

Abstract
B cells use translesion DNA synthesis (TLS) to introduce somatic mutations around genetic lesions caused by activation-induced cytidine deaminase. Monoubiquitination at lysine164 of proliferating cell nuclear antigen (PCNAK164) stimulates TLS. To determine the role of PCNAK164 modifications in somatic hypermutation, PCNAK164R knock-in mice were generated. PCNAK164R/K164R mutants are born at a sub-Mendelian frequency. Although PCNAK164R/K164R B cells proliferate and class switch normally, the mutation spectrum of hypermutated immunoglobulin (Ig) genes alters dramatically. A strong reduction of mutations at template A/T is associated with a compensatory increase at G/C, which is a phenotype similar to polymerase η (Polη) and mismatch repair–deficient B cells. Mismatch recognition, monoubiquitinated PCNA, and Polη likely cooperate in establishing mutations at template A/T during replication of Ig genes.
High-affinity antibodies are generated by somatic hypermutation (SHM) within the variable regions of Ig heavy and light chain genes (1). SHM enables antigen-specific B cells of the germinal center to mutate their Ig genes at a rate of 10−3 base pairs per generation, compared with 10−9 for spontaneous mutations (2). The sequential action of the single B cell–specific DNA lesion inducer activation-induced cytidine deaminase (AID) as well as proteins involved in DNA repair and DNA damage tolerance increase the mutation rate locally by six orders of magnitude (3, 4). AID initiates this process by deamination of cytosine (C) to generate uracil (U) in the variable region of immunoglobulin genes (5). Removal of uracil by the uracil N-glycosylase 2 (UNG2) generates an abasic site. In hypermutating B cells, high fidelity replication over the uracil lesion favors transitions (C to T and G to A), whereas low fidelity replication over abasic sites favors transversions (C to G/A or G to C/T). Consistent with these predictions, mutations at C/G pairs are shifted toward transitions in mice lacking UNG2 (6). These data indicated that lesions, like C to U transitions and noninstructive abasic sites favor the generation of mutations at G/C base pairs (phase I or UNG2-dependent pathway of SHM). Alternatively, the mismatch repair (MMR) complex human mutS homologue 2 (MSH2)–MSH6 can recognize U/G mismatches (7). Lack of either MSH2 or MSH6 results in a reduction of mutations at template A/T, normally accounting for 50% of all mutations generated, and simultaneously increases mutations at G/C (8–10). The increased mutation frequency at template G/C (G/C bias) observed in MSH2-deficient mice implicates that processing of U/G mismatches by MSH2–MSH6 likely triggers an alternative mutagenic repair pathway that is responsible for the establishment of most mutations at template A/T (phase II or MMR-dependent pathway of SHM). Although SHM is perturbed by a single deficiency in either UNG2, MSH2, or MSH6, combined UNG2/MSH2 or UNG2/MSH6 deficiency leads to a total ablation of SHM at A/T pairs, but, as expected, does not affect replication across the initiating U/G mispair, enabling G to A and C to T transitions (6, 8–12). Thus, two repair pathways, base excision repair and MMR, which are normally effective in restoring U/G lesions, provide alternative pathways in generating somatic mutations. What allows base excision repair and MMR to become mutagenic, i.e., what prohibits the completion of these faithful repair pathways to enable programmed mutagenesis? The identification and characterization of damage-tolerant, error-prone replication pathways in yeast provide a possible scenario. Extensive screenings of DNA damage–sensitive mutants led to the identification of the Rad6 epistasis group comprising the ubiquitin-conjugating/ligating complexes Rad6–Rad18 and Mms2–Ubc13–Rad5, the translesion DNA polymerases polymerase η (Polη; Rad30), Rev1, and Polζ (a heterodimer of Rev3 and Rev7), the high fidelity polymerase Polδ, the SRS2 helicase, and proliferating cell nuclear antigen (PCNA; references 13, 14; Fig. 1).
Figure 1.

Role of the Rad6 epistasis group in DNA damage bypass. The ring-shaped PCNA homotrimer encircles DNA, and by tethering DNA Polδ to the template it serves as an important processivity factor for DNA replication. In the presence of DNA damage (star), PCNA becomes mono ubiquitinated at K164 by the ubiquitin-conjugating/ligating complex Rad6–Rad18. PCNA-Ub can directly activate TLS polymerases (like Polη, Rev1, and Polζ), enabling error-prone damage bypass. Alternatively, K63-linked polyubiquitination of PCNA-Ub by the Rad5–Mms2–Ubc13 complex enables template switching and thus an error-free damage bypass. Besides ubiquitination, PCNA can also be SUMOylated at K164. PCNA-SUMO recruits the anti-recombinogenic Srs2 helicase, which prohibits Rad51 filament formation and is thought to favor damage bypass indirectly. The figure was adapted from Hoege et al. (reference 20). Red circle, ubiquitin; black circle, SUMO.
Although the RAD6 epistasis group was first described in the budding yeast, Saccharomyces cerevisiae, functional orthologues have been identified in higher eukaryotic organisms, implying that this pathway is of general importance (15–18). The Rad6 epistasis group provides two alternative pathways to allow stalled DNA replication to continue across damaged templates, such as noninstructive abasic sites (19). Lesions in the DNA template cause replication forks to arrest and triggers Rad6–Rad18–mediated monoubiquitination of PCNA at lysine residue K164 (reference 20; PCNA-Ub). PCNA-Ub serves as a molecular switch for the high fidelity, error-free DNA Polδ and the low-fidelity, error prone translesion DNA synthesis (TLS) polymerases Polη, Rev1, and Polζ (21). TLS polymerases such as Polη and Rev1 can bypass DNA lesions, and Polζ can extend replication from non–Watson Crick base pairs to rescue stalled replication forks, albeit often at the expense of accuracy (22). TLS across lesions or even intact templates can be highly error prone and therefore requires stringent control. The existence of PCNA-interacting peptide box (23) and the ubiquitin binding domains Ub-binding motif and Ub-binding zinc finger in TLS polymerases of the Y family of DNA polymerases provided molecular insights into how TLS polymerase are targeted to stalled replication forks to accomplish polymerase switching and TLS activation (24). PCNA-interacting peptide is likely to provide the PCNA specificity, and Ub-binding motifs increase the avidity of this binding. In fact, several TLS polymerases are shown to become more processive when binding to PCNA-Ub (25–27).
The alternative damage tolerance pathway requires Mms2–Ubc13–Rad5–mediated, K63-linked polyubiquitination of monoubiquitinated PCNAK164. Polyubiquitinated PCNAK164 enables template switching to the intact sister chromatid and as a consequence an error-free damage bypass (19). Besides its role as a processivity factor for high and low fidelity polymerases, the PCNA trimer interacts with multiple damage control and repair factors, including the mismatch recognition proteins MSH6 and MSH3 (23).
