

Abstract
V(D)J recombination and immunoglobulin class switch recombination (CSR) are two somatic rearrangement mechanisms that proceed through the introduction of double-strand breaks (DSBs) in DNA. Although the DNA repair factor XRCC4 is essential for the resolution of DNA DSB during V(D)J recombination, its role in CSR has not been established. To bypass the embryonic lethality of XRCC4 deletion in mice, we developed a conditional XRCC4 knockout (KO) using LoxP-flanked XRCC4 cDNA lentiviral transgenesis. B lymphocyte restricted deletion of XRCC4 in these mice lead to an average two-fold reduction in CSR in vivo and in vitro. Our results connect XRCC4 and the nonhomologous end joining DNA repair pathway to CSR while reflecting the possible use of an alternative pathway in the repair of CSR DSB in the absence of XRCC4. In addition, this new conditional KO approach should be useful in studying other lethal mutations in mice.
In early B and T cells, variable domains of antigen receptors are somatically rearranged from V, D, and J gene loci by V(D)J recombination (1, 2). The lymphoid-specific factors recombination-activating gene 1 (Rag1) and Rag2 initiate V(D)J recombination by introducing a DNA double-strand break (DSB) at the junction between V, D, or J coding sequences and recombination signal sequences. DNA DSBs are then resolved by the nonhomologous end joining (NHEJ) DNA repair machinery, which is the main pathway for DNA DSB repair in mammalian cells. The following seven NHEJ factors have been identified so far: the heterodimer Ku70/Ku80, which forms the DNA–PK complex with the catalytic subunit of the DNA-dependent rotein kinase, Artemis, Cernunnos/XLF, XRCC4 (X4), and DNA ligase IV. The recently identified Cernunnos/XLF factor, whose precise function is not known, is part of the X4–DNA ligase IV complex (3–5). Mutations in genes coding for Artemis, DNA ligase IV, and Cernunnos (3, 6–8) are responsible for human radiosensitive severe combined immunodeficiency. Likewise, gene inactivation of NHEJ factors in mice results in the absence of mature T and B lymphocytes and embryonic lethality in the case of X4 and DNA ligase IV (9–14).
Upon antigen recognition in secondary lymphoid organs, IgM-expressing mature B cells further diversify their repertoire through class switch recombination (CSR), which exchanges the IgM constant region (Cμ) with a downstream CH gene (γ, ɛ, or α), producing antibodies with a different effector function (15). CSR involves large repetitive switch (S) region sequences located upstream of each CH gene, except for Cδ. Breaks are introduced in the DNA flanking the two participating S regions, followed by their fusion, while the intervening sequence, including Cμ, is excised. CSR requires transcription through the target regions, and is initiated by the activation-induced cytidine deaminase (AID) (16, 17). Several lines of evidence suggest that CSR, like V(D)J recombination, involves the generation of DNA DSB, and could potentially involve NHEJ factors for DNA DSB repair: (a) an episomal circle is generated during CSR (15); (b) DNA DSBs have been detected by ligated mediated PCR (LM-PCR) assays in the Sγ3 region in mitogen-activated B cells (18); (c) foci of the phosphorylated histone H2AX (γH2AX), which form around DNA DSBs, occur at the IgH locus in an AID-dependent manner in B cells activated for CSR (19); and (d) CSR is impaired in mice deficient for H2AX, ATM, p53-binding protein 1 (53BP1) and Nbs1, which are early components of the DNA DSB response (20–26).
CSR cannot be directly analyzed in mice deficient in NHEJ factors, as these mice lack a mature immune system. A monoclonal mature B cell compartment has been reconstituted by introducing rearranged IgH and IgL chain knock-in alleles (HL mice) in mice deficient for NHEJ factors, except for X4 and DNA ligase IV, which are embryonic lethal. Ku70 and Ku80-deficient B cells have severely impaired CSR (20, 27, 28). Although this defect could result from a reduced proliferative capacity or an increased apoptosis when induced to undergo CSR (20, 27), Ku mutant Bcl-2 transgenic B cells that divide still do not undergo CSR (20). Conflicting results were reported concerning the role of DNA-PKcs in CSR (29–32). Finally, Artemis deficiency doesn't have any impact on CSR in such monoclonal B cell mice (33). Although some key NHEJ components seem to be required for CSR, one cannot exclude a role outside of NHEJ per se for these factors. X4 and DNA ligase IV are the only factors without a known function apart from NHEJ. Recently, Pan-Hammerström et al. showed that B cells from patients with hypomorphic DNA ligase IV mutations display an altered pattern of junctional resolution, with increased microhomology in Sμ-Sα junctions (34), suggesting a possible implication of the X4–DNA ligase IV complex during CSR in humans.
We developed a conditional X4 KO mouse model. We show that specific inactivation of X4 in mature B cells results in a partial defect in CSR in vivo and in vitro, revealing the possible usage of an alternative pathway in the absence of XRCC4.
RESULTS
Generation of lentiviral transgenic mice expressing mouse X4
X4-deficient mice are embryonic lethal at the embryonic day (E) 16.5 stage of development, owing to defective neurogenesis manifested by extensive neuronal apoptosis (12). Moreover, X4 deficiency in primary mouse cells results in the incapacity to support V(D)J recombination (12). To reverse these defects, we generated a transgenic mouse line expressing mouse X4 and crossed it onto the X4 KO background. To avoid the drawbacks (low efficiency, multiple transgenic concatemers, overexpression, or silencing) associated with classical transgenesis, we used lentiviral gene delivery to one-cell embryos (35, 36).
The lentiviral vector used to produce X4 transgenic mice (pTRIP-X4; Fig. 1 A) has the following characteristics: (a) the human ubiquitin C (UbiC) promoter as internal promoter to drive the transgene expression; (b) a LoxP site into the ΔU3 of 3′LTR, as the duplication of the 3′LTR upon provirus integration results in the mouse X4 transgene being flanked by two LoxP sites (floxed transgene; Fig. 1 B); and (c) the mouse X4 cDNA is cloned in fusion with a tandem affinity purification (TAP) tag, followed by an ires sequence and the GFP reporter gene. Among various promoters tested (PGK and EF1α), the UbiC proved to have the most pleiotropic specificity, including the hematopoietic system, and a high expression in the brain (unpublished data) (37), which is a prerequisite for the complementation of the embryonic lethality in our model. Using this approach, we obtained 83% transgenic chimeras harboring 1–6 integrated proviruses, as determined by Southern blot and PCR analysis (unpublished data). We selected one chimera with a single integration to derive the X4 transgenic (X4T) line.
Figure 1.

Creation of X4T transgenic mice. (A) Schematic representation of the pTRIP-X4 lentiviral vector. Restriction sites: E, EcoNI; A, AvaII. Filled circle, central DNA flap; filled box, X4 Orf. UbiC, human Ubiquitin C promoter. (B) Schematic representation of pTRIP-X4 provirus integrated in the genome of the X4T transgenic mouse line selected for this study. *, the positions of these sites are specific to the integration site. (C) Southern blot analysis of tail DNA shows a single integration of the transgene in the genome of X4T mice. EcoNI–AvaII–digested DNA from X4T-positive and control littermate X4T-negative mice was hybridized with the 5′ flanking probe A, the internal probe B, and the 3′ flanking probe C as indicated in A. Refer to B for expected sizes of bands. (D) Analysis of X4T expression in transgenic mice. Western blot analysis of X4T (top) and endogenous X4 (bottom) expression in purified splenic mature B cells and thymocytes of X4T mice and control X4T-negative mice (all on a wild-type background) revealed by anti-X4 antibody.
