

Abstract
Chondrocyte hypertrophy during endochondral ossification is a well-controlled process in which proliferating chondrocytes stop proliferating and differentiate into hypertrophic chondrocytes, which then undergo apoptosis. Chondrocyte hypertrophy induces angiogenesis and mineralization. This step is crucial for the longitudinal growth and development of long bones, but what triggers the process is unknown. Reactive oxygen species (ROS) have been implicated in cellular damage; however, the physiological role of ROS in chondrogenesis is not well characterized. We demonstrate that increasing ROS levels induce chondrocyte hypertrophy. Elevated ROS levels are detected in hypertrophic chondrocytes. In vivo and in vitro treatment with N-acetyl cysteine, which enhances endogenous antioxidant levels and protects cells from oxidative stress, inhibits chondrocyte hypertrophy. In ataxia telangiectasia mutated (Atm)–deficient (Atm−/−) mice, ROS levels were elevated in chondrocytes of growth plates, accompanied by a proliferation defect and stimulation of chondrocyte hypertrophy. Decreased proliferation and excessive hypertrophy in Atm−/− mice were also rescued by antioxidant treatment. These findings indicate that ROS levels regulate inhibition of proliferation and modulate initiation of the hypertrophic changes in chondrocytes.
Longitudinal growth of long bones occurs through endochondral ossification, which is accompanied by chondrocyte differentiation (1). Initially, mesenchymal cells are committed to become cartilage cells and condense into compact nodules and differentiate into chondrocytes. Chondrocytes then proliferate rapidly to form a model for the bone. As chondrocytes divide, they form a columnar structure and secrete cartilage-specific extracellular matrix proteins, such as type II collagen and aggrecan (1). Chondrocytes then stop dividing and increase their volume dramatically, becoming hypertrophic (2). These large chondrocytes alter the matrix by secreting type X collagen and increased levels of fibronectin to promote mineralization by calcium carbonate. Hypertrophic chondrocytes, which also produce matrix metalloproteinase (MMP) 13, die by apoptosis (1–3). Such events occur in a closed avascular area, suggesting that they are, at least in part, cell-autonomously regulated. Because inappropriate chondrocyte hypertrophy causes skeletal growth abnormalities such as dwarfism, control of hypertrophy is critical for normal longitudinal bone growth (4–6). Hypertrophic chondrocytes express vascular endothelial growth factor, stimulating blood vessels to invade the cartilage model (2). Groups of cells surrounding the cartilage model differentiate into osteoblasts, which begin forming bone matrix on the partially degraded cartilage. However, signals triggering chondrocyte hypertrophy have not been identified.
Chondrocyte hypertrophy is part of a normal differentiation process in which cells undergo apoptosis (1, 2). Reactive oxygen species (ROS), which induce apoptosis in cells such as neurons (7), play critical roles in cell regulation, sometimes as second messengers (8, 9). ROS, including superoxide anion (O2−), hydroxyl radical (OH), and hydrogen peroxide (H2O2), which can diffuse through membranes, are by-products of cellular oxidative metabolism. Nitrogen-containing oxidants such as nitric oxide are called reactive nitrogen species. ROS are also generated by several types of stimulation other than cell metabolism, such as phagocytosis (10). Excess ROS can overwhelm a cell's antioxidant scavenging capacity, causing oxidative damage to DNA, lipids, and proteins, as well as concomitant cellular damage (11). Therefore, the regulation of intracellular ROS is crucial for cell survival.
Ataxia telangiectasia mutated (ATM) functions in oxidative defense, and mice with Atm loss of function show premature aging caused by defects in DNA repair (12, 13). We previously showed that hematopoietic stem cell function was suppressed through elevation of ROS levels, p38 mitogen- activated protein kinase (MAPK) phosphorylation, and p16INK4A expression in ATM-deficient mice (14). In that study, the administration of N-acetyl cysteine (NAC), an antioxidant (15), rescued stem cell function, indicating that ATM regulates intracellular H2O2 levels (14).
In this paper, we demonstrate that increased ROS levels were seen in hypertrophic chondrocytes, and antioxidant treatment blocked chondrocyte hypertrophy and rescued the inhibited proliferation in chondrocytes. In vitro treatment of chondrogenic cells with oxidants directly induced chondrocyte hypertrophy, and induction was inhibited by NAC. ROS activated extracellular signal-regulated kinase (ERK) and p38 MAPK pathways in chondrogenic cells, indicating that ROS transduce a differentiation signal in those cells. Higher levels of ROS were detected in the chondrocytes of growth plates in Atm −/− mice compared with wild types. Significant decreases in proliferation and excessive hypertrophy were observed in Atm −/− growth plates, changes rescued by in vivo NAC treatment. Thus, increasing ROS levels during chondrogenesis inhibit proliferation and initiate chondrocyte hypertrophy in growth plates.
RESULTS
Increased ROS in hypertrophic chondrocytes
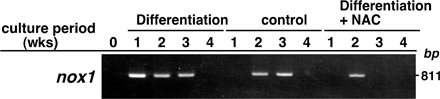
We began by asking whether ROS levels were elevated in vivo during chondrogenesis by undertaking dihydroethidium staining of wild-type E17.5 mouse embryos. Dihydroethidium is intracellularly oxidized by ROS and converted to fluorescent ethidium (Eth). Eth can bind to chromosomal DNA, yielding Eth-DNA; therefore, ROS can be detected by nuclear Eth-DNA accumulation. Higher levels of intracellular ROS were detected in prehypertrophic and hypertrophic chondrocytes compared with proliferating chondrocytes or surrounding tissues (Fig. 1 A). To further analyze ROS levels during chondrogenesis, the chondrogenic cell line ATDC5 was cultured in differentiation medium containing insulin, transferrin, and sodium selenite, as previously described (16). Under this culture condition, ATDC5 cells efficiently undergo a chondrogenic differentiation program similar to that observed in vivo (17). Hypertrophic chondrocyte markers such as type X collagen and mmp13, as well as increased intracellular levels of ROS detected by dichlorodihydrofluorescein diacetate (DCF-DA), were induced in ATDC5 cells cultured in differentiation medium for 2 wk (Fig. 1, B and C). We examined the effect of NAC treatment on intracellular ROS levels during chondrocyte hypertrophy. After NAC treatment, the higher intracellular ROS levels in ATDC5 cells cultured in differentiation medium were effectively reduced to levels comparable to those seen in cells cultured in control medium (Fig. 1 C). Basal ROS levels in cells cultured even in control medium gradually increased and were NAC resistant (Fig. 1 C), suggesting the existence of DCF-sensitive oxidants other than H2O2. Because NAC is a glutathione precursor, which reduces intracellular H2O2 levels, differentiation medium may specifically elevate intracellular H2O2. Nicotinamide adenine dinucleotide phosphate–oxidase–1 generates superoxide and is thus responsible for ROS production (10). H2O2 is generated via dismutation of superoxide. We found that nox1 expression was highly induced in ATDC5 cells cultured in differentiation medium compared with control medium (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20062525/DC1), suggesting that differentiation signals induce expression of an ROS-generating enzyme. Interestingly, nox1 expression in ATDC5 cells cultured in differentiation medium was reduced by antioxidant treatment (Fig. S1). Thus, H2O2 signals may induce expression of ROS-generating enzymes, which in turn stimulate chondrocyte hypertrophy by increasing ROS levels.