Given the findings that Rad6–Rad18–mediated mono ubiquitination of PCNAK164 stimulates the recruitment of TLS polymerases (28) and that the TLS polymerases of the Rad6 epistasis group, Rev1 (29–31), Polζ (32, 33), and Polη (34, 35), have been linked to SHM, we here defined the role of PCNA-dependent damage tolerance in mammalian SHM. Our data strongly indicate that damage tolerance intersects MMR to establish most A/T and other mutations.
RESULTS
Generation of mice carrying a homozygous PCNAK164R mutation
To block PCNA-dependent damage tolerance, a lysine (K) to arginine (R) mutation at residue 164 of PCNA (PCNAK164R) was introduced by targeting an A to G transition in exon 4 of the mouse PCNA locus. The generation of the PCNAK164R targeting construct, targeting of embryonic stem (ES) cells, and development of a high throughput screening system for the simultaneous identification of homologous and nonhomologous recombinants have been published elsewhere (36). ES cells heterozygous for the PCNAK164R mutation were used to derive chimeric mice (37). The capacity of targeted ES cells to transmit the mutation into the mouse germline was tested by intercrossing male chimeras to wild-type mice. Considering the homotrimeric nature of the PCNA sliding clamp and its biallelic expression (Fig. 2 A), monomorph PCNA trimers with a 3K and a 0K composition will assemble in the wild-type and homozygous setting, respectively. In the heterozygous setting two equal pools of wild-type PCNA and mutant PCNAK164R coexist (Fig. 2 B), enabling the formation of trimers with a 0K, 1K, 2K, and 3K composition at a relative frequency of 1:3:3:1, respectively. If a single wild-type PCNA suffices in mediating damage tolerance, seven out of eight replication complexes (1K, 2K, and 3K complexes) are expected to be functional, and, consequently, the phenotype of heterozygous PCNAK164R mutants should be similar to that of wild type. If a 3K composition is required, replication across damaged templates is effective in just one out of eight cases and heterozygous mutants should be similar to homozygous PCNAK164R mutants. Interestingly, chimeras do transmit the PCNAK164R mutation, giving rise to normally developed heterozygous offspring. To test whether a homozygous PCNAK164R mutation is compatible with mammalian life, the offspring (n = 397) from 70 intercrosses between heterozygous mice was genotyped (Fig. 3 A). To our surprise, homozygous mutants are born, albeit at a sub-Mendelian frequency (Fig. 3 B). Overall, only 5% of the progeny carried the PCNAK164R mutation on both alleles compared with the expected 25%. To address whether the PCNAK164R mutation provides a selective disadvantage to homozygous mutant embryos, we genotyped 71 embryonic day (E) 14.5 embryos derived from intercrosses between heterozygous carriers. At day 14.5 of embryonic development 4% homozygous PCNAK164R embryos were found, which is consistent with the 5% observed for the viable offspring. Apparently the selection against homozygous embryos occurs before day E14.5.
Figure 2.

Wild-type and mutant PCNA are expressed at equal levels. (A) RT-MLPA (reference 54) on RNA isolated from B and T cells derived from the spleen of wild-type, heterozygous, and homozygous PCNAK164R mice. The relative mRNA level of wild-type and mutant PCNA mRNA determined by MLPA is shown. Heterozygous mutant mice express the wild-type and mutant allele at equal levels. Actin β and heat shock protein 90 (HSP90) serve as controls. The error bars represent the standard deviation from six independent experiments. (B) PCNA trimer formation in PCNA mutants. Whereas in wild-type and homozygous mice, only monomorph homotrimers of a 3K or 3R composition are formed, respectively, the mixed pool of PCNA molecules in heterozygous mice allows four distinct compositions: 3K, 2K1R, 1K2R, or 3R at a ratio of 1:3:3:1.
Figure 3.

Homozygous PCNAK164R mice are born at sub-Mendelian frequency and are infertile. (A) 397 offspring from 70 intercrosses between heterozygous PCNAK164R mutants were genotyped. In contrast to the 25% expected homozygous mutants, only 5% were observed. Wild-type and heterozygous mice were born at a frequency of 34 and 61%, respectively. (B) Embryos from E14.5 intercrosses between heterozygous PCNAK164R mutants with the respective Applied Biosystems sequencing profile of their PCNA alleles are shown. (C) Failure of germ cell development in homozygous PCNAK164R mice. Histological sections of testis and ovary of 3-mo-old mice. Bars: (small) 500 μm; (large) 200 μm. (top four panels) Control or PCNAK164R testes. The control testis shows all stages of normal spermatogenesis whereas PCNAK164R testes show atrophy of spermatogenesis: only Sertoli cells are found, no sperm is detected. The PCNAK164R testes show strong hyperplasia of Leydig cells. (bottom four panels) The normal ovary contains numerous follicles in all stages of development. The PCNAK164R ovary consists predominantly of interstitial cells.
Homozygous PCNAK164R mice are infertile
Despite the fact that homozygous PCNAK164R mutants are born rather infrequent, survivors develop and grow normally, indicating that the fitness of somatic cells carrying a homozygous PCNAK164R is not drastically altered. In contrast, the finding that homozygous PCNAK164R female and male mice are infertile, in conjunction with a severe hypotrophy of the gonads, suggested a selective defect in germ cell development. A histopathological examination of ovaries and testes revealed a virtual complete absence of germ cells (Fig. 3 C). The selective failure of germ, but not somatic, cell development suggests the existence of a specific PCNA modification essential for germ cells.
PCNAK164R prohibits damage-induced ubiquitination
To test whether the PCNAK164R mutation actually prohibits damage-induced PCNA ubiquitination, primary mouse embryonic fibroblasts (MEFs) were derived and genotyped by multiplex ligation-dependent probe amplification (MLPA) to identify mutant and wild-type alleles (reference 36; Fig. 4 A). The chromatin-associated PCNA fractions from nontreated and UV-irradiated primary wild-type and homozygous mutant MEFs were isolated and analyzed for the presence of ubiquitin-conjugated PCNA (Fig. 4 B). Although in nontreated wild-type MEFs the subfraction of PCNA-Ub is clearly detectable and readily increased upon UV irradiation, monoubiquitination of PCNA is lacking in homozygous PCNAK164R mutants and cannot be detected even after prolonged exposure of the x-ray film. These data confirm that the PCNAK164R mutation prohibits damage-induced mono ubiquitination of PCNA and exclude the existence of alternative damage-induced ubiquitin conjugation sites within mouse PCNA. Because PCNA-Ub is the substrate for Mms2–Ubc13–Rad5–mediated, K63-linked polyubiquitination, homozygous PCNAK164R mutants are expected to be completely defective in PCNA-dependent damage tolerance.
Figure 4.