Southern blot analysis with probe A (Fig. 1, B and C) revealed a 764-bp EcoNI–AvaII band common to all possible integrations, and an equimolar, unique, integration- specific EcoNI band of 860 bp, attesting for the single provirus integration. Additional Southern blot analysis with probes B and C (Fig. 1, B and C) confirmed the single integration (a single 633-bp AvaII band revealed by probe C) and the integrity of the transgene (a 3,089-bp AvaII band revealed by probe B). The integration was mapped to chromosome 16 by inverse PCR analysis, in the third intron of the Spag6 gene (unpublished data). Western blot analysis on lymphoid organs confirmed expression of the transgene in spleen and thymus (Fig. 1 D). Although the iresGFP cassette present in the pTRIP vector was functional in vitro, none of the transgenic chimeras expressed GFP in vivo, irrespective of the integration copy number (unpublished data).
X4T reverts the embryonic lethality and reconstitutes B and T cell development in X4-deficient mice
X4T was introduced in the X4 KO background by crosses to derive X4T X4−/− mice, which were born at Mendelian frequency, thus demonstrating the reversal of X4−/− mice embryonic lethality.
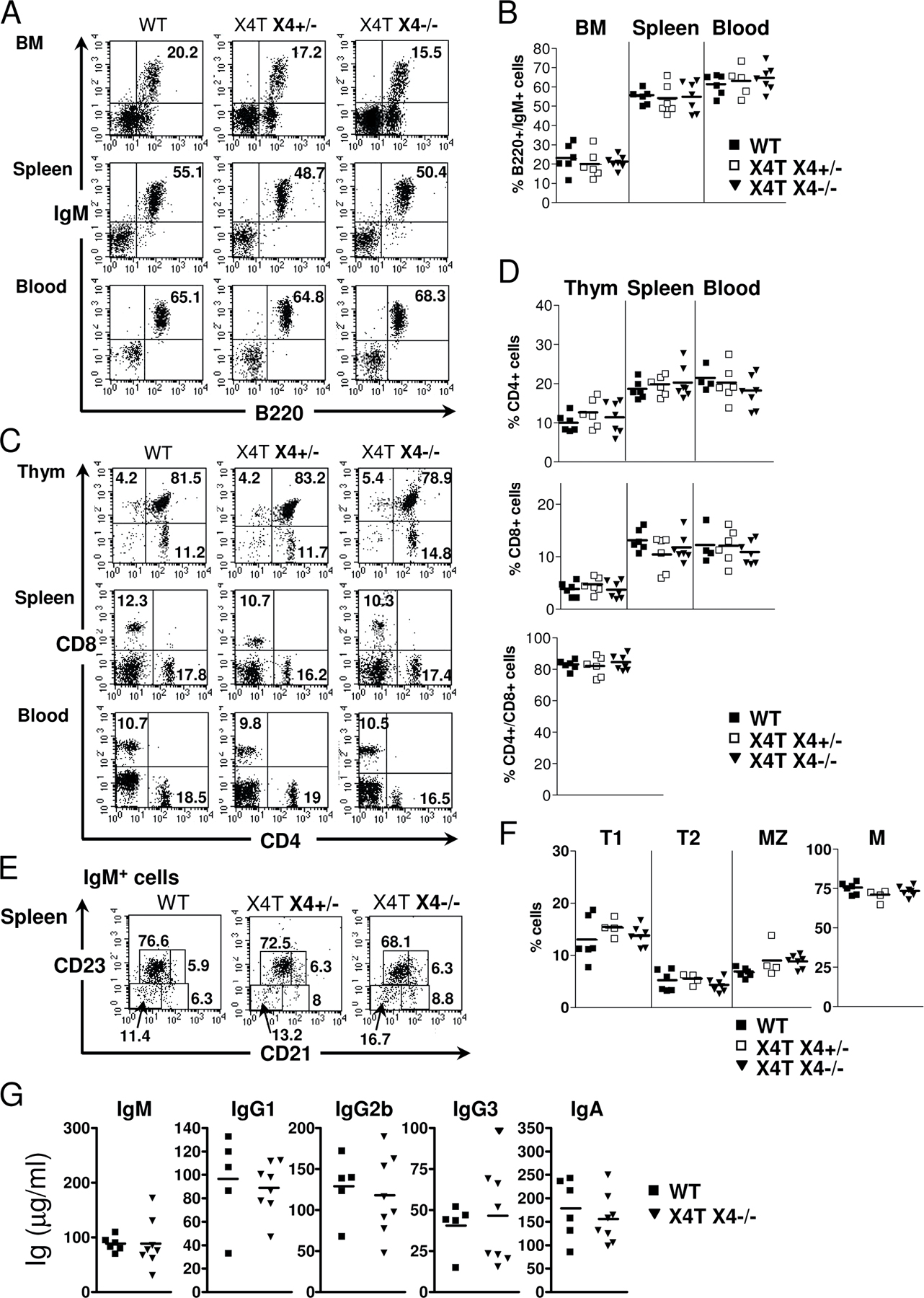
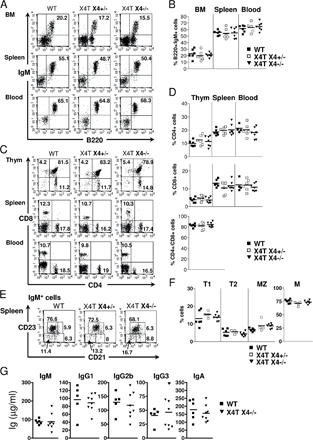
X4−/− mice succumb to massive apoptosis of postmitotic neuron during development (12). This is characterized by a severe acellularity in the mantle layer of the brain, particularly the cortical plate (CP; Fig. 2, A and B), with the presence of many pyknotic nuclei (Fig. 2 C). In contrast, brain tissue sections of X4T X4−/− mice (Fig. 2, D–F) were indistinguishable from that of control X4+/− mice (Fig. 2, G–I), indicating that expression of X4T in the brain had, indeed, complemented the neuronal apoptosis. FACS analyses of BM, spleen, blood, and thymus, demonstrate the complete immune system recovery in X4T X4−/− mice compared with X4T-negative (WT) littermate mice (Fig. S1, A and B [B cells] and C and D [T cells], available at http://www.jem.org/cgi/content/full/jem.20070255/DC1). Because the phenotypes of B and T cells of 8–12-wk-old control littermates X4+/+ and X4+/− mice were not different (as described by Gao et al. [reference 12]), we associated these mice in the WT (X4T-negative) group. The analysis of B cell subpopulations showed that the proportions of transitional T1 (IgM+/CD21−/CD23−), T2 (IgM+/CD21+/CD23+), marginal zone (MZ; IgM+/CD21+/CD23−), and follicular mature (IgM+/CD21low/CD23+) B cells are not different among the three groups of mice (Fig. S1, E and F). Lastly, absolute numbers of B and T cells are comparable in the three groups of mice (unpublished data). The serum Ig levels (Fig. S1 G) in X4T X4−/− and control mice were not statistically different, indicating that X4T expression allows B cells to develop into mature IgM-secreting cells and to secrete Ig of switched isotypes, like IgG1, IgG2b, and IgG3 and IgA. Altogether, these results demonstrate that X4T expression reverts the embryonic lethality and the B and T cell development defect in X4−/− mice.
Figure 2.

X4T reverts neuronal cell death of X4−/− embryos. (A–I) Hematoxylin and eosin staining of littermate X4−/−, X4T X4−/−, and X4+/− brain sections. Coronal brain sections of E14.5 X4−/− (A–C) reveal a severe acellularity in the CP (small arrows) in the cerebral hemisphere. Higher magnifications show normal ventricular zone (VZ) of the mutant cortex, whereas numerous cells with pyknotic nuclei (dense hematoxylin stain, long arrow in C) are present in the intermediate zone (IZ) extending to the CP. In contrast, coronal brain sections of E14.5 X4T X4−/− mice (D–F) do not reveal significant difference in cortical structure with cerebral hemisphere of X4+/− brain (G–I) and an absence of cells with pyknotic nuclei. LV, lateral ventricle; GE, ganglionic eminence. Boxed areas in B, E, and H are enlarged in C, F, and I by image processing. Bars: (A, D, and G) 150 μm; (B, E, and H) 40 μm; (C, F, and I) 20 μm.