Figure 1.

ROS are elevated in hypertrophic chondrocyte zones. (A) Forelimbs of E17.5 embryos were stained with dihydroethidium and observed under a phase contrast (a) or confocal microscope (b). Proliferating and hypertrophic chondrocyte zones were morphologically identified under the phase contrast microscope (a). P, proliferating chondrocyte zone; H, prehypertrophic and hypertrophic chondrocyte zone. Bar, 100 μm. (B) ATDC5 cells were cultured in differentiation medium for 1 or 2 wk, and RT-PCR analysis of the hypertrophic markers type X collagen and mmp13 and the internal control, β-actin, was undertaken. Data representative of at least three repeats are shown. (C) ATDC5 cells were cultured in differentiation medium (open squares), control medium (open circles), or differentiation medium plus antioxidant NAC (open triangles) for 1 or 2 wk, and intracellular ROS concentrations were determined by DCF-DA staining. Results are shown as mean fold increases ± SD in intracellular ROS compared with cells cultured in control medium. *, P < 0.05.
Elevated ROS stimulate chondrocyte hypertrophy in vitro
We next examined the effect of increased ROS on chondrocyte hypertrophy by manipulating intracellular ROS with NAC. Hypertrophic chondrocytes were detected in cartilage nodules, the cellular condensations formed in cultures of ATDC5 cells (17). To determine whether hypertrophic changes in chondrocytes are inhibited by reducing ROS, chondrocyte nodules were analyzed in the presence or absence of NAC (Fig. 2 A). Hypertrophic chondrocytes detected as type X collagen–positive cells were more effectively induced in the nodules by differentiation medium compared with control medium, and such hypertrophic changes were inhibited by NAC treatment. However, there was a recovery in the formation of type X collagen–positive cell–containing nodules even after NAC treatment at 3 and 4 wk of culture. This is possibly caused by an elevation in the levels of basal oxidants resistant to NAC, as shown in Fig. 1 C.
Figure 2.

Hypertrophic changes are associated with elevated ROS. (A, top) ATDC5 cells cultured in control medium (a), differentiation medium (b), or differentiation medium plus NAC (c) for 3 wk were stained using rabbit anti–type X collagen antibody, followed by Alexa Fluor 488–conjugated anti–rabbit Ig antibody, and observed under a fluorescence microscope. (bottom) Data are mean numbers ± SD of type X collagen–positive hypertrophic chondrocyte–containing nodules. Bar, 50 μm. (B) ATDC5 cells were cultured in differentiation medium, control medium, or differentiation medium plus antioxidant NAC for 1–4 wk, and the expression of mmp13, type X collagen, runx2, and β-actin was determined by RT-PCR.
It has been reported that mmp13 and type X collagen are late hypertrophic chondrocyte differentiation markers, and their expression is not detected in proliferating chondrocyte zones in vivo (2, 3). Runx2 is a transcription factor essential for osteoblast differentiation, and Runx2-deficient mice show defective hypertrophic chondrocyte maturation, whereas forced Runx2 expression induces chondrocyte hypertrophy (5, 18). To evaluate chondrocyte hypertrophy in ATDC5 cells cultured in various conditions with or without NAC, we examined the expression of hypertrophic chondrocyte markers such as mmp13, type X collagen, and runx2 by RT-PCR. Expression of mmp13, type X collagen, and runx2 was reduced by NAC treatment, and the hypertrophic changes in chondrocytes were inhibited by reducing ROS (Fig. 2 B). Again, there was a recovery in expression of these genes at late culture stages after NAC treatment.
Elevated ROS stimulate chondrocyte hypertrophy in vivo
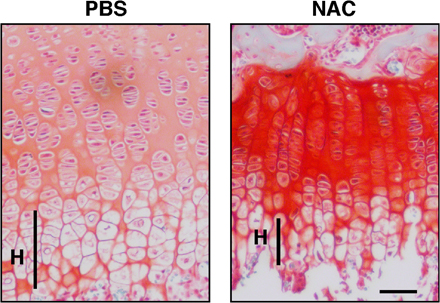
To analyze the role of ROS in chondrocyte hypertrophy in vivo, we analyzed chondrogenesis in mice treated with NAC. To do so, we injected NAC into mice every other day from the day of birth until 2 or 3 wk of age, when animals were analyzed. ROS levels in growth-plate chondrocytes were effectively reduced by this treatment (Fig. 3 A). To analyze chondrocyte hypertrophy in growth plates treated with NAC, proteoglycans were stained by Alcian blue or Safranin-O, as previously described (19, 20). Proteoglycans are mainly produced in proliferating chondrocytes and down-regulated in hypertrophic chondrocytes. NAC treatment reduced the size of the hypertrophic chondrocyte zone in growth plates (Fig. 3 B). Interestingly, the proteoglycan synthesis indicated by Alcian blue staining was up-regulated by NAC treatment (Fig. 3 B), and the reduced hypertrophy and up-regulated proteoglycan production were confirmed by Safranin-O staining (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20062525/DC1).
Figure 3.

Elevated ROS stimulate chondrocyte hypertrophy in vivo. (A) Three mice were treated subcutaneously with PBS and three were treated with NAC (1 g/kg, every 2 d) for 2 wk from the day of birth, and intracellular ROS concentration was evaluated by dihydroethidium staining in growth plates. Representative data are shown. Growth plates are shown between the dotted lines. (right) Data are mean relative concentrations ± SD of ROS in chondrocytes in mice treated with NAC compared with PBS-treated mice. A reduction of intracellular ROS in chondrocytes by NAC treatment was observed. *, P < 0.01. Bar, 100 μm. (B) Three mice were treated subcutaneously with PBS and three were treated with NAC (1 g/kg, every 2 d) for 3 wk from the day of birth, and chondrocyte hypertrophy was analyzed by Alcian blue staining. The hypertrophic zone (H) was reduced in NAC-treated compared with PBS-treated mice. (left) Representative data are shown. (right) Data are mean relative lengths ± SD of the hypertrophic zone in growth plates treated with NAC for 3 wk compared with that of PBS-treated mice. *, P < 0.01. Bar, 50 μm. (C) Three mice were treated subcutaneously with PBS and three were treated with NAC (1 g/kg, every 2 d) for 3 wk from the day of birth, and chondrocyte proliferation was evaluated by BrdU staining. TOTO3 was used as a nuclear stain. (left) Representative data are shown. Growth plates are shown between the dotted lines. (right) Data are mean relative numbers ± SD of BrdU-positive chondrocytes in mice treated with NAC compared with PBS-treated mice. *, P < 0.01. Bar, 100 μm. (D) Three mice were treated subcutaneously with PBS and three were treated with NAC (1 g/kg, every 2 d) for 2 wk from the day of birth, and angiogenesis was analyzed by PECAM staining. Frozen sections were stained with rat anti-PECAM antibody, followed by Alexa Fluor 488–conjugated goat anti–rat Ig antibody, and were observed under a phase contrast (left) or confocal (middle and right) microscopes. Representative data are shown. Bar, 50 μm.
