo. J. Immunol. 174:1374–1384. [DOI] [PubMed] [Google Scholar]
- 29.Stevceva, L., P. Pavli, A.J. Husband, and W.F. Doe. 2001. The inflammatory infiltrate in the acute stage of the dextran sulphate sodium induced colitis: B cell response differs depending on the percentage of DSS used to induce it. BMC Clin. Pathol. 1:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wei, B., P. Velazquez, O. Turovskaya, K. Spricher, R. Aranda, M. Kronenberg, L. Birnbaumer, and J. Braun. 2005. Mesenteric B cells centrally inhibit CD4+ T cell colitis through interaction with regulatory T cell subsets. Proc. Natl. Acad. Sci. USA. 102:2010–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Becker, C., M.C. Fantini, C. Schramm, H.A. Lehr, S. Wirtz, A. Nikolaev, J. Burg, S. Strand, R. Kiesslich, S. Huber, et al. 2004. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 21:491–501. [DOI] [PubMed] [Google Scholar]
- 32.Rubin, D.T., and N. Parekh. 2006. Colorectal cancer in inflammatory bowel disease: molecular and clinical considerations. Curr. Treat. Options Gastroenterol. 9:211–220. [DOI] [PubMed] [Google Scholar]
- 33.Maggio-Price, L., H. Bielefeldt-Ohmann, P. Treuting, B.M. Iritani, W. Zeng, A. Nicks, M. Tsang, D. Shows, P. Morrissey, and J.L. Viney. 2005. Dual infection with Helicobacter bilis and Helicobacter hepaticus in p-glycoprotein-deficient mdr1a−/− mice results in colitis that progresses to dysplasia. Am. J. Pathol. 166:1793–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka, T., H. Kohno, R. Suzuki, Y. Yamada, S. Sugie, and H. Mori. 2003. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 94:965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bienz, M., and H. Clevers. 2000. Linking colorectal cancer to Wnt signaling. Cell. 103:311–320. [DOI] [PubMed] [Google Scholar]
- 36.Ju, T., K. Brewer, A. D'Souza, R.D. Cummings, and W.M. Canfield. 2002. Cloning and expression of human core 1 beta1,3-galactosyltransferase. J. Biol. Chem. 277:178–186. [DOI] [PubMed] [Google Scholar]
- 37.Ten Hagen, K.G., T.A. Fritz, and L.A. Tabak. 2003. All in the family: the UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases. Glycobiology. 13:1R–16R. [DOI] [PubMed] [Google Scholar]
- 38.Smithson, J.E., A. Campbell, J.M. Andrews, J.D. Milton, R. Pigott, and D.P. Jewell. 1997. Altered expression of mucins throughout the colon in ulcerative colitis. Gut. 40:234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Podolsky, D.K., and D.A. Fournier. 1988. Alterations in mucosal content of colonic glycoconjugates in inflammatory bowel disease defined by monoclonal antibodies. Gastroenterology. 95:379–387. [DOI] [PubMed] [Google Scholar]
- 40.Renes, I.B., J.A. Boshuizen, D.J. Van Nispen, N.P. Bulsing, H.A. Buller, J. Dekker, and A.W. Einerhand. 2002. Alterations in Muc2 biosynthesis and secretion during dextran sulfate sodium-induced colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 282:G382–G389. [DOI] [PubMed] [Google Scholar]
- 41.Van der Sluis, M., B.A. De Koning, A.C. De Bruijn, A. Velcich, J.P. Meijerink, J.B. Van Goudoever, H.A. Buller, J. Dekker, I. Van Seuningen, I.B. Renes, and A.W. Einerhand. 2006. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 131:117–129. [DOI] [PubMed] [Google Scholar]
- 42.Strober, W., I.J. Fuss, and R.S. Blumberg. 2002. The immunology of mucosal models of inflammation. Annu. Rev. Immunol. 20:495–549. [DOI] [PubMed] [Google Scholar]
- 43.Ju, T., and R.D. Cummings. 2005. Protein glycosylation: chaperone mutation in Tn syndrome. Nature. 437:1252. [DOI] [PubMed] [Google Scholar]
- 44.Williams, C.N., K. Kocher, E.S. Lander, M.J. Daly, and J.D. Rioux. 2002. Using a genome-wide scan and meta-analysis to identify a novel IBD locus and confirm previously identified IBD loci. Inflamm. Bowel Dis. 8:375–381. [DOI] [PubMed] [Google Scholar]
- 45.Olson, T.S., B.K. Reuter, K.G. Scott, M.A. Morris, X.M. Wang, L.N. Hancock, T.L. Burcin, S.M. Cohn, P.B. Ernst, F. Cominelli, et al. 2006. The primary defect in experimental ileitis originates from a nonhematopoietic source. J. Exp. Med. 203:541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin, H.M., B.J. Campbell, C.A. Hart, C. Mpofu, M. Nayar, R. Singh, H. Englyst, H.F. Williams, and J.M. Rhodes. 2004. Enhanced Escherichia coli adherence and invasion in Crohn's disease and colon cancer. Gastroenterology. 127:80–93. [DOI] [PubMed] [Google Scholar]