Conditional X4 deletion in mature B cells
To analyze the role of X4 during CSR, we crossed X4T X4−/− mice with CD21-Cre transgenic mice in which the Cre recombinase expression is driven by the CD21 promoter, specifically in mature B cells (38), to create ΔX4T X4−/−, which will be called ΔX4T from this point forward and will always be compared with Cre-negative X4T X4−/−, hereafter named X4T. PCR assay using primers flanking the X4T integration site revealed a ΔX4T-specific PCR mainly in CD43-negative purified splenic B cells and, to a much lesser extent, in total BM, but not in thymus or in DNA from Cre-negative animals (Fig. 3, A and B). Quantitative Southern blot analysis further established that X4T deletion was almost complete in isolated mature splenic B cells from mice expressing Cre recombinase, as demonstrated by the disappearance of the 5.7-kb EcoNI–NdeI X4T-specific band and the concomitant appearance of a 2.3-kb ΔX4T-specific band in a mean proportion of 89% compared with TgWT allele (Fig. 3 C, lanes 7–9). No deletion was observed in liver (Fig. 3 C, lanes 3 and 4), thymus (lanes 11 and 12), or in B cells in the absence of Cre (lanes 5 and 6). These results demonstrate a specific deletion of X4T in mature B cells in ΔX4T mice.
Figure 3.

Conditional X4 deletion in mature B cells. (A) Schematic representation of the X4T locus before (X4T) and after (ΔX4T) Cre recombination and of a WT allele. Restriction sites: E, EcoNI; N, NdeI. (B) Analysis of X4T deletion by PCR using primers 1 and 2 located in A. Genomic DNA from total BM, purified splenic mature B cells, and thymocytes from X4T X4−/− mice expressing Cre (+) or not (−) were used in the PCR reaction. ΔX4T and WT fragments are indicated. Because of its large size, the X4T fragment is not detected in these PCR conditions. GAPDH-specific PCR was used as loading control. (C) Southern blot analysis of X4T deletion. DNA was prepared from liver, thymus, and from purified splenic mature B cells from X4T X4−/− (CD21-Cre-), ΔX4T X4−/− (CD21-Cre+), and nontransgenic (WT) mice, digested with EcoNI–NdeI, and hybridized with chromosome 16 integration site-specific probe D (refer to A for expected sizes of bands). Quantification of deletion is indicated below lanes 7, 8, and 9 as the percentage of the amount of ΔX4T compared with WT allele.
CSR is reduced in conditional X4−/− mice in vivo
To explore the role of X4 in CSR, we quantified the basal serum Ig levels of different isotypes by ELISA in 8–12-wk-old ΔX4T mice. IgM concentration was not affected (Fig. 4), indicating that X4T deletion in mature B cells does not modify their ability to undergo IgM secretion. In contrast, ΔX4T mice had lower levels of IgG1, IgG2b, and IgG3 (Fig. 4), the difference being statistically significant for IgG2b and IgG3 isotypes. Altogether, the titers were 76, 60, and 41% that of control mice, respectively. No difference in IgA secretion was noted in ΔX4T mice. These results demonstrate a CSR defect in ΔX4T mice in vivo.
Figure 4.

Ig production is impaired in conditional X4 KO mice. Sera from ΔX4T and control X4T mice were collected and total IgM, IgG1, IgG2b, IgG3, and IgA were determined by ELISA (microgram/milliliter). The results of statistical tests are indicated; * indicates a statistically significant difference (P < 0.05, two-tailed Mann-Whitney test).
CSR is reduced in ΔX4T mice in vitro
Given the specific CD21-Cre–mediated deletion in mature B cells, the CSR defect in ΔX4T mice is probably entirely caused by an intrinsic inability of B cells to undergo CSR. To verify this hypothesis, we induced CSR to different isotypes in vitro by stimulating purified splenic CD43-negative mature B cells from ΔX4T mice with various polyclonal B cell activators. Surface Ig expression on B cells was analyzed by FACS after 4 d of stimulation, and Ig secretion was quantified in day 5 culture supernatants. The frequency of switched mature B cells after 4 d activation was reduced in ΔX4T mice for all the isotypes analyzed (Fig. 5 A). The difference between ΔX4T mice and control mice was statistically significant for IgG1 (P = 0.001) and for IgG2b and IgG3 (P = 0.05). The decrease in cell surface Ig expression on B cells from ΔX4T mice was accompanied by a reduction of the respective Ig secretion in day 5 culture supernatants (Fig. 5 B). We noted a decrease in IgG1 (65% decrease), IgG2b (35%), IgG3 (63%), IgG2a (66%), and IgE (49%) secretion in cultures from X4-deficient B cells, without any decrease in IgM secretion (Fig. 5 B). We conclude that CSR is impaired in ΔX4T mice because of a B cell intrinsic defect.
Figure 5.

CSR defect in X4-deficient B lymphocytes in vitro. Surface expression and secretion of switched isotypes. Purified splenic mature B cells from ΔX4T and control X4T mice were labeled with CFSE and stimulated for 4 or 5 d with various polyclonal B cell activators for switching. (A) Percentage of total B cells expressing indicated isotypes of Ig, as determined by flow cytometry analysis, after 4 d of in vitro stimulation. (B) Secreted Ig (nanogram/milliliter) was analyzed in supernatants from splenic B cells cultures after 5-d in vitro stimulation. IgA was undetectable in supernatants. For A and B, the results of statistical tests are indicated; * indicates a statistically significant difference (P < 0.05, two-tailed Mann-Whitney test). (C–G) Analysis of CSR in the various subsets of proliferative B cells according to CFSE intensity. (C) Plots show IgG1 (top) and IgG3 (bottom) staining together with CFSE, on viable (PI−) lymphoid cells. The percentages of switched cells are given. (D–F) Percentages of total (D and F) and IgG1-switched (E, top) or IgG3-switched (E, bottom) B cells that have undergone the indicated numbers of cell division, for ΔX4T (white) and X4T (gray) mice. (G) In each cell division, the decrease of CSR to IgG1, IgG2a, IgG2b, IgG3, and IgA, in B cells from ΔX4T compared with X4T mice is represented as a percentage of CSR defect.
X4 deficiency does not alter B cell proliferation in vitro
Because the ability to undergo CSR is linked to B cell proliferation (39), we analyzed the consequence of X4 deletion on B cell proliferation in vitro, first through CFSE labeling coupled with Ig isotype determination (Fig. 5, C–G). The frequency of cells expressing various Ig isotypes was determined by flow cytometry after 4 d of stimulation in the overall B220+ B cell population, as well as among the various subsets of proliferative B cells according to CFSE intensity. The overall proliferative capacity of B cells was not altered by the X4 deletion in culture stimulated with LPS/IL-4 and LPS (Fig. 5 D) or with anti-CD40/IL-4/IFNγ and anti-CD40/IL-4/TGFβ (Fig. 5 F), even in cells that underwent several cycles of cell division. Most importantly, the decrease in CSR to IgG1 and IgG3 (Fig. 5, E and G), IgG2a, IgG2b, and IgA (Fig. 5 G) was not restricted to the cells that had proliferated less. Interestingly, the CSR defect, indeed, appears to increase with the extent of proliferation (see Discussion; Fig. 5, E and G). Moreover, cell survival, determined by propidium iodide staining on day 4, was not reduced in ΔX4T versus control mice (P = 0.86 for LPS stimulation; P = 0.45 for LPS/IL-4 stimulation, two-tailed Mann-Whitney test).
Altogether, these results demonstrate a diminution in CSR in B cells deficient for X4 both in vitro and in vivo, which is not attributable to a reduced capacity of clonal expansion or a decrease in cell survival.