The number of BrdU-positive proliferating cells also increased in growth-plate chondrocytes after NAC treatment (Fig. 3 C). Chondrocyte hypertrophy induces vascular endothelial growth factor expression, resulting in angiogenesis, and the hypertrophic chondrocytes then undergo apoptosis. NAC administration every other day suppressed growth of endothelial cells, as detected by platelet-endothelial cell adhesion molecule PECAM staining (Fig. 3 D). The number of apoptotic cells was also reduced by this treatment (Fig. S3 A, available at http://www.jem.org/cgi/content/full/jem.20062525/DC1).
Treatment of ATDC5 cells with H2O2 significantly reduced the number of cells, a change abolished by addition of NAC to the culture medium (Fig. S3 B). H2O2 treatment also induced apoptosis, as judged by the appearance of Annexin V–positive and propidium iodide–negative ATDC5 cells, which was reversed by NAC (Fig. S3 C). Thus, increased ROS inhibit proliferation and induce both chondrocyte hypertrophy and apoptosis. Such changes mediated by H2O2 were reversed by addition of NAC to the culture. It is assumed that physiologically increasing ROS inhibits proliferation and stimulates the differentiation of proliferating chondrocytes into hypertrophic chondrocytes, which eventually undergo apoptosis.
During endochondral ossification, hypertrophic chondrocytes induce angiogenesis and mineralization below the hypertrophic chondrocyte zone. Mineralization below the growth plates was weakly reduced by NAC treatment (unpublished data). Collectively, these results indicate that increasing ROS in chondrocytes physiologically induce hypertrophy and further regulate the apoptosis of chondrocytes, which are followed by angiogenesis and mineralization in growth plates.
ROS transduce differentiation signals in chondrocytes via ERK and p38 MAPK pathways
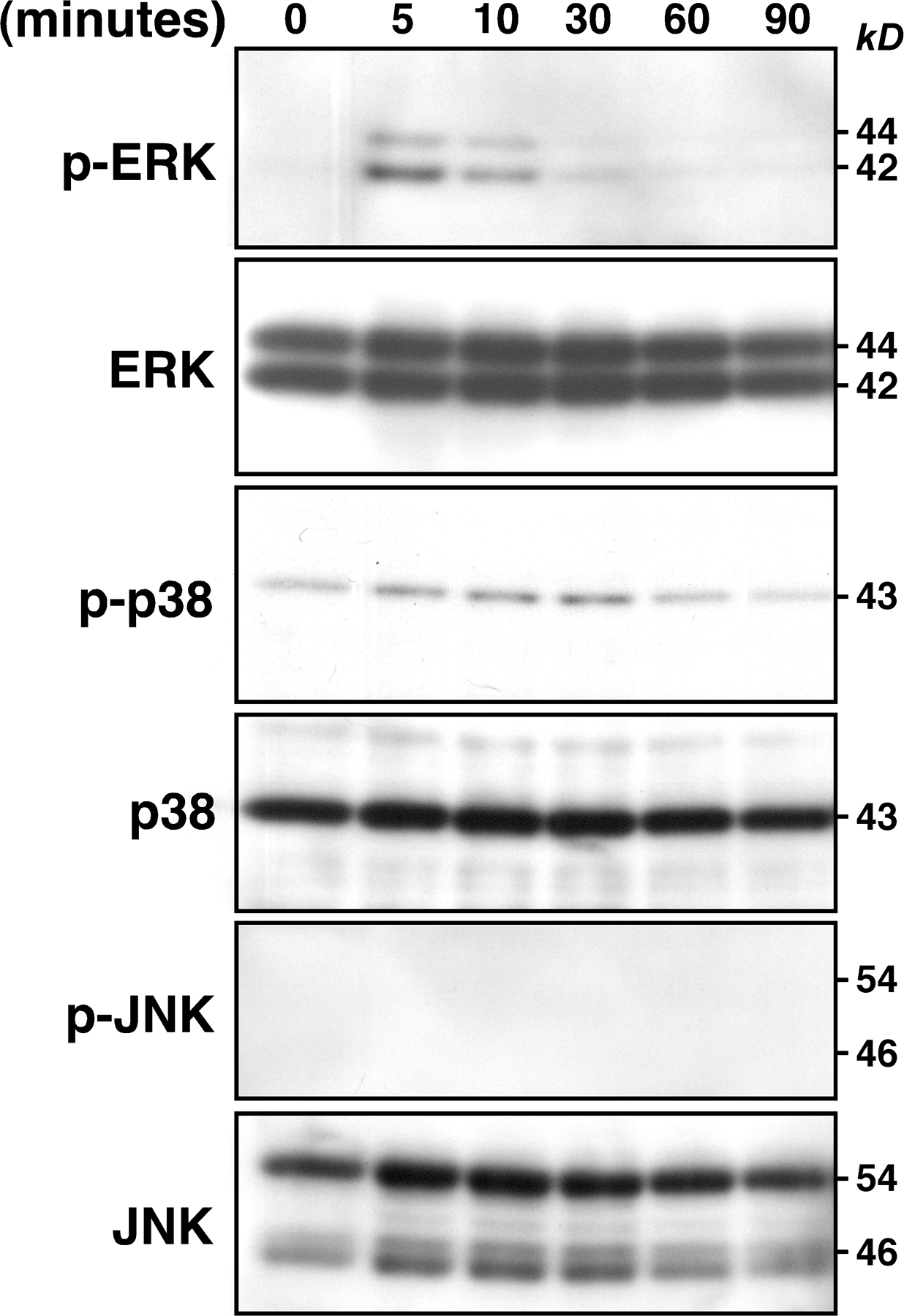
To analyze ROS signaling in chondrocyte hypertrophy directly, we elevated ROS levels by treating ATDC5 cells with H2O2 in control medium for 48 h and assayed expression of differentiation markers by RT-PCR (Fig. 4 A). H2O2 treatment effectively induced expression of these markers after 48 h of culture (Fig. 4 A). Induction of mmp13 by 48 h of treatment with H2O2 was dose dependent (Fig. 4 B). Runx2- deficient mice show defective hypertrophic chondrocyte maturation, and forced Runx2 expression induces chondrocyte hypertrophy (4, 5, 18, 21). Induction of both mmp13 and runx2 after treatment with H2O2 for 48 h was inhibited by NAC (Fig. 4 C).
Figure 4.