The PCNAK164R mutation prohibits damage-induced ubiquitination. (A) Primary MEFs were genotyped by MLPA to identify wild-type (top), heterozygous (middle), and homozygous (bottom) PCNAK164R mutant cell lines. The top capillary electrophoresis pattern only detects wild-type PCNA. (middle) Besides wild type, capillary electrophoresis pattern also detects mutant PCNA at the expected ratio of 1:1. The bottom capillary electrophoresis pattern detects only mutant PCNA. 1, 2, and 3 are the three control peaks. (B) To test if PCNA ubiquitination is prohibited in homozygous mutant cells, primary MEFs from wild-type and homozygous mutants were analyzed for the presence of chromatin-associated PCNA-Ub in the absence or presence of UV-induced damage. Whereas in wild-type MEFs PCNA-Ub is readily increased upon UV-induced damage, neither the untreated nor the UV-irradiated chromatin fraction of PCNA is ubiquitinated in homozygous MEFs.
Survival and proliferation capacity of LPS-activated B cell blasts
The proliferative capacity and survival of PCNAK164R mutant cells were determined ex vivo for LPS-activated B cell blasts. B cells isolated from the spleen of wild-type, heterozygous, and homozygous mutant mice were loaded intracellularly with the fluorescent cell tracker CFSE and stimulated polyclonally with LPS for 3 d. Comparing the overlay histograms of the CFSE dilution profiles, no substantial differences in the percentage of B cells triggered to divide upon LPS activation (responder frequency), the mean of divisions among those cells that divided at least once (burst size), and the relative frequency of cells with n divisions (proliferative capacity) were found (Fig. 5 A). The survival of LPS-activated B cell blasts was measured by staining phosphatidyl serine in the outer leaflet of the plasma membrane with fluorochrome-conjugated Annexin V (AnV) and the uptake of the fluor escent, DNA binding molecule propidium iodine (PI) was used to measure the permeabilization of the plasma membrane for small molecules. The frequency of live (AnV−, PI−), apoptotic (AnV+, PI−), and dead (AnV+, PI+) cells at defined time points after polyclonal LPS activation remained indistinguishable (Fig. 5 B). In line with the growth capacity of homozygous PCNAK164R mice, these data suggest that PCNA-dependent damage tolerance plays no major role in determining the proliferative capacity and survival of PCNAK164R/K164R cells in the presence of spontaneous damage.
Figure 5.

Proliferation and survival of in vitro LPS-stimulated B cells. (A) Proliferation of B cells of all genotypes stimulated for 3 d with LPS was determined by CFSE dilution. No substantial differences were observed. (B) 3-d survival of LPS-stimulated B cells as determined by AnV staining and PI uptake. The analysis of live (AnV−, PI−), apoptotic (AnV+, PI−), and dead (AnV+, PI+) cells revealed no differences in the survival capacity of LPS blasts. The error bars represent the standard deviation from three independent experiments.
Class switch recombination is unaltered in PCNAK164R mutant mice
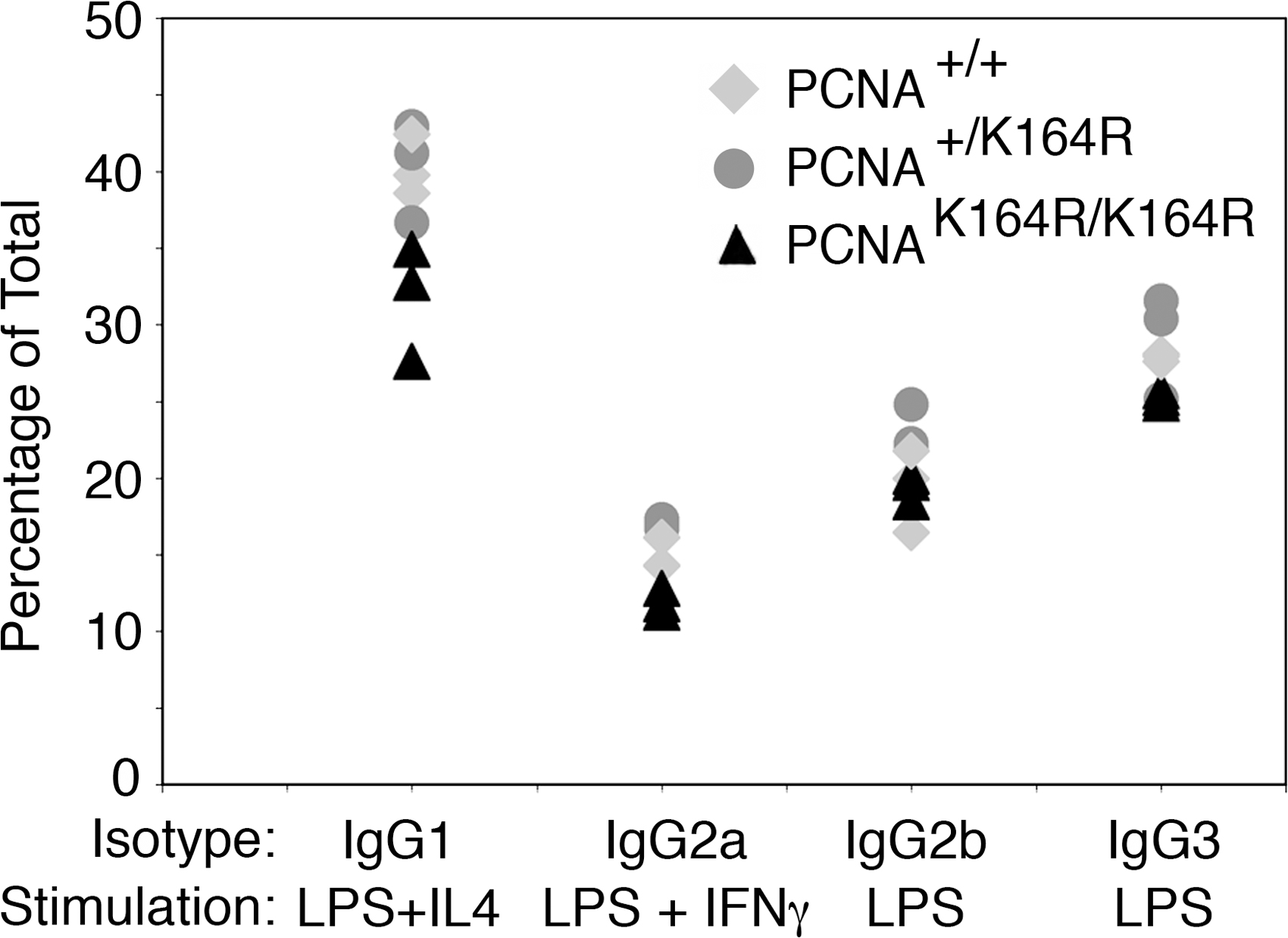
Besides SHM, AID is critical in initiating class switch recombination by the deamination of cytosines in Ig switch regions. Subsequent processing of the uracil by UNG2 or recognition of the U/G mismatch by MSH2–MSH6 is thought to generate double strand DNA breaks triggering a deletional recombination associated with class switch recombination to alter the effector function of the antibody (1). To study the role of PCNA modification in class switch recombination, B cells from the spleen were stimulated ex vivo for 4 d in the presence of different stimuli that induce switching to different Ig classes. Using LPS, LPS/IL-4, and LPS/IFN-γ, no difference in switching was observed between wild-type, heterozygous, or homozygous PCNAK164R–stimulated B cells. These data clearly indicate that modification of PCNA does not play a role in class switch recombination (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20070902/DC1).