X4 deficiency reduces CSR at the level of DNA recombination
The function of X4 in ligation of DNA DSB suggests that the observed defect in CSR could intervene at the level of DNA recombination, i.e., after induction of AID expression and germline transcription of the CH genes targeted for recombination, but before Ig circle transcripts production. To explore this hypothesis, we cultured purified splenic CD43-negative mature B cells from conditional X4 KO and control mice for 4 d with LPS and IL-4, which induces CSR to IgG1. Total RNA was isolated and analyzed by real-time quantitative RT-PCR to quantify AID transcripts, μ and γ1 germline (sterile) transcripts (GT), and γ1 circle transcripts (CT). Fig. 6 shows the level for these transcripts in ΔX4T mice relative to the X4T control mice. Whereas the expression of μGT, γ1GT, and AID transcripts were not affected by the X4 deletion, the level of γ1CT transcript showed a mean threefold reduction in ΔX4T B cells compared with control B cells. This indicates that the defect of CSR observed in B cells from conditional X4 KO mice is the consequence of a decreased efficiency for ligation of DNA DSB produced in CSR after the induction of AID and germline transcription of CH loci targeted for recombination.
Figure 6.

Ig γ1 circle transcript production is impaired in X4-deficient B cells. Purified splenic mature B cells from ΔX4T and X4T mice were stimulated with LPS and IL-4 for 4 d, and μGT, γ1GT, γ1CT, and AID transcripts were quantified by real-time RT-PCR. Each point represents the level for the indicated transcript in ΔX4T mice (which has been normalized to CD19 transcripts) relative to X4T mice analyzed in the same experiment.
X4 deficiency slightly increases the use of short microhomologies in Sμ-Sγ1 junctions
Although CSR is impaired in the absence of X4, some cells undergo switching, suggesting that an alternative pathway might develop to resolve CSR junctions when X4 function is impaired. As a possibility for an alternative pathway, Pan-Hammarström et al. recently showed that patients carrying hypomorphic mutations in DNA ligase IV notably display a slight increase in donor/acceptor homology at Sμ-Sγ1 junctions (34). We compared IgG1 CSR junctions from ΔX4T and X4T B cells after 4 d of stimulation with LPS and IL-4 (Table I and Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20070255/DC1). Although the proportion of Sμ-Sγ1 junctions without any donor/acceptor homology was slightly decreased in ΔX4T mice (46 vs. 60%), the proportion of junctions with short (1–3 bp) homologies was increased (48 vs. 34%), compared with X4T control mice, although not to statistical significance (P = 0.22, Fisher's exact test). We conclude that the resolution of IgG1 CSR junctions is slightly shifted to the use of short microhomologies in the absence of X4.
Table I.
Increased microhomology at Sμ-Sγ1 junctions in B cells deficient for X4
| Perfectly matched homology | Total n° of S junctions | |||||
|---|---|---|---|---|---|---|
| Sμ-Sγ1 junctions | 0 bp | 1–3 bp | 4–6 bp | 7–9 bp | ≥10 bp | |
| X4T | 30 (60%) | 17 (34%) | 2 (4%) | 1 (2%) | 0 (0%) | 50 |
| ΔX4T | 23 (46%) | 24 (48%) | 2 (4%) | 1 (2%) | 0 (0%) | 50 |
Purified splenic mature B cells from ΔX4T and X4T mice were stimulated for 4 d with LPS and IL-4, and then Sμ-Sγ1 junctions were amplified by PCR and sequenced. Microhomology was determined by identifying the longest region of perfect donor/acceptor identity at the switch junction. See Fig. S2 for sequence data, available at http://www.jem.org/cgi/content/full/jem.20070255/DC1.
Why is the defect of CSR in ΔX4T B cells partial?
The CSR defect observed in the absence of X4 both in vitro and in vivo is partial. As shown in Fig. 3 C, X4T deletion efficiency is almost complete and so likely does not account for the partial nature of the CSR defect in ΔX4T mice. Given the structure of the X4T transgene, with LoxP sites flanking the transcriptional unit upon virus integration (Fig. 7 A), the partial CSR defect observed in ΔX4T B cells could reflect the persistence of X4T expression from the Cre-excised X4T cassette in mature B cells. To explore this possibility, we performed a Southern blot analysis using a probe specific for the third exon of the XRCC4 gene (Fig. 7 A, probe E), which is deleted in X4 KO mice (12). The 2.8-kb NdeI band revealed by probe E and specific for the X4 WT allele (Fig. 7 B, lane 13) was absent in all samples from mice deficient for endogenous X4, as expected (X4T and ΔX4T mice; Fig. 7 B, lanes 1 to 12). In contrast, hybridization with probe E revealed the integrated X4T transgene as a 5.7-kb EcoNI–NdeI band in Cre-negative, X4T mice (Fig. 7 B, lanes 1, 2, 5, 6, and 10), as well as in liver and thymus from ΔX4T mice (lanes 3, 4, 11, and 12), but not in purified mature B cells from ΔX4T mice (lanes 7–9), where it is replaced by a 4.7-kb band. The size of this band was compatible with that of the Cre-excised X4T cassette as a circular episome (Fig. 7 A, episomal X4T and eX4T). The X4T episome was further detected by PCR (Fig. 7 A, primers 1 and 2), specifically in purified splenic mature B cells from mice expressing Cre recombinase, but not in thymus or liver from these mice (Fig. 7 C, left). The reduced intensity of the eX4T PCR product in B cells after 4-d activation (Fig. 7 C, right) suggests that although this episome is not degraded upon Cre-mediated excision, it is diluted out with cellular proliferation, which is a situation reminiscent of the TCR-α rearrangement excision circles in thymocytes (40). These results were confirmed by a real-time quantitative RT-PCR using a probe specific for the second and third exon of the XRCC4 gene, which demonstrates that, whereas the level of X4 transcripts is comparable in purified splenic mature B cells from X4T and ΔX4T mice (Fig. 7 D, d0), the amount of X4 transcripts sharply decreases in B cells from ΔX4T mice after 4-d activation in vitro (Fig. 7 D, d4).
Figure 7.

Production of episomal DNA carrying X4T upon Cre-mediated deletion. (A) Schematic representation of X4T locus before (X4T) and after (ΔX4T) Cre recombination, of episomal form of X4T (eX4T) and of X4 genomic locus (WT). Restriction sites: E, EcoNI; N, NdeI. (B) Southern blot analysis of X4T deletion and eX4T detection, using X4 third exon–specific probe E depicted in A. EcoNI–NdeI–digested DNA from total liver, thymus, or purified splenic mature B cells from X4T X4−/− (CD21-Cre-), ΔX4T X4−/− (CD21-Cre+) and non transgenic (WT) mice, was hybridized with probe E (refer to A for expected sizes of bands). (C) Detection of eX4T by PCR using primers 1 and 2 located in (A), on genomic DNA from the following: (left) purified splenic mature B cells, liver and thymocytes from ΔX4T mice; (right) purified splenic mature B cells from ΔX4T mice before (d0) and after (d4) 4 d of stimulation with LPS and IL-4. eX4T fragment is indicated. Artemis- (left) or GAPDH-specific (right) PCR was used as loading control. (D) Real-time quantitative RT-PCR analysis of X4 transcripts in purified splenic mature B cells from ΔX4T and X4T mice before (d0) and after stimulation with LPS and IL-4 for 4 d (d4). Bars represent the level of X4 transcript in ΔX4T B cells relative to X4T B cells. Results are means of two independent experiments.
Together, these results indicate that whereas the X4T transgene is almost completely deleted from chromosome in mature B cells expressing Cre, the X4T transcriptional unit is not degraded and still expressed in early deleted cells. Even though this expression is lost upon episome dilution that occurs during B cell proliferation, it could mask or attenuate the CSR defect and a differential resolution of CSR junctions in B cells from ΔX4T mice.
DISCUSSION
Although mutant mice have implicated the NHEJ pathway in CSR (27, 28, 30), the direct demonstration of the participation of X4 in this process was lacking. This question cannot be addressed easily in X4-deficient mice because of their embryonic lethality and V(D)J recombination defect (12). To overcome this limitation, we developed a lentiviral transgenic mouse line expressing one copy of the floxed mouse X4 transgene (named X4T), and crossed it onto a X4-deficient background. The X4T transgene reverts embryonic lethality of X4-deficient mice and allows for the normal development of a mature immune system. The transgenic line thus obtained is amenable to conditional inactivation through Cre-mediated excision of the LoxP-flanked cDNA transgene.