Elevated ROS induce chondrocyte hypertrophy. (A) ATDC5 cells were cultured in control medium with 100 μM H2O2 for 1–48 h, and expression of the hypertrophic markers type X collagen and mmp13 and of internal control β-actin was analyzed by RT-PCR. (B) ATDC5 cells were treated with various concentrations of H2O2 in control medium for 48 h, and mmp13 and β-actin expression was analyzed by RT-PCR. (C) ATDC5 cells were cultured in control medium with or without 100 μM H2O2 in the presence or absence of 100 μM NAC for 48 h, and mmp13 and runx2 expression was analyzed by RT-PCR. Blocked up-regulation of mmp13 and runx2 by treatment with 100 μM NAC in ATDC5 cells cultured for 48 h under 100 μM H2O2-induced oxidative stress conditions was observed. (D) ATDC5 cells were starved for 24 h and treated with 100 μM H2O2 for the indicated periods. Representative data determined by Western blot analysis are shown among three experiments. H2O2 treatment induced phosphorylation of ERK and p38 MAPK but not JNK. (E) ATDC5 cells were cultured in control medium with 100 μM H2O2 in the presence or absence (vehicle) of an MEK inhibitor (U0126), a p38 MAPK inhibitor (SB203580), or a JNK inhibitor (SP600125) for 48 h, and mmp13 and β-actin expression was analyzed by RT-PCR. Blocked mmp13 up-regulation by the MEK or p38 MAPK inhibitor but not by the JNK inhibitor under 100 μM H2O2-induced oxidative stress conditions was observed. Data were determined by RT-PCR. NC, no template control. (F) The effects of elevated ROS on chondrogenesis were evaluated in metatarsal bone explants after treatment in the presence or absence of 500 μM H2O2 with or without 500 μM NAC for 5 d. Metatarsal bone sections were stained with H &E. Representative data among three independent experiments are shown. Arrows indicate proliferating chondrocyte zones, and proliferating chondrocyte zones are highlighted (bottom). Bar, 50 μm.
It has been reported that the MAPK/ERK activating kinase (MEK)–ERK pathway plays a role in hypertrophic differentiation in chondrocytes, whereas p38 signaling is required for the transition from a prehypertrophic to a completely hypertrophic chondrocyte phenotype (for review see reference 22). To determine whether elevated ROS directly transduce a signal through MAPK pathways, we performed a Western blot analysis to analyze phosphorylation of ERK, c-Jun N-terminal kinase (JNK), and p38 MAPK in H2O2-treated ATDC5 cells. H2O2 treatment induced ERK and p38 MAPK phosphorylation but not that of JNK (Fig. 4 D). ERK and p38 MAPK but not JNK phosphorylation were also induced in ATDC5 cells treated with insulin (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20062525/DC1). Because insulin was added to control medium to yield differentiation medium, H2O2 transduced signals similar to those mediated by differentiation medium. U016 (an MEK inhibitor that thereby inhibits the ERK pathway) and SB203580 (a p38 inhibitor) but not SP600125 (a JNK inhibitor) suppressed mmp13 up-regulation by H2O2, indicating that ROS transduce a differentiation signal via the ERK and p38 MAPK pathways (Fig. 4 E). These results show that elevation of ROS in chondrocytes during differentiation activates ERK and p38 MAPK, and that activation of these MAPKs is required for chondrocyte hypertrophy.
Next, we performed metatarsal culture, an ex vivo explant culture, to examine how ROS elevation induces hypertrophy. To do so, we dissected three middle metatarsals from the hindlimb of E17.5 mouse embryos and cultured them in the presence or absence of H2O2 with or without NAC for 5 d. Treatment of metatarsal bones by H2O2 disrupted formation of a columnar structure in the proliferating chondrocyte zone (Fig. 4 F). Mineralization and apoptosis of the metatarsal bones were also stimulated by H2O2 treatment (Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20062525/DC1).
Disruption of the columnar structure in proliferating zones and elongated hypertrophic zones in the growth plates of Atm−/− mice
Increased ROS levels are detected in various cells in ataxia-telangiectasia patients (13). Because chondrogenesis in growth plates plays a pivotal role in longitudinal growth, we analyzed the expression and activation of ATM in chondrocytes from growth plates of Atm −/− mice. Phosphorylated ATM, the active form of the protein, was detected in growth-plate chondrocytes in wild-type mice (Fig. 5 A). Next, we assayed ROS concentration in growth-plate chondrocytes. Intracellular ROS concentrations, evaluated by dihydroethidium staining, were higher in chondrocytes from Atm −/− mice than from wild-type controls (Fig. 5 B). To analyze whether ROS elevation affects chondrocyte proliferation, we undertook BrdU analysis in Atm −/− and wild-type mice. The number of proliferating, BrdU-positive chondrocytes was significantly reduced in Atm −/− mice compared with controls (Fig. 5 C), suggesting that ROS levels are regulated by ATM in growth-plate chondrocytes and that such ROS accumulation may affect chondrocyte proliferation in growth plates.
Figure 5.

ATM functions in the development of growth plates. (A) Tibial sections of 3-wk-old wild-type mice were stained with rabbit antiphosphorylated ATM (pATM) antibody, followed by Alexa Fluor 488–conjugated anti–rabbit Ig antibody, and observed under a confocal microscope. Nuclei were stained with TOTO3. Representative data are shown. Growth plates are indicated between the dotted lines. pATM was detected in the chondrocytes of growth plates. Bar, 100 μm. (B) Increased intracellular ROS was detected by dihydroethidium staining in growth-plate chondrocytes of 2-wk-old Atm−/− mice compared with those seen in wild-type (Atm+/+) mice. Bar, 100 μm. (C) Chondrocyte proliferation was evaluated by BrdU staining in 2-wk-old Atm−/− or wild-type (Atm+/+) mice. (left) Representative data are shown. (right) Data are mean relative numbers ± SD of BrdU-positive cells in Atm −/− compared with Atm+/+ chondrocytes. Growth plates are shown between the dotted lines. Atm −/− deficiency caused defective chondrocyte proliferation. *, P < 0.01. Bar, 50 μm. (D) Disruption of columnar formation in proliferating zones (P) and enlargement of hypertrophic zones (H) were observed in growth plates of 3-wk-old Atm −/− compared with Atm+/+ mice by H&E (top) and Alcian blue (bottom) staining. Bar, 50 μm. (E) Mineralization was evaluated by alizarin red staining in growth plates of 3-wk-old Atm −/− or wild-type (Atm+/+) mice. Frozen sections that had not been decalcified were stained with alizarin red. Bar, 100 μm.
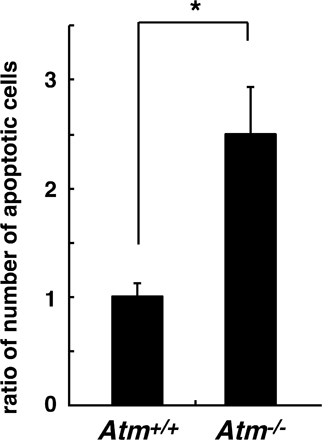
In wild-type mice, proliferating chondrocytes line up along the long axis of the bone, which shows a columnar structure (Fig. 5 D, highlighted in the small boxes). However, in the growth plates of Atm −/− mice, the formation of such columnar structures characteristic of proliferating chondrocyte zones is disrupted (Fig. 5 D). Proteoglycan synthesis was also reduced, and hypertrophic zones were enlarged in Atm −/− mice compared with controls (Fig. 5 D). Induction of vascular endothelial cells under the hypertrophic chondrocyte zone and induction of apoptosis in hypertrophic chondrocytes were stimulated in Atm −/− mice compared with wild-type mice (Fig. S6, available at http://www.jem.org/cgi/content/full/jem.20062525/DC1; and not depicted). Furthermore, mineralization under the growth-plate region, as evaluated by alizarin red staining, increased in Atm −/− compared with control mice (Fig. 5 E). Thus, ATM may regulate the process whereby proliferating chondrocytes differentiate into hypertrophic chondrocytes.