- 47.Zolotarevsky, Y., G. Hecht, A. Koutsouris, D.E. Gonzalez, C. Quan, J. Tom, R.J. Mrsny, and J.R. Turner. 2002. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology. 123:163–172. [DOI] [PubMed] [Google Scholar]
- 48.Blair, S.A., S.V. Kane, D.R. Clayburgh, and J.R. Turner. 2006. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab. Invest. 86:191–201. [DOI] [PubMed] [Google Scholar]
- 49.Fuss, I.J., F. Heller, M. Boirivant, F. Leon, M. Yoshida, S. Fichtner-Feigl, Z. Yang, M. Exley, A. Kitani, R.S. Blumberg, et al. 2004. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J. Clin. Invest. 113:1490–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yen, D., J. Cheung, H. Scheerens, F. Poulet, T. McClanahan, B. McKenzie, M.A. Kleinschek, A. Owyang, J. Mattson, W. Blumenschein, et al. 2006. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Invest. 116:1310–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown, S.J., A.M. Miller, P.J. Cowan, J. Slavin, W.R. Connell, G.T. Moore, S. Bell, P.R. Elliott, P.V. Desmond, and A.J. d'Apice. 2004. Altered immune system glycosylation causes colitis in alpha1,2-fucosyltransferase transgenic mice. Inflamm. Bowel Dis. 10:546–556. [DOI] [PubMed] [Google Scholar]
- 52.Sartor, R.B. 2003. Targeting enteric bacteria in treatment of inflammatory bowel diseases: why, how, and when. Curr. Opin. Gastroenterol. 19:358–365. [DOI] [PubMed] [Google Scholar]
- 53.Panwala, C.M., J.C. Jones, and J.L. Viney. 1998. A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J. Immunol. 161:5733–5744. [PubMed] [Google Scholar]
- 54.Banner, K.H., C. Cattaneo, J.L. Le Net, A. Popovic, D. Collins, and J.D. Gale. 2004. Macroscopic, microscopic and biochemical characterisation of spontaneous colitis in a transgenic mouse, deficient in the multiple drug resistance 1a gene. Br. J. Pharmacol. 143:590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.ten Hove, T., P. Drillenburg, J. Wijnholds, A.A. Te Velde, and S.J. van Deventer. 2002. Differential susceptibility of multidrug resistance protein-1 deficient mice to DSS and TNBS-induced colitis. Dig. Dis. Sci. 47:2056–2063. [DOI] [PubMed] [Google Scholar]
- 56.Lobe, C.G., K.E. Koop, W. Kreppner, H. Lomeli, M. Gertsenstein, and A. Nagy. 1999. Z/AP, a double reporter for cre-mediated recombination. Dev. Biol. 208:281–292. [DOI] [PubMed] [Google Scholar]
- 57.Furuta, G.T., J.R. Turner, C.T. Taylor, R.M. Hershberg, K. Comerford, S. Narravula, D.K. Podolsky, and S.P. Colgan. 2001. Hypoxia-inducible factor 1–dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J. Exp. Med. 193:1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corless, C.E., M. Guiver, R. Borrow, V. Edwards-Jones, E.B. Kaczmarski, and A.J. Fox. 2000. Contamination and sensitivity issues with a real-time universal 16S rRNA PCR. J. Clin. Microbiol. 38:1747–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nadkarni, M.A., F.E. Martin, N.A. Jacques, and N. Hunter. 2002. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology. 148:257–266. [DOI] [PubMed] [Google Scholar]
- 60.Ott, S.J., M. Musfeldt, U. Ullmann, J. Hampe, and S. Schreiber. 2004. Quantification of intestinal bacterial populations by real-time PCR with a universal primer set and minor groove binder probes: a global approach to the enteric flora. J. Clin. Microbiol. 42:2566–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Araki, A., T. Kanai, T. Ishikura, S. Makita, K. Uraushihara, R. Iiyama, T. Totsuka, K. Takeda, S. Akira, and M. Watanabe. 2005. MyD88-deficient mice develop severe intestinal inflammation in dextran sodium sulfate colitis. J. Gastroenterol. 40:16–23. [DOI] [PubMed] [Google Scholar]
- 62.Van der Heijden, P.J., and W. Stok. 1987. Improved procedure for the isolation of functionally active lymphoid cells from the murine intestine. J. Immunol. Methods. 103:161–167. [DOI] [PubMed] [Google Scholar]
- 63.Sun, J., M.A. Blaskovich, D. Knowles, Y. Qian, J. Ohkanda, R.D. Bailey, A.D. Hamilton, and S.M. Sebti. 1999. Antitumor efficacy of a novel class of non-thiol-containing peptidomimetic inhibitors of farnesyltransferase and geranylgeranyltransferase I: combination therapy with the cytotoxic agents cisplatin, Taxol, and gemcitabine. Cancer Res. 59:4919–4926. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}