PCNA modification controls A/T mutagenesis
To explore the role of PCNA-dependent damage tolerance in programmed mutagenesis, i.e., the intentional introduction of AID lesions into Ig variable regions and the subsequent recruitment of TLS polymerases to subserve mutator functions (4, 38), the JH4 intronic regions of memory B cells isolated from wild-type, heterozygous, and homozygous PCNAK164R mutants were amplified, sequenced, and ana lyzed for mutations. Interestingly, the frequency of point mutations in the JH4 intron of memory B cells from these mice (n = 5/genotype) remains quite similar: 1.06% (PCNA+/+), 1.10% (PCNA+/K164R), and 0.79% (PCNAK164R/K164R). The decrease in the mutation frequency is linked to a decrease in the mean number of point mutations per sequence, which is 4.5 in homozygous, 5.6 in heterozygous, and 5.8 in wild-type B cells. The frequency of mutated sequences with n mutations is shown in Fig. 6 A. The distributions of point mutations along the JH4 intron were comparable for all genotypes (Fig. 6 B). Striking alterations were observed when comparing the base exchange pattern of wild-type, heterozygous, and homozygous PCNAK164R mutants. The failure to modify PCNAK164 in homozygous mutant B cells resulted in a 10-fold reduction of transitions and transversions at template A/T—normally accounting for 50% of all mutations generated. The selective failure to mutate template A/T is compensated by an increase in mutations at template G/C, with a noted exception of C to G transversions (Fig. 6, C and D). The decrease of mutations at template A/T and simultaneous increase at template G/C further indicate the existence of two alternative pathways, a major PCNAK164-dependent A/T mutator pathway and a major PCNAK164-independent G/C mutator pathway. Interestingly, no significant differences were observed in the base exchange pattern when comparing heterozygous PCNAK164R and wild-type B cells (p-values are provided in Table S1, available at http://www.jem.org/cgi/content/full/jem.20070902/DC1).
Figure 6.

SHM is altered in the absence of PCNAK164 modification. Mutations found in memory B cells isolated from five individual mice of each genotype as indicated in the legend were pooled. (A) The mutation load is provided as the frequency of mutated sequences carrying n mutations (x axis). The mean mutation load (number of point mutations in the 5′ region of the JH4 intron) was 5.8, 5.6, and 4.5 per sequence in wild-type, heterozygous, and homozygous PCNAK164R mutants, respectively. (B) Distribution of mutations in the 5′ region of the JH4 intron, starting with the splice donor. The distribution of the mutations was found to be similar between wild-type, heterozygous, and homozygous PCNAK164R mice. -, location of RGYW/WRCY mutation hotspots; triplet, three nucleotides. (C) Base exchange pattern. Point mutations from wild-type (n = 502), heterozygous (n = 255), and homozygous (n = 503) PCNAK164R mice are analyzed. The absolute number (left) and the relative number (right) of defined base exchanges are shown. No major differences were observed between wild-type and heterozygous PCNAK164R mice. In homozygous mice, A/T mutations are virtually lacking. The decrease in generating mutations at template A/T is associated with a relative and absolute increase of G/C mutations, with the noted exception of C to G transversions. The prohibition of PCNA ubiquitination in homozygous mutants causes a twofold increase of G to A and C to T transitions. (D) To simplify the comparison of the mutation spectra, the base exchange patterns were normalized to wild type. The percentage of each base exchange in the wild type is set to one and the relative ratio of mutant to wild type is shown. With the exception of C to G transversions, all other exchanges found in homozygous mutant B cells differ significantly as determined by the χ2 test (P < 0.05). No significant changes are observed in the heterozygous mice. The individual p-values are provided in Table S1 (available at http://www.jem.org/cgi/content/full/jem.20070902/DC1).
DISCUSSION
Here we report on the generation of PCNAK614R mutant mice. This mutation prohibits site-specific modifications of PCNAK164 required for PCNA-dependent DNA damage tolerance (20). Surprisingly, homozygous PCNAK164R mice are born, albeit at a sub-Mendelian frequency. Only 5% of the offspring from heterozygous parents were homozygous for the PCNAK164R allele, suggesting a counter selection of homozygous mutants during embryonic development. The counter selection occurs before E14.5, as the same frequency is found at this stage of mouse development. Despite the initial counter selection, our data indicate that mammals can develop in the absence of PCNA-dependent DNA damage tolerance. We are currently addressing the possible activation of compensatory DNA repair or damage tolerance pathways enabling the survival of some homozygous mutants in the presence of spontaneous DNA damage. With the exception of germ cells, somatic cells appear to develop normally in surviving homozygous mice. The infertility, caused by the lack of germ cells, suggests the existence of a PCNAK164 modification that is essential for a germ cell–specific process, possibly linked to meiosis. Besides ubiquitination, the alternative conjugation of the small ubiquitin-like modifier (SUMO) to PCNAK164 has to be considered as an essential PCNA modification for germ cells.
Monoubiquitination of PCNAK164 has been proposed to regulate the recruitment and activity of TLS polymerases, enabling direct replication across damaged as well as nondamaged templates (39–41). As TLS polymerases were found to play a role in SHM (29–35) and some TLS polymerases are activated by PCNA-Ub (25–27), we determined the requirement for PCNA-Ub in establishing somatic mutations. Because the mutation load is highest in memory B cells, we chose to analyze SHM in small, class switched CD19+, IgM−, and Igκhigh B cells of the spleen. As these B cells are isolated from nonimmunized mice they are unlikely to be recently activated and therefore are referred to as memory B cells. The normal frequency of these memory B cells in homozygous mutants (unpublished data) already suggested that the PCNA mutation does not affect the ability of B cells to switch their Ig isotype. This is further supported by in vitro observations indicating that PCNAK164R mutant B cells switch normally in response to LPS, LPS/IL-4, and LPS/IFN-γ stimulation. To determine the impact of the PCNAK164R mutation on SHM, nonselected mutations from the 5′ region of the JH4 intron were analyzed. PCNAK164R B cells are able to mutate their Ig genes, but at a reduced mutation frequency of 0.79% compared with 1.06 and 1.10% for wild-type and heterozygous mutants. This finding contrasts the phenotype observed in a PCNAK164R chicken DT40 clone (31). Although the overall mutation frequency in the DT40 clone is reduced sevenfold to 15% of wild-type levels, homozygous PCNAK164R mutant B cells maintain the mutation frequency at 75%.