Increased ROS cause reduced proliferation in chondrocytes of Atm−/− growth plates
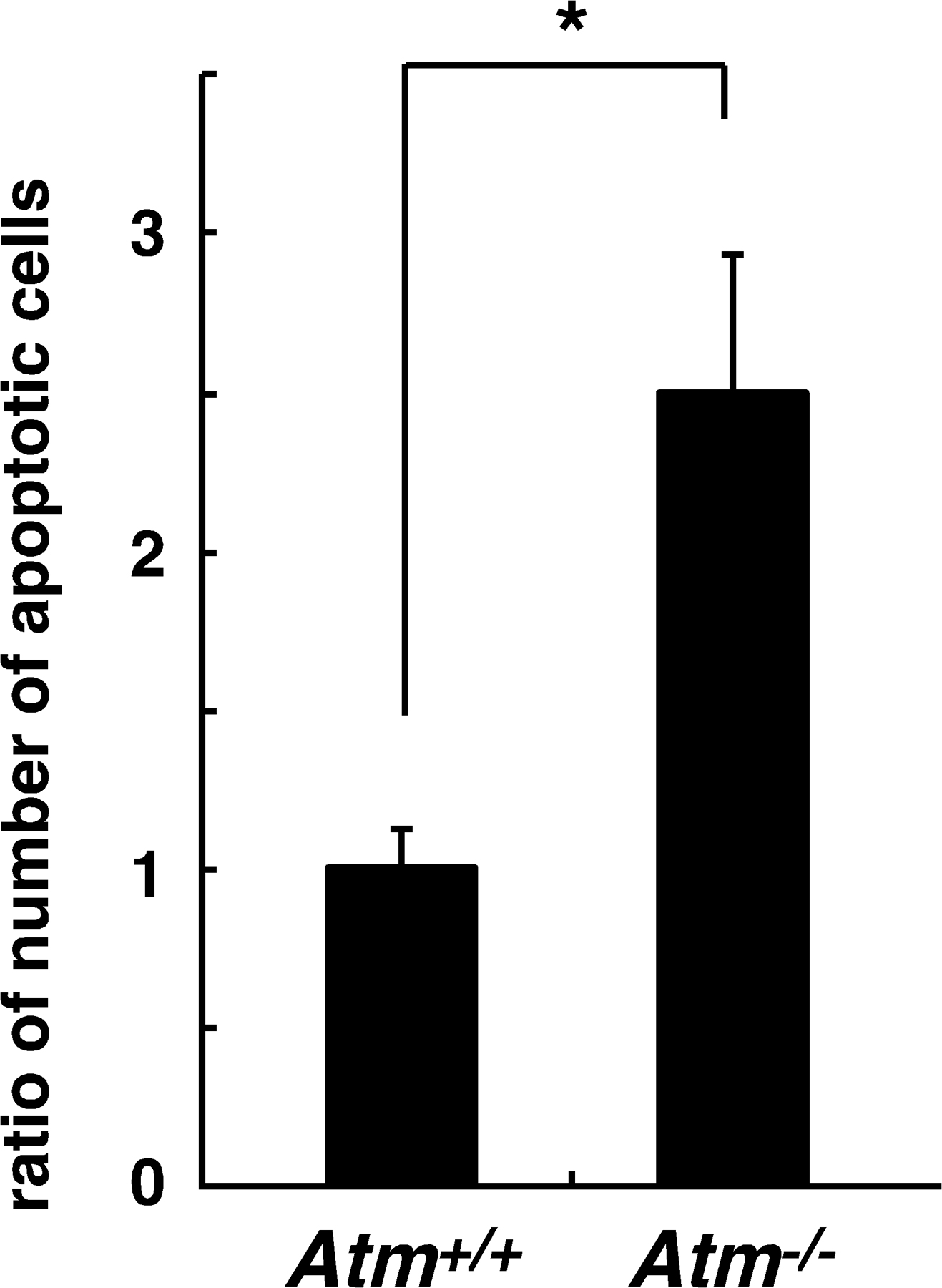
To determine whether altered chondrogenesis in Atm −/− mice is related to oxidative stress, we injected NAC into Atm −/− mice every other day from the day of birth until 2 wk of age, when animals were analyzed. The high ROS levels seen in growth-plate chondrocytes were effectively reduced by such treatment in Atm −/− mice (Fig. 6 A). Inhibited proliferation was rescued by NAC treatment in growth-plate chondrocytes of Atm −/− mice (Fig. 6 B). The elongated hypertrophic chondrocyte zone seen in the growth plates of Atm −/− mice and reduced proteoglycan synthesis were rescued by NAC treatment (Fig. 6 C). The increased number of apoptotic cells in Atm −/− hypertrophic chondrocytes was also significantly rescued by NAC treatment (Fig. 6 D), as was angiogenesis stimulated below the hypertrophic chondrocyte zone in Atm −/− mice (Fig. 6 E). Collectively, our findings suggest that elevated intracellular ROS transduce a signal suppressing proliferation and induce a hypertrophic state in chondrocytes, and that control of ROS levels by ATM is required for appropriate chondrocyte proliferation and differentiation in the growth plate.
Figure 6.

Elevated ROS levels cause reduced proliferation and an elongated hypertrophic zone in growth plates of Atm−/− mice. (A) Three Atm −/− mice were treated subcutaneously with PBS and three were treated with NAC (1 g/kg, every 2 d) for 2 wk from the day of birth, and intracellular ROS concentration was evaluated by dihydroethidium staining in growth plates. Growth plates are shown between the dotted lines. (right) Data are mean relative concentrations ± SD of ROS in chondrocytes in mice treated with NAC compared with PBS-treated mice. Increased intracellular ROS and its reduction by NAC treatment were observed in the chondrocytes of Atm −/− mice. *, P < 0.01. Bar, 100 μm. (B) Three Atm −/− mice were treated subcutaneously with PBS and three were treated with NAC (1 g/kg, every 2 d) for 2 wk from the day of birth, and chondrocyte proliferation was evaluated by BrdU staining. (left) Representative data are shown. Growth plates are shown between the dotted lines. (right) Data are mean relative numbers ± SD of BrdU-positive chondrocytes in Atm −/− mice treated with NAC compared with PBS-treated Atm −/− mice. *, P < 0.01. Bar, 100 μm. (C) Three Atm −/− mice were treated subcutaneously with PBS and three were treated with NAC (1 g/kg, every 2 d) for 3 wk from the day of birth, and chondrocyte hypertrophy was analyzed by Alcian blue staining. The hypertrophic zone (H) was reduced in NAC-treated compared with PBS-treated mice. (left) Representative data are shown. (right) Data are mean relative lengths ± SD of the hypertrophic zone in Atm −/− growth plates treated with NAC for 3 wk compared with that of PBS-treated Atm −/− mice. *, P < 0.01. Bar, 50 μm. (D) Three Atm −/− mice were treated subcutaneously with PBS and three were treated with NAC (1 g/kg, every 2 d) for 3 wk from the day of birth, and chondrocyte apoptosis was evaluated by TOTO3 nuclear staining. The number of apoptotic cells was reduced in NAC-treated compared with PBS-treated mice. Data are mean relative numbers ± SD of apoptotic chondrocytes in Atm −/− mice treated with NAC compared with those of PBS-treated Atm −/− mice. *, P < 0.01. (E) Three Atm −/− mice were treated subcutaneously with PBS and three were treated with NAC (1 g/kg, every 2 d) for 2 wk from the day of birth, and angiogenesis was analyzed by PECAM-positive endothelial cells. Frozen sections were stained with rat anti-PECAM antibody, followed by Alexa Fluor 488–conjugated goat anti–rat Ig antibody, and observed by phase contrast (left) or confocal (middle and right) microscopes. Representative data are shown. Bar, 50 μm.