This experimental scheme presents several advantages over the classical conditional KO strategy by introducing LoxP sites in the genome. First, the lentiviral transgenesis is very efficient and much less effort/time demanding than homologous recombination in embryonic stem cells. It can be used in conjunction with all the already existing KO mice that were not primarily designed for conditional KO (which was the case for the X4 KO mice we used in this study). Second, the same strategy can also be applied to the ever-growing list of mutant mice obtained either through ENU (N-ethyl-N-nitrosourea) or insertional mutagenesis. Third, the use of TAP-Tag fused to cDNAs makes it possible to perform ex vivo proteomic studies from these transgenic animals. Lastly, the introduction of an iresGFP cassette should allow tracking of cells that have undergone Cre-mediated excision of the transgene, although, in our case, the ires sequence used in the X4T transgenic mice proved to be nonfunctional in vivo. One limitation of this strategy comes from the location of the LoxP site in the 3′LTR of the lentiviral vector. The Cre-mediated, excised episome is not degraded, and it still expresses the transgene in cells that carry it. It is ultimately lost by dilution upon cell proliferation. Although this may not represent a problem for rapidly dividing cells, it certainly constitutes a major drawback for studies in nonproliferating tissues (such as postmitotic neurons), and it may result in a hypomorphic CSR phenotype in ΔX4T mice. The repositioning of LoxP sites between the UbiC promoter and the cDNA in the lentiviral vector should eliminate this problem.
To study the role of X4 in CSR, we crossed X4T X4−/− mice to CD21-Cre transgenic mice (38), which lead to an almost complete deletion of the floxed X4T from the genome, specifically in mature B cells from the resulting ΔX4T mice. These mice demonstrate reduced CSR in vivo and in vitro, thus confirming an intrinsic failure of X4-deficient B cells to undergo CSR. Several studies have linked CSR to the capacity of B cell to proliferate (39, 41, 42). The proliferative capacity of B cells from ΔX4T mice was not affected, arguing for the role of X4 during the CSR rearrangement process. The inverse correlation between the proliferation and CSR levels in B cells from ΔX4T mice, owing to the progressive loss of the eX4T episome (see below), further strengthens this conclusion.
CSR is strictly dependent on AID enzyme, and on the production of sterile germline transcripts (GTs) from the CH genes that are to be recombined, which may provide the accessibility of DNA to the recombination machinery. CSR leads to the production of post-switch circle transcripts (15). Induction of AID and expression of μGT and γ1GT transcripts are normal in X4-deficient B cells after stimulation in vitro, whereas the amount of CSR as measured by circular γ1 transcripts is reduced, indicating that X4 deficiency impairs the ligation of broken S regions during CSR.
To explore the possibility that an alternative, X4-independent pathway could account for the partial resolution in CSR junctions in ΔX4T mice, we analyzed Sμ-Sγ1 junction sequences in purified mature splenic B cells after stimulation with LPS and IL-4, and found a slight increase in the use of microhomologies in ΔX4T B cells. Although this increase concerns only short microhomologies and is not statistically significant, these results are reminiscent of the CSR junctions from a DNA ligase IV–deficient patient (34) and from ATM-deficient mice (22) and of the existence of alternative end-joining mechanisms using microhomologies, in V(D)J junctions in Ku86-deficient mice, or in plasmid assays in DNA ligase IV or XRCC4-deficient systems (43–47). Notably, Verkaik et al. developed an assay in which a DNA substrate is linearized in such a way that joining on a particular microhomology creates a novel restriction enzyme site; in this assay, XRCC4-deficient XR-1 cells and DNA ligase IV–deficient N114 or 180BR cells showed an increased microhomology use (44).
A residual expression of X4 transcripts after Cre deletion could also participate in the partial CSR defect in this setting. Indeed, the Cre recombination process generates an episomal DNA that carries the X4T transcriptional unit (eX4T), which persists in nondividing mature B cells, can be transcribed, and is diluted out upon cell proliferation in vitro, leading to the concomitant decrease of X4 transcription. Moreover, the CSR defect increases in cells that have undergone several rounds of cell division as assessed by CFSE labeling coupled to Ig surface expression analysis (Fig. 5, E and G); as an example, B lymphocytes that have undergone zero versus six cycles of cell division show 28 and 57% CSR defect to IgG3, respectively. This was true for all analyzed isotypes. These observations support the idea that the CSR process is efficient to some extent in ΔX4T B cells ongoing few divisions because of the persistence of eX4T, but becomes less and less efficient as cells go through several cycles of proliferation and lose the episome. We propose that the partial defect of CSR observed after 4 d of stimulation in vitro is the result of the sum of these events. This interpretation implies that we may underestimate the role of X4 in CSR in our experimental system.
A requirement for the NHEJ factors Ku70 and Ku80 during CSR has been established in complemented KO mice harboring monoclonal B cell population (20, 27, 28). However, these factors also carry NHEJ-independent functions in DNA repair, probably through their interaction with ATM (48). In contrast, X4 is not known to be involved in pathways other than NHEJ. The experimental system we describe in this study provides the first evidences for a role of X4 in CSR. X4 belongs to the of X4/DNA ligase IV complex, together with the recently identified Cernunnos/XLF factor (3, 5). These three factors are essential for DNA end joining through NHEJ. X4, and probably DNA ligase IV and Cernunnos as well, play a role in CSR by promoting the final joining of the two broken end regions, in agreement with the results from Pan-Hammarström in patient with hypomorphic mutations in DNA ligase IV gene (34). This is also in accord with our observation that some Cernunnos-deficient patients present a hyper-IgM syndrome, a hallmark of a CSR defect in humans (3). The development of DNA ligase IV and Cernunnos conditional KO mice should definitely accredit this conclusion. Our study also highlights the likely existence of an alternative pathway, which, in the absence of XRCC4, allows CSR to proceed to some extent.
MATERIALS AND METHODS
DNA constructs.
The self-inactivating lentiviral vector pTRIP-Ubi-X4-TAP-iresGFP-ΔU3-LoxP (Fig. 1 A) used to produce transgenic mice was constructed as follows: the lentiviral pTRIP-Ubi-GFP-U3 was obtained by substituting the EF1α promoter for the human ubiquitin C promoter from pUbi-JunB (provided by P. Angel, Deutshces Krebforschugszentrum, Heidelberg, Germany [reference 37]) in MluI and BamHI sites of pTRIP-EF1α-GFP-U3 (49). A LoxP site (ATAACTTCGTATAATGTATGCTATACGAAGTTAT) was inserted in the lentiviral ΔU3–R–U5 3′LTR fragment in PL plasmid (provided by P. Charneau, Institut Pasteur, Paris, France). This ΔU3–R–U5 LoxP fragment was used to replace the U3/R/U5 3′LTR in pTRIP-Ubi-GFP-U3, producing pTRIP-Ubi-GFP-ΔU3-LoxP. The GFP fragment was replaced by a TAP-iresGFP cassette to obtain pTRIP-Ubi-TAP-iresGFP-ΔU3-LoxP. The TAP sequence consists in the Ig-binding fragment of protein A fused to calmodulin-binding protein (50). Finally, the pTRIP-Ubi-X4-TAP-iresGFP-ΔU3-LoxP lentiviral vector was constructed by introducing the mouse X4 ORF inframe with the TAP tag. We will refer to this vector as pTRIP-X4.
Virus production and titration.
Lentiviral particles were produced by transient transfection of 293T cells, as previously described (49).
Mice.
A volume of 10 pl of virus at 5 × 108 IU/ml was injected into the perivitelline space of one-cell stage B6D2F1 mouse embryos. Embryos were reimplanted into pseudopregnant females. A single-copy X4 founder line (see following paragraph) was selected for further analysis. Conditional X4 KO mice were obtained by breeding the X4 transgenic line to X4 KO mice (provided by F. Alt, Harvard Medical School, Boston, MA) (12) and CD21-Cre transgenic mice (provided by K. Rajewsky, Harvard Medical School, Boston, MA) (38). Progeny were screened by PCR on tail DNA. Mice were bred and maintained under specific pathogen-free conditions. All mice used in this study were 8–12 wk old. Mouse experiments were with the approval of institutional committee from the french ministry of agriculture.