Considerable changes in the base exchange pattern were found in homozygous but not heterozygous PCNAK164R B cells. We observed a 90% decrease in mutations at template A/T, which is a phenotype lacking in a gene conversion-defective chicken DT40 cell line with a PCNAK164R alteration (31). The lack of an A/T mutator activity in chicken DT40 cell lines is consistent with other observations indicating that this activity is low in hypermutation-proficient cell lines (42–45). Our in vivo system clearly reveals the existence of a major PCNA-dependent A/T mutator pathway.
Because most A/T mutations (normally accounting for half of all mutations) are lacking in homozygous PCNAK164R B cells, the mutation frequency is expected to be reduced by 50%. The finding that the overall mutation frequency is reduced by only 25% indicates a partial compensation by G/C mutator activities, with the noted exception of C to G transversions. As reported previously, the deoxycytidyl-transferase Rev1 is involved in the generation of both C to G and G to C transversions in mutated Ig genes (29, 30). Remarkably, both TLS polymerases Polη and Rev1 were shown to depend on binding to PCNA-Ub for effective damage bypass (26, 46). The relative reduction in C to G transversions observed in PCNA mutant B cells may therefore be explained by an impaired Rev1 activity. The striking observation that G/C mutations are not impaired in the absence of PCNAK164 modification strongly implicates the existence of an alternative pathway that allows other TLS polymerases (mainly G/C mutators) to become activated. In fact, the heterotrimeric Rad9–Rad1–Hus1 complex (also known as the 9–1–1 complex), which is structurally similar to PCNA (47), was shown in yeast to interact with TLS polymerases (48, 49) and may thereby provide a platform for PCNA-independent TLS.
Considering the trimeric nature of the PCNA sliding clamp, it is interesting that the mutation frequency, mutation load, and base exchange pattern in heterozygous PCNAK614R mice do not differ considerably from wild type. These observations have great implications regarding the dependence of Polη activity on the ubiquitination status of the PCNA trimer. Analysis of PCNA messenger RNA (mRNA) of heterozygous B and T cell blasts reveal that both PCNA alleles are transcribed at equal levels. Therefore, in this setting, only one out of eight PCNA trimers will have a 3K composition and become ubiquitinated at all three K164 residues. Although normally all monomers within the PCNA trimer can become ubiquitinated (28, 39), the unimpaired generation of A/T mutations in heterozygous PCNAK164R B cells favors the idea that a single PCNA-Ub within the PCNA trimer suffices in activating Polη.
Our data clearly indicate that mutations at template A/T, which resembles 50% of all mutations in hypermutated Ig genes of mammals, strongly depend on PCNAK164 modification. Interestingly, a very similar phenotype was observed in B cells from Polη−/− and MMR-deficient MSH2−/− or MSH6−/− mice (8–10, 34, 35). By normalizing that published data on MSH2-, MSH6-, and Polη-deficient mice, we have compared the hypermutation phenotype between PCNA mutant, Polη-deficient, and MMR-deficient mice for substantial differences in the frequency of A/T mutations as well as base-exchange pattern. Except for Polη-deficient mice, which have a slightly higher remaining frequency of A/T mutations, the aforementioned parameters remain very similar between the different mutants. This suggests that mismatch recognition, PCNA-Ub, and Polη act in concert to establish A/T mutations during SHM in mammalian B cells. The somewhat higher frequency of A/T mutations in the Polη-deficient B cells is likely to be attributed to remaining A to C and T to G transversions. This suggests the existence of a TLS polymerase other than Polη that depends on MMR and PCNA ubiquitination to generate these transversions.
PCNA is highly expressed during S phase and becomes ubiquitinated upon DNA damage to allow replication across DNA lesions (20, 50). These observations implicate that the introduction of mutations at template A/T occurs during replication, which is consistent with the notion that Rad6- dependent damage tolerance is linked to replication (51). This assumption is further supported by the observations that G to A and C to T transitions are increased not just in homozygous PCNAK164R mutant B cells but also in Polη- and MMR- deficient B cells (8, 34, 35). The failure to modify PCNAK164 prohibits Polη and Rev1 activation and thereby favors replicative bypass of uracils by the high fidelity Polδ. Although Polδ can easily explain the increased frequency of G to A and C to T transitions, transversions at G and C likely relate to an UNG2-dependent G/C mutator activity. We speculate that the 25% reduction in the mutation frequency observed in homozygous PCNAK164R mice might therefore be attributed to the differential processing of AID-induced lesions, i.e., allowing relatively more A/T mutations to be introduced during long patch MMR-dependent resynthesis compared with G/C mutations in UNG2-dependent short patch resynthesis or UNG2-independent replicative bypass (52).
In conclusion, to explore the role of PCNA monoubiquiti nation in SHM, we have generated PCNAK164R mutant mice. Homozygous mice show a marked reduction in mutations at template A/T and a compensatory increase of mutations at template G/C. The normal base exchange pattern in heterozygous mice implies that a single PCNA-Ub suffices to activate the A/T mutator Polη. The striking similarities regarding the alterations in the base exchange patterns derived from Polη−/−, MSH2−/−, MSH6−/−, and PCNAK164R mice strongly indicate that mismatch recognition, PCNA-Ub, and Polη cooperate during replication to establish mutations at template A/T within mammalian Ig genes. This favors a model for phase II mutagenesis in which the MMR pathway is intersected by PCNA-dependent damage tolerance, enabling the recruitment and activation of specific PCNA-Ub–dependent TLS polymerases (preferentially Polη) to generate somatic mutations during replication.
MATERIALS AND METHODS
Generation of PCNAK164R mice.
The generation of the targeting construct and the targeting of the ES cells have been described previously (36). The pFlexible construct was provided by A. Bradley (Wellcome Trust Sanger Institute, Cambridge, UK). ES cells were introduced in C57BL/6J blastocysts according to standard procedures (53). Male PCNAK164R chimeras were bred to C57BL/6J females to generate heterozygous PCNAK164R offspring (F1). To determine germline transmission, tail DNA was analyzed for the presence of PCNAK164R by MLPA, as well as sequence analysis. The sequences of the MLPA probes have been published elsewhere (36). PCR primers used for sequence analysis are wild-type PCNAK164 forward (5′ GCTGAGCCTTCCCCCTTTTCTAGACT 3′) and reverse (5′ GCCGAGGCTCCATCCCTGCTTCA 3′). The PCR program used was as follows: 3 min at 95°C, 5 min at 75°C, and 1.5 min at 72°C, followed by 30 cycles of 1 min at 94°C, 1 min at 63°C, and 1.5 min at 72°C. The final extension was 10 min at 72°C. The resulting PCR fragments were cloned into a TOPO TA cloning kit (Invitrogen) and sequenced. F1 as well as F2 heterozygous PCNAK164R mice were intercrossed to generate homozygous PCNAK164R offspring. Wild-type and heterozygous littermates of homozygous PCNAK164R mice served as controls for all analyses. All experiments were performed according to national ethical guidelines and all required permissions were obtained.