DISCUSSION
Chondrogenesis occurs via three steps: (a) commitment to chondrocyte differentiation by mesenchymal cells, seen as mesenchymal condensation; (b) chondrocyte proliferation in the growth plate to facilitate longitudinal development; and (c) differentiation of proliferating chondrocytes to hypertrophic chondrocytes (for review see references 1, 2). During endochondral ossification, chondrocyte hypertrophy is critical, because cells alter the extracellular matrix and induce vascular invasion. Extrinsic factors such as bone morphogenetic proteins, Indian hedgehog, and modulators such as Sox9, Runx2, Smads, and histone deacetylase 4 are reportedly essential for chondrogenesis (1, 2). Loss of histone deacetylase 4 causes early onset of hypertrophy, followed by growth retardation (6). Overexpression of Runx2 under the control of a type II collagen promoter/enhancer induces dwarfism because of precocious endochondral ossification and accelerated chondrocyte differentiation (4, 5). Thus, regulation of hypertrophy is critical for normal growth (4–6), and abnormalities in chondrocyte hypertrophy cause skeletal growth retardation.
We propose that ROS play an essential role as signal transducers for chondrocyte hypertrophy. We demonstrate that ROS mediated inhibition of proliferation in proliferating chondrocytes and induction of hypertrophic chondrocytes. Because chondrocyte hypertrophy is associated with apoptosis, angiogenesis, and mineralization, regulation of ROS levels in chondrocytes is crucial for normal longitudinal bone growth. We show that ROS was specifically elevated in chondrocytes during the hypertrophic changes. Chondrocyte hypertrophy in ATDC5 cells, a chondrogenic cell line, was inhibited by antioxidant NAC treatment, whereas H2O2 treatment stimulated hypertrophic activity. Inhibition of chondrocyte hypertrophy was observed in vivo after administration of NAC to normal mice, indicating that physiological levels of ROS determine chondrocyte hypertrophy. We also show that ROS transduced a differentiation signal via ERK and p38 MAPK pathways, suggesting that ROS act as a biological messenger in chondrocyte hypertrophy. Chondrocyte apoptosis, angiogenesis, and mineralization were inhibited by NAC treatment, suggesting that these events result from chondrocyte hypertrophy induced by ROS. To further clarify the in vivo role of ROS in chondrogenesis, we analyzed the chondrogenesis of Atm −/− mice, which show higher intracellular accumulation of ROS compared with wild-type mice in growth-plate chondrocytes. ATM likely functions in oxidative defense by inducing major antioxidative systems, and this study shows that ATM loss increased ROS levels in chondrocytes and induced premature hypertrophy in chondrocytes. Because apoptosis is stimulated in the chondrocytes of ATM-deficient mice, elongation of the hypertrophic chondrocyte zone in ATM-deficient mice is not particularly marked compared with other mutant animals such as MMP13-deficient mice (19). We demonstrate that in vivo administration of NAC to Atm −/− mice down-regulated intracellular H2O2 levels and up-regulated proliferation and proteoglycan synthesis in chondrocytes, thereby rescuing premature hypertrophy. ROS-induced tissue damage is implicated in diseases such as osteoarthritis (23, 24), Alzheimer's disease (25, 26), and ataxia-telangiectasia (13) and in aging (27–29) and stem cell dysfunction (14). We underscored a physiological role for ROS in chondrocyte hypertrophy and observed abnormalities of chondrocyte hypertrophy after an increase in ROS levels.
ROS accumulation is caused by two mechanisms: increased ROS generation and decreased ROS degradation. Intracellular ROS may accumulate as a by-product of a high cellular metabolism, because proliferating chondrocytes show high proliferation activity. In addition, differentiation signals induced the expression of nox1, which is responsible for ROS production, including H2O2. This suggests that chondrocyte hypertrophy was stimulated by the expression of an ROS-producing gene. Further studies are needed to clarify the signals responsible for the increased generation of ROS during chondrocyte hypertrophy. Meanwhile, it has been shown that chondrocyte maturation is accompanied by a progressive decrease in catalase activity in the growth cartilage (30). Catalase plays key roles in protecting cells from oxidative stress by destruction of H2O2. Thus, chondrocyte hypertrophy is mediated by intracellular ROS levels, which are regulated by ATM. In fact, intracellular ROS were down-regulated by NAC, which reduced ROS levels in the chondrocytes of wild-type and ATM-deficient mice, and NAC treatment was accompanied by inhibition of chondrocyte hypertrophy. Collectively, we conclude that ROS accumulation beyond threshold levels may initiate inhibition of proliferation in late-proliferating chondrocytes and stimulate hypertrophic differentiation.
MATERIALS AND METHODS
Mice.
Animals were purchased from CLEA Japan, kept under pathogen-free conditions, and cared for in accordance with the guidelines of Keio University School of Medicine. Pregnant C57BL/6 mice were purchased, and embryos between E15.5–17.5 were isolated from the uteri of pregnant females. Atm+/− mice were a gift from P.J. Mckinnon (St. Jude Children's Research Hospital, Memphis, TN). Genotyping was performed by PCR-based assays of mouse-tail DNA. Littermates served as controls in all experiments.
Cell culture.
ATDC5 cells were purchased from the Institute of Physical and Chemical Research Cell Bank. Cells were plated at 6 × 104 cells/well in six-well plates and cultured in DMEM/Ham's F12 (1:1; DMEM/F12) medium (Invitrogen) supplemented with 5% FBS (Equitech-Bio, Inc.), 10 μg/ml of human transferrin (Roche Molecular Biochemicals), and 3 × 10−8 M sodium selenite (Sigma-Aldrich) as control medium, or in 10 μg/ml of human transferrin, 3 × 10−8 M sodium selenite, and 10 mg/ml of bovine insulin as differentiation medium. Cells were also cultured in DMEM/F12 containing 5% FBS with various concentrations of H2O2 (Wako) instead of differentiation medium for 48 h. For experiments with NAC and MAPK inhibitors, ATDC5 cells were cultured in the presence of 100 μM H2O2 with 500 μM NAC (Sigma-Aldrich), 10 μM U0126 (MEK1/2 inhibitor), 10 μM SB203580 (p38 MAPK inhibitor), or 10 μM SP600125 (JNK inhibitor) for 48 h. To analyze apoptosis, ATDC5 cells cultured in the presence or absence of 100 μM H2O2 with or without 500 μM NAC in control medium for 24 h were stained with FITC-conjugated Annexin V and propidium iodide (BD Biosciences) and analyzed by FACSCalibur (Beckton Dickinson).