Analysis of the number of proviral integrations by Southern blotting.
The number of proviral integrations was analyzed by Southern blot (Fig. 1) on AvaII- and EcoNI-digested tail DNA, as previously described (49), using the following vector probes: 1,027-bp 5′ flanking probe A (49), 638-bp internal GFP probe B, and 253-bp 3′ flanking probe C (Fig. 1 B). The signal was quantified using ImageQuant software.
Analysis of X4T deletion by Southern blotting.
DNA from liver, thymus, and purified splenic mature B cells was digested with EcoNI and NdeI. Southern blots were hybridized with the 795-bp probe D (Fig. 3 A) and the 327-bp probe E (Fig. 7 A) and quantified using ImageQuant software.
Determination of provirus site integration by inverse PCR.
1 μg tail DNA from a X4T transgenic mouse was digested with MfeI and ligated with T4 DNA ligase (Invitrogen), and inverse PCR was performed with Platinium Taq DNA Polymerase High Fidelity (Invitrogen) in two-round reactions. The first round used provirus-specific oligonucleotide primers (5′-GACTCGGCTTGCTGAAGC-3′ and 5′-CGAGAGAGCTCCTCTGGTTTC-3′). Amplification conditions were 4 cycles at 94°C (30 s), 65°C (30 s), and 68°C (2 min); 4 cycles at 94°C (30 s), 63°C (30 s), 68°C (2 min); and 27 cycles at 94°C (30 s), 60°C (30 s), 68°C (30 s + 10 s/cycle). 5 μl of the first PCR product was reamplified with an internal set of primers (5′-GGGGGAGAATTAGATCGC-3′ and 5′-CTTTCAAGTCCCTGTTCGG-3′), with 4 cycles at 94°C (30 s), 62°C (30 s), 68°C (2 min); 4 cycles at 94°C (30 s), 60°C (30 s), 68°C (2 min); and 27 cycles at 94°C (30 s), 57°C (30 s), 68°C (30 s + 10 s/cycle). The resulting 2-kb PCR fragment was gel purified, sequenced, and blasted against the mouse genome.
Analysis of X4T deletion and detection of episomal X4T by PCR.
X4T deletion was analyzed by PCR on DNA from BM, thymus, and purified splenic mature B cells using primers designed in the genomic sequence flanking the X4T integration site (K16F, K16R). GAPDH-specific PCR was used as loading control. The Cre-excised X4T episome was detected by PCR using primers 1 and 2 depicted in Fig. 7 A (3′F1, 5′R1). Artemis- and GAPDH-specific PCRs were used as loading controls. The PCR assay was performed on 20 ng of DNA (and one-third serial dilutions of DNA when indicated). The primers used were as follows: K16-F (5′-GCTGCTGGCCTGGTCTTAGT-3′), K16-R (5′-AAGCATGGCAGGACTCTCAT-3′), GAPDH-F (5′-AGTATGATGACAAGAAGG-3′), and GAPDH-R (5′-ATGGTATTCAAGAGAGTAGGG-3′); 3′F1 (5′-CCTGGCTAGAAGCACAAGAG-3′) and 5′R1 (5′-CGAGAGAGCTCCTCTGGTTTC-3′); Artemis K5-5 (5′-TCCATGACCTTATCCACAGTGAGGC-3′) and K5.4 (5′-TTCCTCCTTCCCTTCCCCCACATAG-3′).
Analysis of transgene expression by Western blotting.
Whole-cell extracts were prepared from thymus and spleen according to standard protocols. X4T and endogenous X4 were detected with rabbit polyclonal anti-XRCC4 antibody (AHP387; Serotec), followed by HRP-goat anti–rabbit antibody (GE Healthcare).
Histological analysis of mouse brain sections.
Embryos were fixed in 4% paraformaldehyde, embedded in 15% sucrose and 7.5% gelatine in PBS, serially sectioned (16 μm), and stained with hematoxylin and eosin, as previously described (12).
Flow cytometry analysis of lymphocyte populations and Ig CSR.
Cell phenotype was performed on blood, thymic, splenic, and BM lymphoid populations by four-color fluorescence analysis according to standard protocols. The following antibodies were used: FITC, PE, or APC anti–mouse B220, CD21, CD23, CD3, CD8, and CD4 (all from BD Biosciences); Cy5 anti–mouse IgM (Jackson Immunoresearch Laboratories). For analysis of CSR, cells were stained with PE anti–mouse B220 (BD Biosciences); biotin anti–mouse IgG1, IgG2a, IgG2b, IgG3, IgA, and IgE (Southern Biotechnology Associates); and APC-streptavidin (BD Biosciences). Cells were analyzed using a FACSCalibur immediately after incubation with 5 μg/ml propidium iodide to exclude dead cells.
Purification and activation of splenic mature B cells in vitro.
Mature B cells were purified from spleen and labeled with CFDA-SE, as previously described (20), and stimulated for 4 or 5 d with 25 μg/ml LPS from Escherichia coli (Sigma-Aldrich), 20 ng/ml IL-4 (R&D Systems), and 0.5 μg/ml anti–mouse CD40 (BD Biosciences), 1 ng/ml human TGFβ1 (R&D Systems), 0.5 μg/ml recombinant mouse IFNγ (PeproTech France) for 4 and 5 d, in the following combinations: LPS for IgG2b and IgG3 switching, LPS/IL-4 for IgG1 and IgE switching, anti–mouse CD40/ IL-4/IFNγ for IgG2a switching, and anti–mouse CD40/IL-4/TGFβ for IgA switching.
Antibody detection by ELISA.
Total IgA, IgG1, IgG2a, IgG2b, IgG3, IgE, or IgM levels were measured in serum from 8–12-wk-old mice, and in supernatants after 5 d of stimulation, as previously described (22). The wells were developed with Fast OPD substrate (Sigma-Aldrich). Absorbance was measured at 490 nm. Levels of Ig were determined by comparison with a standard curve using purified IgA, IgG1, IgG2a, IgG2b, IgG3, IgE (all from Southern Biotechnology Associates), and IgMκ (Sigma-Aldrich).
Sequencing of μ/γ1 switch recombination junctions.
Genomic DNA was prepared from sorted IgG1+ B cells after 4 d of stimulation with LPS and IL-4, and Sμ/Sγ1 switch junctions were amplified by PCR as previously described (20). PCR products were cloned using pGEM-T vector kit (Promega) and sequenced using SP6 and T7 universal primers. Sequence analysis was performed using Blast2 sequences in comparison with mouse Sμ (MUSIGCD07) and Sγ1 (MUSIGHANB) regions.
Quantitative real-time RT-PCR analysis of CSR transcripts.
cDNA was prepared form total RNA extracted from B cells after 4 d of stimulation with LPS and IL-4, and μGT, γ1GT, γ1CT, and AID transcripts were quantified using GAPDH for normalization, as previously described (20, 22, 51). For each sample, the level of CSR transcripts was normalized to CD19 transcript. Primers used for CD19 transcripts quantification were CD19F (5′-CCATCGAGAGGCACGTGAA-3′) and CD19R (5′-TCCAATCCACCAGTTCTCAACAG-3′) (52).
Quantitative real-time RT-PCR analysis of X4 transcripts.
Quantitative real-time PCR was performed on 20 ng cDNA using Taqman Universal Mastermix (Applied Biosystems) and Assays-on-Demand reaction (Applied Biosystems) including forward and reverse primers and the 6-carboxy-fluorescein (FAM)–labeled probe for the target gene (assay for mouse X4 exons 2–3 was no. Mm01283065_m1; for 18S, it was Hs99999901_s1). Conditions for PCR amplification and detection were: 50°C (2 min), 95°C (10 min), and 40 cycles at 95°C (15 s), 60°C (60 s). Each sample was amplified in quadriplicate. mRNA levels were calculated with the sequence detector SDS2.1 software (Applied Biosystems), using the comparative cycle threshold method, and normalized to the endogenous control 18S.