Histological analysis.
Mice were killed and isolated organs were fixed in 4% paraformaldehyde in PBS, pH 7.8, embedded in paraffin, sectioned, and stained with hematoxylin and eosin. These organs include testes, ovary, uterus, mammary glands, prostate, skin, kidney, liver, intestines, thymus, heart, lungs, salivary gland, eye, brain, and pituitary gland.
Characterization of PCNAK164R expression.
To compare the expression of wild-type PCNA and mutant PCNAK164R mRNA in wild-type, heterozygous, and homozygous mice, total RNA was extracted from Con A and LPS blasts using an RNeasy kit (QIAGEN). The RNA was used for RT-MLPA analysis. Synthetic MLPA probes consisting of either two (for the controls) or three (for PCNA) oligonucleotides were developed according to Langerak et al. (36). All probes, apart from the PCNAK164R-specific MLPA probes, were designed to span an intron to omit DNA-generated background. In addition, a specific RT primer was developed for each MLPA probe. Because PCNA is expressed at high levels, competitor oligonucleotides lacking the 5′ universal primer sequence had to be added to allow a direct comparison with the control mRNAs. A list containing the sequences and amount of all oligonucleotides used can be found in Table S2 (available at http://www.jem.org/cgi/content/full//jem.20070902/DC1). RT-MLPA was performed according to Eldering et al. (54) apart from the ligation reaction, which was performed at 62°C instead of 54°C. To quantify relative expression, each MLPA probe peak height was divided by the combined peak height from the actin β and heat shock protein 90.
To reveal posttranslational modification of PCNA, we isolated MEFs from day 14.5 embryos and performed Western blotting on chromatin-associated PCNA. Non- and UV-irradiated (50 J/m2) MEFs were lysed in buffer A (100 mM NaCl, 300 mM sucrose, 3 μM MgCl2, 50 mM Hepes, pH 6.8, 1 mM EGTA, pH 8.0, 0.5% Triton-X100, and protease inhibitors) on ice for 15 min. The nuclear pellet was washed once in lysis buffer A and 50 μl of 0.1% SDS buffer was added per 350 μg of cytoplasmic protein (50 mM TrisHCl, pH 7.5, 150 mM NaCl, and 0.1% SDS), followed by sonication. The chromatin fraction was separated on a NuPAGE 12% Bis-Tris gel (Invitrogen), blotted on nitrocellulose transfer membrane (Protran; Whatman), incubated with anti-PCNA antibody (1:2,000) conjugated to horseradish peroxidase (sc-56 HRP; Santa Cruz Biotechnology, Inc.), and visualized with Supersignal (Pierce Chemical Co.).
Proliferation and survival of LPS-stimulated B cells.
CFSE labeling and FACS analysis (Becton Dickinson) was performed as described previously (29).
Isolation of memory B cells and analysis of SHM.
Viable (DAPI negative), class switched IgD− and IgM− (FITC−), CD19+ (APC+), and Igκ+ (PE+) memory B cells were sorted from spleens of 3–5-mo-old mice. DNA was isolated by proteinase K treatment and ethanol precipitation, and the equivalent of 5,000 memory B cells was used as a template to amplify the JH4 intron by PCR with pfu-polymerase (Promega) as described previously (55). The PCR products were cloned into the Zero Blunt TOPO PCR cloning kit (Invitrogen). Single bacterial colonies were sequenced using the T7 primer.
Class switch recombination assay.
The assay was performed as described previously (56). In short, T cell–depleted splenocytes were grown per 100,000 cells in a 24-well plate in RPMI medium containing 8% FBS, nonessential amino acids, sodiumpyruvate, 50 μM 2-mercaptoethanol, penicillin/streptomycin, and 25 μg/ml LPS alone (Salmonella typhimurium DIFCO) or in combination with 50 U/ml of recombinant mouse IL-4 (PeproTech) or 100 ng/ml IFN-γ (R&D Systems) for 3 or 4 d. Switching was determined by FACS analysis.
Mutation analysis and statistics.
Clonally related sequences (based on their identical third complementarity determining region rearrangements) and duplicated sequences were excluded from the analysis. Statistical analysis of the base exchange pattern was performed using the χ2 test.
Online supplemental material.
Fig. S1 shows class switch recombination in the absence and presence of PCNAK164 modification. Table S1 shows the p-values as determined by the χ2 test for the base exchanges shown in Fig. 6. Table S2 shows the oligonucleotide sequences, probe lengths, and competitor to oligonucleotide ratios as used for the RT-MLPA reactions. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20070902/DC1.
Supplemental Material
Acknowledgments
The authors wish to thank Rahmen Bin Ali for ES cell injection, Allan Bradley for providing the targeting vector pFlexible, Frank van Diepen for cell sortings, Martin van der Valk for mouse pathology, Titia Sixma, Bradley Wouters, Joyce Lebbink, and Jannie Borst for discussion, and the animal caretaker team of the Netherlands Cancer Institute-Antonie van Leeuwenhoek Hospital.
This work was made possible by financial support from the Netherlands Organization for Health Research and Development (VIDI program 917.56.328) and a startup grant from the Netherlands Cancer Institute (2.129 SFN to H. Jacobs).
The authors have no conflicting financial interests.
Abbreviations used: AID, activation-induced cytidine deaminase; AnV, Annexin V; ES, embryonic stem; K, lysine; MEF, mouse embryonic fibroblast; MLPA, multiplex ligation-dependent probe amplification; MMR, mismatch repair; mRNA, messenger RNA; MSH, mutS homologue; PCNA, proliferating cell nuclear antigen; PI, propidium iodine; Polη, polymerase η; R, arginine; SHM, somatic hypermutation; SUMO, small ubiquitin-like modifier; TLS, translesion DNA synthesis; UNG2, uracil N-glycosylase 2.