Metatarsal culture.
Three middle metatarsals of each hindlimb of E17.5 mouse embryos were dissected and cultured for 5 d on a culture plate insert (Millicell; Millipore) in 1 ml BGJb medium (Invitrogen) supplemented with 10% FCS. 500 μM H2O2 was added with or without 100 μM NAC. After cultivation, metatarsals were fixed in 4% paraformaldehyde/PBS, embedded in paraffin, sectioned to 4 μm, and stained with hematoxylin and eosin (H&E).
Analysis of intracellular ROS.
Cell samples were either stained with DCF-DA (Sigma-Aldrich) at 4°C for 30 min and analyzed by FACSCalibur or loaded with 5 μM DCF-DA and incubated on a shaker at 37°C for 30 min. The peak excitation and emission wavelengths for oxidized DCF were 488 and 525 nm, respectively. For dihydroethidium staining, the forelimb of E17.5 mouse embryos was dissected, embedded in minced rat liver, and frozen in 2-methylbutane (Sigma-Aldrich) plus liquid nitrogen. Frozen sections of forelimb that had not been decalcified were cut on a cryostat (model HM505; MICROME), stained with dihydroethidium (Invitrogen) at room temperature for 30 min, and observed under a confocal microscope (FV1000; Olympus). ROS production in chondrocytes was quantified by signal intensity using a confocal image analyzer (FV10-ASW; Olympus).
Immunohistochemical analysis.
Tibias were dissected from 2–3-wk-old Atm −/− or control mice treated with or without PBS or NAC (1 g/kg, every 2 d) for 2–3 wk. Frozen sections that had not been decalcified were prepared as previously described (31). Specimens were stained with antiphosphorylated ATM antibody (Rockland), followed by Alexa Fluor 488–conjugated goat anti–rabbit IgG antibody (Invitrogen) and TOTO3 (Invitrogen) for nuclear staining. For BrdU staining, 200 μl BrdU solution was injected into the peritoneal cavity of 2-wk-old Atm −/− or control mice. Mice were killed 2.5 h after injection, and the tibias were extracted. Frozen sections were prepared as previously described (31) and subjected to BrdU immunohistochemistry analysis (Calbiochem). Fluorescent images were obtained using a confocal laser scanning microscope. For H&E and Alcian blue staining, tibias were dissected from 3-wk-old Atm −/− or control mice treated with or without PBS or NAC (1 g/kg, every 2 d) for 3 wk, fixed in 10% formalin, decalcified, embedded in paraffin, and cut into 4-μm sections. Deparaffinized sections of paraffin-embedded samples were stained with H&E (Muto Pure Chemical Co. Ltd.) or Alcian blue (Sigma-Aldrich).
Western blot analysis.
ATDC5 cells were starved in control medium without FBS for 12 h and stimulated with 100 μM H2O2 for various minutes. Cell lysates were collected, and Western blot analysis was performed using polyclonal antibodies to detect ERK, phosphorylated ERK, p38 MAPK, phosphorylated p38 MAPK, JNK, and phosphorylated JNK. All antibodies were obtained from Cell Signaling Technology.
RT-PCR.
Total RNA was extracted from cultured ATDC5 cells, and cDNA was synthesized by RT. RT-PCR analysis was performed using the following primer sets: mmp13-sense, 5′-AGAAGTCTACAGTGACCTCCACAGTT-3′; mmp13-antisense, 5′-GACTCTCACAATGCGATTACTCC-3′; type X collagen–sense, 5′-TTCATGGGATGTTTTATGCTGAACG-3′; type X collagen–antisense, 5′-TTTAGGTCCTTGGGGTCCCATATTC-3′; runx2- sense, 5′-AGAAGGCTCTGGCGTTTAAATGGTT-3′; runx2-antisense, 5′-AAAAGGACTTGGTGCAGAGTTCAGG-3′; β-actin–sense, 5′-TCGTGCGTGACATCAAAGAG-3′; and β-actin–antisense, 5′-TGGACAGTGAGGCCAGGATG-3′.
Statistical analysis.
p-values were calculated by the unpaired Student's t test.
Online supplemental material.
Fig. S1 shows the expression of nox1 in chondrocytes. Fig. S2 shows the Safranin-O staining of chondrocytes in mice treated with PBS or NAC. Fig. S3 shows the induction of chondrocyte apoptosis by ROS. Fig. S4 shows the activation of MAPKs by insulin in chondrocytes. Fig. S5 shows the induction of mineralization and apoptosis in chondrocytes by ROS. Fig. S6 shows the chondrocyte apoptosis in ATM-deficient mice. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20062525/DC1.
Supplemental Material
Acknowledgments
We thank P.J. McKinnon for providing Atm +/− mice, and Y. Sato and A. Kumakubo for technical support.
T. Miyamoto was supported by a grant-in-aid for Young Scientists (B), Japan. T. Suda was supported by a grant-in-aid from Specially Promoted Research of the Ministry of Education, Culture, Sports, Science and Technology, Japan.
The authors have no conflicting financial interests.
Abbreviations used: ATM, ataxia telangiectasia mutated; DCF-DA, dichlorodihydrofluorescein diacetate; ERK, extracellular signal-regulated kinase; Eth, ethidium; H&E, hematoxylin and eosin; JNK, c-Jun-N-terminal kinase; MAPK, mitogen-activated protein kinase; MEK, MAPK/ERK activating kinase; MMP, matrix metalloproteinase; NAC, N-acetyl cysteine; PECAM, platelet-endothelial cell adhesion molecule; ROS, reactive oxygen species.