Online supplemental material.
Fig. S1 shows B and T cell development in X4−/− mice complemented with X4T. FACS analysis and serum Ig level determination show the complete immune recovery in X4T X4−/− mice compared with X4T-negative (WT) mice. Fig. S2 illustrates the Sμ-Sγ1 junction sequence data (25 bp upstream and downstream of each junction) in X4-deficient B cells. Sequence analysis was performed using Blast2 sequences in comparison with mouse Sμ (MUSIGCD07) and Sγ1 (MUSIGHANB) regions. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20070255/DC1.
Supplemental Material
Acknowledgments
We thank Dr. F. Alt for X4 KO mice and Dr. K. Rajewsky for providing the CD21-Cre transgenic mice; A. Rossard and M. Gaillard for excellent animal care; P. Souque for technical help in virus production; and B Reina-San-Martin for critical reading of the manuscript.
This work was supported by grants from Institut National de la Santé et de la Recherche Médicale (INSERM), the Ligue Nationale contre le Cancer (Equipe labellisée LA LIGUE 2005), the Commissariat à l'Energie Atomique (LRC-CEA N°40V), and the Institut National de Cancer/Cancéroplole IdF. P. Soulas-Sprauel was supported by INSERM, Electricité de France (EDF), and the Fondation Singer Polignac.
The authors have no conflicting financial interests.
Abbreviations used: AID, activation-induced cytidine deaminase; CP, cortical plate; CSR, class switch recombination; DSB, double-strand break; E, embryonic day; GT, germline transcript; NHEJ, nonhomologous end joining; RAG, recombination-activating gene; TAP, tandem affinity purification.
References
- 1.de Villartay, J.P., A. Fischer, and A. Durandy. 2003. The mechanisms of immune diversification and their disorders. Nat. Rev. Immunol. 3:962–972. [DOI] [PubMed] [Google Scholar]
- 2.Revy, P., D. Buck, F. le Deist, and J.P. de Villartay. 2005. The repair of DNA damages/modifications during the maturation of the immune system: lessons from human primary immunodeficiency disorders and animal models. Adv. Immunol. 87:237–295. [DOI] [PubMed] [Google Scholar]
- 3.Buck, D., L. Malivert, R. de Chasseval, A. Barraud, M.C. Fondaneche, O. Sanal, A. Plebani, J.L. Stephan, M. Hufnagel, F. le Deist, et al. 2006. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 124:287–299. [DOI] [PubMed] [Google Scholar]
- 4.Callebaut, I., L. Malivert, A. Fischer, J.P. Mornon, P. Revy, and J.P. de Villartay. 2006. Cernunnos interacts with the XRCC4 x DNA-ligase IV complex and is homologous to the yeast nonhomologous end-joining factor Nej1. J. Biol. Chem. 281:13857–13860. [DOI] [PubMed] [Google Scholar]
- 5.Ahnesorg, P., P. Smith, and S.P. Jackson. 2006. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 124:301–313. [DOI] [PubMed] [Google Scholar]
- 6.Moshous, D., I. Callebaut, R. de Chasseval, B. Corneo, M. Cavazzana-Calvo, F. Le Deist, I. Tezcan, O. Sanal, Y. Bertrand, N. Philippe, et al. 2001. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 105:177–186. [DOI] [PubMed] [Google Scholar]
- 7.Buck, D., D. Moshous, R. de Chasseval, Y. Ma, F. le Deist, M. Cavazzana-Calvo, A. Fischer, J.L. Casanova, M.R. Lieber, and J.P. de Villartay. 2006. Severe combined immunodeficiency and microcephaly in siblings with hypomorphic mutations in DNA ligase IV. Eur. J. Immunol. 36:224–235. [DOI] [PubMed] [Google Scholar]
- 8.O'Driscoll, M., K.M. Cerosaletti, P.M. Girard, Y. Dai, M. Stumm, B. Kysela, B. Hirsch, A. Gennery, S.E. Palmer, J. Seidel, et al. 2001. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol. Cell. 8:1175–1185. [DOI] [PubMed] [Google Scholar]
- 9.Barnes, D.E., G. Stamp, I. Rosewell, A. Denzel, and T. Lindahl. 1998. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr. Biol. 8:1395–1398. [DOI] [PubMed] [Google Scholar]
- 10.Frank, K.M., J.M. Sekiguchi, K.J. Seidl, W. Swat, G.A. Rathbun, H.L. Cheng, L. Davidson, L. Kangaloo, and F.W. Alt. 1998. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature. 396:173–177. [DOI] [PubMed] [Google Scholar]
- 11.Rooney, S., J. Sekiguchi, C. Zhu, H.L. Cheng, J. Manis, S. Whitlow, J. DeVido, D. Foy, J. Chaudhuri, D. Lombard, and F.W. Alt. 2002. Leaky Scid phenotype associated with defective V(D)J coding end processing in Artemis-deficient mice. Mol. Cell. 10:1379–1390. [DOI] [PubMed] [Google Scholar]
- 12.Gao, Y., Y. Sun, K.M. Frank, P. Dikkes, Y. Fujiwara, K.J. Seidl, J.M. Sekiguchi, G.A. Rathbun, W. Swat, J. Wang, et al. 1998. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 95:891–902. [DOI] [PubMed] [Google Scholar]
- 13.Bosma, M.J., and A.M. Carroll. 1991. The SCID mouse mutant: definition, characterization, and potential uses. Annu. Rev. Immunol. 9:323–350. [DOI] [PubMed] [Google Scholar]
- 14.Bassing, C.H., W. Swat, and F.W. Alt. 2002. The mechanism and regulation of chromosomal V(D)J recombination. Cell. 109(Suppl):S45–S55. [DOI] [PubMed] [Google Scholar]
- 15.Chaudhuri, J., and F.W. Alt. 2004. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat. Rev. Immunol. 4:541–552. [DOI] [PubMed] [Google Scholar]
- 16.Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. [DOI] [PubMed] [Google Scholar]
- 17.Revy, P., T. Muto, Y. Levy, F. Geissmann, A. Plebani, O. Sanal, N. Catalan, M. Forveille, R. Dufourcq-Labelouse, A. Gennery, et al. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 102:565–575. [DOI] [PubMed] [Google Scholar]
- 18.Wuerffel, R.A., J. Du, R.J. Thompson, and A.L. Kenter. 1997. Ig Sgamma3 DNA-specific double strand breaks are induced in mitogen-activated B cells and are implicated in switch recombination. J. Immunol. 159:4139–4144. [PubMed] [Google Scholar]
- 19.Petersen, S., R. Casellas, B. Reina-San-Martin, H.T. Chen, M.J. Difilippantonio, P.C. Wilson, L. Hanitsch, A. Celeste, M. Muramatsu, D.R. Pilch, et al. 2001. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 414:660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reina-San-Martin, B., S. Difilippantonio, L. Hanitsch, R.F. Masilamani, A. Nussenzweig, and M.C. Nussenzweig. 2003. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J. Exp. Med. 197:1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reina-San-Martin, B., H.T. Chen, A. Nussenzweig, and M.C. Nussenzweig. 2004. ATM is required for efficient recombination between immunoglobulin switch regions. J. Exp. Med. 200:1103–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lumsden, J.M., T. McCarty, L.K. Petiniot, R. Shen, C. Barlow, T.A. Wynn, H.C. Morse III, P.J. Gearhart, A. Wynshaw-Boris, E.E. Max, and R.J. Hodes. 2004. Immunoglobulin class switch recombination is impaired in Atm-deficient mice. J. Exp. Med. 200:1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manis, J.P., J.C. Morales, Z. Xia, J.L. Kutok, F.W. Alt, and P.B. Carpenter. 2004. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat. Immunol. 5:481–487. [DOI] [PubMed] [Google Scholar]