References
- 1.Chaudhuri, J., and F.W. Alt. 2004. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat. Rev. Immunol. 4:541–552. [DOI] [PubMed] [Google Scholar]
- 2.Rajewsky, K. 1996. Clonal selection and learning in the antibody system. Nature. 381:751–758. [DOI] [PubMed] [Google Scholar]
- 3.Muramatsu, M., V.S. Sankaranand, S. Anant, M. Sugai, K. Kinoshita, N.O. Davidson, and T. Honjo. 1999. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J. Biol. Chem. 274:18470–18476. [DOI] [PubMed] [Google Scholar]
- 4.Casali, P., Z. Pal, Z. Xu, and H. Zan. 2006. DNA repair in antibody somatic hypermutation. Trends Immunol. 27:313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neuberger, M.S., R.S. Harris, J. Di Noia, and S.K. Petersen-Mahrt. 2003. Immunity through DNA deamination. Trends Biochem. Sci. 28:305–312. [DOI] [PubMed] [Google Scholar]
- 6.Rada, C., G.T. Williams, H. Nilsen, D.E. Barnes, T. Lindahl, and M.S. Neuberger. 2002. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 12:1748–1755. [DOI] [PubMed] [Google Scholar]
- 7.Wilson, T.M., A. Vaisman, S.A. Martomo, P. Sullivan, L. Lan, F. Hanaoka, A. Yasui, R. Woodgate, and P.J. Gearhart. 2005. MSH2–MSH6 stimulates DNA polymerase η, suggesting a role for A:T mutations in antibody genes. J. Exp. Med. 201:637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rada, C., M.R. Ehrenstein, M.S. Neuberger, and C. Milstein. 1998. Hot spot focusing of somatic hypermutation in MSH2-deficient mice suggests two stages of mutational targeting. Immunity. 9:135–141. [DOI] [PubMed] [Google Scholar]
- 9.Jacobs, H., Y. Fukita, G.T. van der Horst, J. de Boer, G. Weeda, J. Essers, N. de Wind, B.P. Engelward, L. Samson, S. Verbeek, et al. 1998. Hypermutation of immunoglobulin genes in memory B cells of DNA repair-deficient mice. J. Exp. Med. 187:1735–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiesendanger, M., B. Kneitz, W. Edelmann, and M.D. Scharff. 2000. Somatic hypermutation in MutS homologue (MSH)3-, MSH6-, and MSH3/MSH6-deficient mice reveals a role for the MSH2–MSH6 heterodimer in modulating the base substitution pattern. J. Exp. Med. 191:579–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen, H.M., A. Tanaka, G. Bozek, D. Nicolae, and U. Storb. 2006. Somatic hypermutation and class switch recombination in Msh6(−/−)Ung(−/−) double-knockout mice. J. Immunol. 177:5386–5392. [DOI] [PubMed] [Google Scholar]
- 12.Rada, C., J.M. Di Noia, and M.S. Neuberger. 2004. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol. Cell. 16:163–171. [DOI] [PubMed] [Google Scholar]
- 13.Kunz, B.A., A.F. Straffon, and E.J. Vonarx. 2000. DNA damage-induced mutation: tolerance via translesion synthesis. Mutat. Res. 451:169–185. [DOI] [PubMed] [Google Scholar]
- 14.Watts, F.Z. 2006. Sumoylation of PCNA: wrestling with recombination at stalled replication forks. DNA Repair (Amst.). 5:399–403. [DOI] [PubMed] [Google Scholar]
- 15.Motegi, A., R. Sood, H. Moinova, S.D. Markowitz, P.P. Liu, and K. Myung. 2006. Human SHPRH suppresses genomic instability through proliferating cell nuclear antigen polyubiquitination. J. Cell Biol. 175:703–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franko, J., C. Ashley, and W. Xiao. 2001. Molecular cloning and functional characterization of two murine cDNAs which encode Ubc variants involved in DNA repair and mutagenesis. Biochim. Biophys. Acta. 1519:70–77. [DOI] [PubMed] [Google Scholar]
- 17.Jansen, J.G., and N. de Wind. 2003. Biological functions of translesion synthesis proteins in vertebrates. DNA Repair (Amst.). 2:1075–1085. [DOI] [PubMed] [Google Scholar]
- 18.Chiu, R.K., J. Brun, C. Ramaekers, J. Theys, L. Weng, P. Lambin, D.A. Gray, and B.G. Wouters. 2006. Lysine 63-polyubiquitination guards against translesion synthesis-induced mutations. PLoS Genet. 2:e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao, W., B.L. Chow, S. Broomfield, and M. Hanna. 2000. The Saccharomyces cerevisiae RAD6 group is composed of an error-prone and two error-free postreplication repair pathways. Genetics. 155:1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoege, C., B. Pfander, G.L. Moldovan, G. Pyrowolakis, and S. Jentsch. 2002. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 419:135–141. [DOI] [PubMed] [Google Scholar]
- 21.Plosky, B.S., and R. Woodgate. 2004. Switching from high-fidelity replicases to low-fidelity lesion-bypass polymerases. Curr. Opin. Genet. Dev. 14:113–119. [DOI] [PubMed] [Google Scholar]
- 22.Prakash, S., R.E. Johnson, and L. Prakash. 2005. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem. 74:317–353. [DOI] [PubMed] [Google Scholar]
- 23.Maga, G., and U. Hubscher. 2003. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J. Cell Sci. 116:3051–3060. [DOI] [PubMed] [Google Scholar]
- 24.Bienko, M., C.M. Green, N. Crosetto, F. Rudolf, G. Zapart, B. Coull, P. Kannouche, G. Wider, M. Peter, A.R. Lehmann, et al. 2005. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 310:1821–1824. [DOI] [PubMed] [Google Scholar]
- 25.Garg, P., and P.M. Burgers. 2005. Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases eta and REV1. Proc. Natl. Acad. Sci. USA. 102:18361–18366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parker, J.L., A.B. Bielen, I. Dikic, and H.D. Ulrich. 2007. Contributions of ubiquitin- and PCNA-binding domains to the activity of polymerase eta in Saccharomyces cerevisiae. Nucleic Acids Res. 35:881–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo, C., T.S. Tang, M. Bienko, J.L. Parker, A.B. Bielen, E. Sonoda, S. Takeda, H.D. Ulrich, I. Dikic, and E.C. Friedberg. 2006. Ubiquitin-binding motifs in REV1 protein are required for its role in the tolerance of DNA damage. Mol. Cell. Biol. 26:8892–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haracska, L., I. Unk, L. Prakash, and S. Prakash. 2006. Ubiquitylation of yeast proliferating cell nuclear antigen and its implications for translesion DNA synthesis. Proc. Natl. Acad. Sci. USA. 103:6477–6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jansen, J.G., P. Langerak, A. Tsaalbi-Shtylik, P. van den Berk, H. Jacobs, and N. de Wind. 2006. Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice. J. Exp. Med. 203:319–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ross, A.L., and J.E. Sale. 2006. The catalytic activity of REV1 is employed during immunoglobulin gene diversification in DT40. Mol. Immunol. 43:1587–1594. [DOI] [PubMed] [Google Scholar]