K. Morita and T. Miyamoto contributed equally to this work.
References
- 1.Kronenberg, H.M. 2003. Developmental regulation of the growth plate. Nature. 423:332–336. [DOI] [PubMed] [Google Scholar]
- 2.Goldring, M.B., K. Tsuchimochi, and K. Ijiri. 2006. The control of chondrogenesis. J. Cell. Biochem. 97:33–44. [DOI] [PubMed] [Google Scholar]
- 3.Ortega, N., D.J. Behonick, and Z. Werb. 2004. Matrix remodeling during endochondral ossification. Trends Cell Biol. 14:86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takeda, S., J.P. Bonnamy, M.J. Owen, P. Ducy, and G. Karsenty. 2001. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 15:467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ueta, C., M. Iwamoto, N. Kanatani, C. Yoshida, Y. Liu, M. Enomoto-Iwamoto, T. Ohmori, H. Enomoto, K. Nakata, K. Takada, et al. 2001. Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J. Cell Biol. 153:87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vega, R.B., K. Matsuda, J. Oh, A.C. Barbosa, X. Yang, E. Meadows, J. McAnally, C. Pomajzl, J.M. Shelton, J.A. Richardson, et al. 2004. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 119:555–566. [DOI] [PubMed] [Google Scholar]
- 7.Rego, A.C., and C.R. Oliveira. 2003. Mitochondrial dysfunction and reactive oxygen species in excitotoxicity and apoptosis: implications for the pathogenesis of neurodegenerative diseases. Neurochem. Res. 28:1563–1574. [DOI] [PubMed] [Google Scholar]
- 8.Dawson, T.M., and S.H. Snyder. 1994. Gases as biological messengers: nitric oxide and carbon monoxide in the brain. J. Neurosci. 14:5147–5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forman, H.J., J.M. Fukuto, and M. Torres. 2004. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am. J. Physiol. Cell Physiol. 287:C246–C256. [DOI] [PubMed] [Google Scholar]
- 10.Bedard, K., and K.H. Krause. 2007. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87:245–313. [DOI] [PubMed] [Google Scholar]
- 11.Bredesen, D.E. 1995. Neuronal apoptosis. Ann. Neurol. 38:839–851. [DOI] [PubMed] [Google Scholar]
- 12.Savitsky, K., A. Bar-Shira, S. Gilad, G. Rotman, Y. Ziv, L. Vanagaite, D.A. Tagle, S. Smith, T. Uziel, and S. Sfez. 1995. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 268:1749–1753. [DOI] [PubMed] [Google Scholar]
- 13.Barzilai, A., G. Rotman, and Y. Shiloh. 2002. ATM deficiency and oxidative stress: a new dimension of defective response to DNA damage. DNA Repair (Amst.). 1:3–25. [DOI] [PubMed] [Google Scholar]
- 14.Ito, K., A. Hirao, F. Arai, S. Matsuoka, K. Takubo, I. Hamaguchi, K. Nomiyama, K. Hosokawa, K. Sakurada, N. Nakagata, et al. 2004. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 431:997–1002. [DOI] [PubMed] [Google Scholar]
- 15.Jayalakshmi, K., M. Sairam, S.B. Singh, S.K. Sharma, G. Ilavazhagan, and P.K. Banerjee. 2005. Neuroprotective effect of N-acetyl cysteine on hypoxia-induced oxidative stress in primary hippocampal culture. Brain Res. 1046:97–104. [DOI] [PubMed] [Google Scholar]
- 16.Shukunami, C., C. Shigeno, T. Atsumi, K. Ishizeki, F. Suzuki, and Y. Hiraki. 1996. Chondrogenic differentiation of clonal mouse embryonic cell line ATDC5 in vitro: differentiation-dependent gene expression of parathyroid hormone (PTH)/PTH-related peptide receptor. J. Cell Biol. 133:457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shukunami, C., K. Ishizeki, T. Atsumi, Y. Ohta, F. Suzuki, and Y. Hiraki. 1997. Cellular hypertrophy and calcification of embryonal carcinoma-derived chondrogenic cell line ATDC5 in vitro. J. Bone Miner. Res. 12:1174–1188. [DOI] [PubMed] [Google Scholar]
- 18.Komori, T., H. Yagi, S. Nomura, A. Yamaguchi, K. Sasaki, K. Deguchi, Y. Shimizu, R.T. Bronson, Y.H. Gao, M. Inada, et al. 1997. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 89:755–764. [DOI] [PubMed] [Google Scholar]
- 19.Stickens, D., D.J. Behonick, N. Ortega, B. Heyer, B. Hartenstein, Y. Yu, A.J. Fosang, M. Schorpp-Kistner, P. Angel, and Z. Werb. 2004. Altered endochondral bone development in matrix metalloproteinase 13- deficient mice. Development. 131:5883–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horiki, M., T. Imamura, M. Okamoto, M. Hayashi, J. Murai, A. Myoui, T. Ochi, K. Miyazono, H. Yoshikawa, and N. Tsumaki. 2004. Smad6/Smurf1 overexpression in cartilage delays chondrocyte hypertrophy and causes dwarfism with osteopenia. J. Cell Biol. 165:433–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Enomoto, H., M. Enomoto-Iwamoto, M. Iwamoto, S. Nomura, M. Himeno, Y. Kitamura, T. Kishimoto, and T. Komori. 2000. Cbfa1 is a positive regulatory factor in chondrocyte maturation. J. Biol. Chem. 275:8695–8702. [DOI] [PubMed] [Google Scholar]
- 22.Stanton, L.A., T.M. Underhill, and F. Beier. 2003. MAP kinases in chondrocyte differentiation. Dev. Biol. 263:165–175. [DOI] [PubMed] [Google Scholar]
- 23.Henrotin, Y., B. Kurz, and T. Aigner. 2005. Oxygen and reactive oxygen species in cartilage degradation: friends or foes? Osteoarthritis Cartilage. 13:643–654. [DOI] [PubMed] [Google Scholar]
- 24.Henrotin, Y.E., P. Bruckner, and J.P. Pujol. 2003. The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthritis Cartilage. 11:747–755. [DOI] [PubMed] [Google Scholar]
- 25.Manton, K.G., S. Volovik, and A. Kulminski. 2004. ROS effects on neurodegeneration in Alzheimer's disease and related disorders: on environmental stresses of ionizing radiation. Curr. Alzheimer Res. 1:277–293. [DOI] [PubMed] [Google Scholar]
- 26.Milton, N.G. 2004. Role of hydrogen peroxide in the aetiology of Alzheimer's disease: implications for treatment. Drugs Aging. 21:81–100. [DOI] [PubMed] [Google Scholar]
- 27.Andressoo, J.O., and J.H. Hoeijmakers. 2005. Transcription-coupled repair and premature ageing. Mutat. Res. 577:179–194. [DOI] [PubMed] [Google Scholar]
- 28.Schriner, S.E., N.J. Linford, G.M. Martin, P. Treuting, C.E. Ogburn, M. Emond, P.E. Coskun, W. Ladiges, N. Wolf, H. Van Remmen, et al. 2005. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 308:1909–1911. [DOI] [PubMed] [Google Scholar]
- 29.Miller, R.A. 2005. Biomedicine. The anti-aging sweepstakes: catalase runs for the ROSes. Science. 308:1875–1876. [DOI] [PubMed] [Google Scholar]
- 30.Matsumoto, H., S.F. Silverton, K. Debolt, and I.M. Shapiro. 1991. Superoxide dismutase and catalase activities in the growth cartilage: relationship between oxidoreductase activity and chondrocyte maturation. J. Bone Miner. Res. 6:569–574. [DOI] [PubMed] [Google Scholar]
- 31.Fujita, N., T. Miyamoto, J. Imai, N. Hosogane, T. Suzuki, M. Yagi, K. Morita, K. Ninomiya, K. Miyamoto, H. Takaishi, et al. 2005. CD24 is expressed specifically in the nucleus pulposus of intervertebral discs. Biochem. Biophys. Res. Commun. 338:1890–1896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}