- 24.Ward, I.M., B. Reina-San-Martin, A. Olaru, K. Minn, K. Tamada, J.S. Lau, M. Cascalho, L. Chen, A. Nussenzweig, F. Livak, et al. 2004. 53BP1 is required for class switch recombination. J. Cell Biol. 165:459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reina-San-Martin, B., M.C. Nussenzweig, A. Nussenzweig, and S. Difilippantonio. 2005. Genomic instability, endoreduplication, and diminished Ig class-switch recombination in B cells lacking Nbs1. Proc. Natl. Acad. Sci. USA. 102:1590–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kracker, S., Y. Bergmann, I. Demuth, P.O. Frappart, G. Hildebrand, R. Christine, Z.Q. Wang, K. Sperling, M. Digweed, and A. Radbruch. 2005. Nibrin functions in Ig class-switch recombination. Proc. Natl. Acad. Sci. USA. 102:1584–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manis, J.P., Y. Gu, R. Lansford, E. Sonoda, R. Ferrini, L. Davidson, K. Rajewsky, and F.W. Alt. 1998. Ku70 is required for late B cell development and immunoglobulin heavy chain class switching. J. Exp. Med. 187:2081–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Casellas, R., A. Nussenzweig, R. Wuerffel, R. Pelanda, A. Reichlin, H. Suh, X.F. Qin, E. Besmer, A. Kenter, K. Rajewsky, and M.C. Nussenzweig. 1998. Ku80 is required for immunoglobulin isotype switching. EMBO J. 17:2404–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cook, A.J., L. Oganesian, P. Harumal, A. Basten, R. Brink, and C.J. Jolly. 2003. Reduced switching in SCID B cells is associated with altered somatic mutation of recombined S regions. J. Immunol. 171:6556–6564. [DOI] [PubMed] [Google Scholar]
- 30.Manis, J.P., D. Dudley, L. Kaylor, and F.W. Alt. 2002. IgH class switch recombination to IgG1 in DNA-PKcs-deficient B cells. Immunity. 16:607–617. [DOI] [PubMed] [Google Scholar]
- 31.Bosma, G.C., J. Kim, T. Urich, D.M. Fath, M.G. Cotticelli, N.R. Ruetsch, M.Z. Radic, and M.J. Bosma. 2002. DNA-dependent protein kinase activity is not required for immunoglobulin class switching. J. Exp. Med. 196:1483–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kiefer, K., J. Oshinsky, J. Kim, P.B. Nakajima, G.C. Bosma, and M.J. Bosma. 2007. The catalytic subunit of DNA-protein kinase (DNA-PKcs) is not required for Ig class-switch recombination. Proc. Natl. Acad. Sci. USA. 104:2843–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rooney, S., F.W. Alt, J. Sekiguchi, and J.P. Manis. 2005. Artemis-independent functions of DNA-dependent protein kinase in Ig heavy chain class switch recombination and development. Proc. Natl. Acad. Sci. USA. 102:2471–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan-Hammarström, Q., A.M. Jones, A. Lahdesmaki, W. Zhou, R.A. Gatti, L. Hammarstrom, A.R. Gennery, and M.R. Ehrenstein. 2005. Impact of DNA ligase IV on nonhomologous end joining pathways during class switch recombination in human cells. J. Exp. Med. 201:189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lois, C., E.J. Hong, S. Pease, E.J. Brown, and D. Baltimore. 2002. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 295:868–872. [DOI] [PubMed] [Google Scholar]
- 36.Pfeifer, A., M. Ikawa, Y. Dayn, and I.M. Verma. 2002. Transgenesis by lentiviral vectors: lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. Proc. Natl. Acad. Sci. USA. 99:2140–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schorpp, M., R. Jager, K. Schellander, J. Schenkel, E.F. Wagner, H. Weiher, and P. Angel. 1996. The human ubiquitin C promoter directs high ubiquitous expression of transgenes in mice. Nucleic Acids Res. 24:1787–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kraus, M., M.B. Alimzhanov, N. Rajewsky, and K. Rajewsky. 2004. Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell. 117:787–800. [DOI] [PubMed] [Google Scholar]
- 39.Hodgkin, P.D., J.H. Lee, and A.B. Lyons. 1996. B cell differentiation and isotype switching is related to division cycle number. J. Exp. Med. 184:277–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Villey, I., D. Caillol, F. Selz, P. Ferrier, and J.P. de Villartay. 1996. Defect in rearrangement of the most 5′ TCR-J alpha following targeted deletion of T early alpha (TEA): implications for TCR alpha locus accessibility. Immunity. 5:331–342. [DOI] [PubMed] [Google Scholar]
- 41.Severinson, E., S. Bergstedt-Lindqvist, W. van der Loo, and C. Fernandez. 1982. Characterization of the IgG response induced by polyclonal B cell activators. Immunol. Rev. 67:73–85. [DOI] [PubMed] [Google Scholar]
- 42.Kenter, A.L., and J.V. Watson. 1987. Cell cycle kinetics model of LPS-stimulated spleen cells correlates switch region rearrangements with S phase. J. Immunol. Methods. 97:111–117. [DOI] [PubMed] [Google Scholar]
- 43.Bogue, M.A., C. Wang, C. Zhu, and D.B. Roth. 1997. V(D)J recombination in Ku86-deficient mice: distinct effects on coding, signal, and hybrid joint formation. Immunity. 7:37–47. [DOI] [PubMed] [Google Scholar]
- 44.Verkaik, N.S., R.E. Esveldt-van Lange, D. van Heemst, H.T. Bruggenwirth, J.H. Hoeijmakers, M.Z. Zdzienicka, and D.C. van Gent. 2002. Different types of V(D)J recombination and end-joining defects in DNA double-strand break repair mutant mammalian cells. Eur. J. Immunol. 32:701–709. [DOI] [PubMed] [Google Scholar]
- 45.van Heemst, D., L. Brugmans, N.S. Verkaik, and D.C. van Gent. 2004. End-joining of blunt DNA double-strand breaks in mammalian fibroblasts is precise and requires DNA-PK and XRCC4. DNA Repair (Amst.). 3:43–50. [DOI] [PubMed] [Google Scholar]
- 46.Smith, J., E. Riballo, B. Kysela, C. Baldeyron, K. Manolis, C. Masson, M.R. Lieber, D. Papadopoulo, and P. Jeggo. 2003. Impact of DNA ligase IV on the fidelity of end joining in human cells. Nucleic Acids Res. 31:2157–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kabotyanski, E.B., L. Gomelsky, J.O. Han, T.D. Stamato, and D.B. Roth. 1998. Double-strand break repair in Ku86- and XRCC4-deficient cells. Nucleic Acids Res. 26:5333–5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sekiguchi, J., D.O. Ferguson, H.T. Chen, E.M. Yang, J. Earle, K. Frank, S. Whitlow, Y. Gu, Y. Xu, A. Nussenzweig, and F.W. Alt. 2001. Genetic interactions between ATM and the nonhomologous end-joining factors in genomic stability and development. Proc. Natl. Acad. Sci. USA. 98:3243–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zennou, V., C. Petit, D. Guetard, U. Nerhbass, L. Montagnier, and P. Charneau. 2000. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell. 101:173–185. [DOI] [PubMed] [Google Scholar]
- 50.Rigaut, G., A. Shevchenko, B. Rutz, M. Wilm, M. Mann, and B. Seraphin. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17:1030–1032. [DOI] [PubMed] [Google Scholar]
- 51.Sayegh, C.E., M.W. Quong, Y. Agata, and C. Murre. 2003. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat. Immunol. 4:586–593. [DOI] [PubMed] [Google Scholar]
- 52.Shoham, T., R. Rajapaksa, C. Boucheix, E. Rubinstein, J.C. Poe, T.F. Tedder, and S. Levy. 2003. The tetraspanin CD81 regulates the expression of CD19 during B cell development in a postendoplasmic reticulum compartment. J. Immunol. 171:4062–4072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}