- 31.Arakawa, H., G.L. Moldovan, H. Saribasak, N.N. Saribasak, S. Jentsch, and J.M. Buerstedde. 2006. A role for PCNA ubiquitination in immuno globulin hypermutation. PLoS Biol. 4:e366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zan, H., A. Komori, Z. Li, A. Cerutti, A. Schaffer, M.F. Flajnik, M. Diaz, and P. Casali. 2001. The translesion DNA polymerase zeta plays a major role in Ig and bcl-6 somatic hypermutation. Immunity. 14:643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diaz, M., L.K. Verkoczy, M.F. Flajnik, and N.R. Klinman. 2001. Decreased frequency of somatic hypermutation and impaired affinity maturation but intact germinal center formation in mice expressing antisense RNA to DNA polymerase zeta. J. Immunol. 167:327–335. [DOI] [PubMed] [Google Scholar]
- 34.Zeng, X., D.B. Winter, C. Kasmer, K.H. Kraemer, A.R. Lehmann, and P.J. Gearhart. 2001. DNA polymerase eta is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat. Immunol. 2:537–541. [DOI] [PubMed] [Google Scholar]
- 35.Delbos, F., A. De Smet, A. Faili, S. Aoufouchi, J.C. Weill, and C.A. Reynaud. 2005. Contribution of DNA polymerase η to immunoglobulin gene hypermutation in the mouse. J. Exp. Med. 201:1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langerak, P., A.O. Nygren, J.P. Schouten, and H. Jacobs. 2005. Rapid and quantitative detection of homologous and non-homologous recombination events using three oligonucleotide MLPA. Nucleic Acids Res. 33:e188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Capecchi, M.R. 1989. Altering the genome by homologous recombination. Science. 244:1288–1292. [DOI] [PubMed] [Google Scholar]
- 38.Friedberg, E.C., P.L. Fischhaber, and C. Kisker. 2001. Error-prone DNA polymerases: novel structures and the benefits of infidelity. Cell. 107:9–12. [DOI] [PubMed] [Google Scholar]
- 39.Kannouche, P.L., J. Wing, and A.R. Lehmann. 2004. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell. 14:491–500. [DOI] [PubMed] [Google Scholar]
- 40.Watanabe, K., S. Tateishi, M. Kawasuji, T. Tsurimoto, H. Inoue, and M. Yamaizumi. 2004. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 23:3886–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plosky, B.S., A.E. Vidal, A.R. Fernandez de Henestrosa, M.P. McLenigan, J.P. McDonald, S. Mead, and R. Woodgate. 2006. Controlling the subcellular localization of DNA polymerases iota and eta via interactions with ubiquitin. EMBO J. 25:2847–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bachl, J., and M. Wabl. 1996. An immunoglobulin mutator that targets G.C base pairs. Proc. Natl. Acad. Sci. USA. 93:851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woo, C.J., A. Martin, and M.D. Scharff. 2003. Induction of somatic hypermutation is associated with modifications in immunoglobulin variable region chromatin. Immunity. 19:479–489. [DOI] [PubMed] [Google Scholar]
- 44.Zan, H., A. Cerutti, P. Dramitinos, A. Schaffer, Z. Li, and P. Casali. 1999. Induction of Ig somatic hypermutation and class switching in a human monoclonal IgM+ IgD+ B cell line in vitro: definition of the requirements and modalities of hypermutation. J. Immunol. 162:3437–3447. [PMC free article] [PubMed] [Google Scholar]
- 45.Sale, J.E., and M.S. Neuberger. 1998. TdT-accessible breaks are scattered over the immunoglobulin V domain in a constitutively hyper mutating B cell line. Immunity. 9:859–869. [DOI] [PubMed] [Google Scholar]
- 46.Guo, C., E. Sonoda, T.S. Tang, J.L. Parker, A.B. Bielen, S. Takeda, H.D. Ulrich, and E.C. Friedberg. 2006. REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol. Cell. 23:265–271. [DOI] [PubMed] [Google Scholar]
- 47.Majka, J., and P.M. Burgers. 2004. The PCNA-RFC families of DNA clamps and clamp loaders. Prog. Nucleic Acid Res. Mol. Biol. 78:227–260. [DOI] [PubMed] [Google Scholar]
- 48.Sabbioneda, S., B.K. Minesinger, M. Giannattasio, P. Plevani, M. Muzi-Falconi, and S. Jinks-Robertson. 2005. The 9-1-1 checkpoint clamp physically interacts with polzeta and is partially required for spontaneous polzeta-dependent mutagenesis in Saccharomyces cerevisiae. J. Biol. Chem. 280:38657–38665. [DOI] [PubMed] [Google Scholar]
- 49.Kai, M., and T.S. Wang. 2003. Checkpoint activation regulates mutagenic translesion synthesis. Genes Dev. 17:64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pfander, B., G.L. Moldovan, M. Sacher, C. Hoege, and S. Jentsch. 2005. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature. 436:428–433. [DOI] [PubMed] [Google Scholar]
- 51.Ulrich, H.D. 2005. The RAD6 pathway: control of DNA damage bypass and mutagenesis by ubiquitin and SUMO. ChemBioChem. 6:1735–1743. [DOI] [PubMed] [Google Scholar]
- 52.Akbari, M., M. Otterlei, J. Pena-Diaz, P.A. Aas, B. Kavli, N.B. Liabakk, L. Hagen, K. Imai, A. Durandy, G. Slupphaug, and H.E. Krokan. 2004. Repair of U/G and U/A in DNA by UNG2-associated repair complexes takes place predominantly by short-patch repair both in proliferating and growth-arrested cells. Nucleic Acids Res. 32:5486–5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thompson, S., A.R. Clarke, A.M. Pow, M.L. Hooper, and D.W. Melton. 1989. Germ line transmission and expression of a corrected HPRT gene produced by gene targeting in embryonic stem cells. Cell. 56:313–321. [DOI] [PubMed] [Google Scholar]
- 54.Eldering, E., C.A. Spek, H.L. Aberson, A. Grummels, I.A. Derks, A.F. de Vos, C.J. McElgunn, and J.P. Schouten. 2003. Expression profiling via novel multiplex assay allows rapid assessment of gene regulation in defined signalling pathways. Nucleic Acids Res. 31:e153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jolly, C.J., N. Klix, and M.S. Neuberger. 1997. Rapid methods for the analysis of immunoglobulin gene hypermutation: application to transgenic and gene targeted mice. Nucleic Acids Res. 25:1913–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schrader, C.E., W. Edelmann, R. Kucherlapati, and J. Stavnezer. 1999. Reduced isotype switching in splenic B cells from mice deficient in mismatch repair enzymes. J. Exp. Med. 190:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}