

Abstract
Listeria monocytogenes is a food-borne bacterial pathogen that causes systemic infection by traversing the intestinal mucosa. Although MyD88-mediated signals are essential for defense against systemic L. monocytogenes infection, the role of Toll-like receptor and MyD88 signaling in intestinal immunity against this pathogen has not been defined. We show that clearance of L. monocytogenes from the lumen of the distal small intestine is impaired in MyD88−/− mice. The distal ileum of wild-type (wt) mice expresses high levels of RegIIIγ, which is a bactericidal lectin that is secreted into the bowel lumen, whereas RegIIIγ expression in MyD88−/− mice is nearly undetectable. In vivo depletion of RegIIIγ from the small intestine of wt mice diminishes killing of luminal L. monocytogenes, whereas reconstitution of MyD88-deficient mice with recombinant RegIIIγ enhances intestinal bacterial clearance. Experiments with bone marrow chimeric mice reveal that MyD88-mediated signals in nonhematopoietic cells induce RegIIIγ expression in the small intestine, thereby enhancing bacterial killing. Our findings support a model of MyD88-mediated epithelial conditioning that protects the intestinal mucosa against bacterial invasion by inducing RegIIIγ.
L. monocytogenes is a Gram-positive bacterium that causes sepsis and meningoencephalitis in immunocompromised adults and life-threatening maternal/fetal infections during pregnancy. In humans, systemic infection results from ingestion of L. monocytogenes–contaminated foods and requires bacterial invasion of the intestinal mucosa and dissemination to deep tissues (1, 2). On its surface, L. monocytogenes expresses internalin A, which is a protein that associates with E-cadherin and promotes bacterial uptake by epithelial cells, particularly on the tips of intestinal villi (3, 4). Once internalized, L. monocytogenes expresses listeriolysin O (LLO) to escape the vacuole and actin assembly-inducing protein ActA to move within the host cell cytoplasm (5, 6). Within the mammalian host, successful innate immune defense against systemic L. monocytogenes infection requires Toll-like receptor (TLR)–mediated signals (7–10). Mice lacking MyD88, which is an intracellular adaptor protein involved in most TLR-mediated signaling, have markedly increased susceptibility to intravenous L. monocytogenes infection, a defect that is partially attributable to deficient TNF and iNOS production by recruited monocytes (9). Although TNF, iNOS, and IFN-γ each play an essential role in innate immune defense against L. monocytogenes infection, the precise mechanism of in vivo bacterial killing in spleen and liver remains unclear.
Although invasion of the intestinal mucosa is a critical step in the pathogenesis of mammalian L. monocytogenes infection, little is known about intestinal immune defenses against this pathogen. Oral infection of mice with high doses of L. monocytogenes results in systemic spread of bacteria to spleen and liver, with pathogen-specific T cell activation in central and mucosal lymphoid compartments (11, 12). The efficiency of mouse intestinal infection can be increased by transgenically expressing human E-cadherin in intestinal epithelial cells because the affinity of bacterial internalin A is higher for human than for mouse E-cadherin (3). A recent study demonstrated that L. monocytogenes induces a dramatic innate inflammatory response in intestinal epithelial cells of germ-free, human E-cadherin transgenic mice (13). Remarkably, the inflammatory response was similar upon infection with internalin A–deficient L. monocytogenes, indicating that innate immune activation results from an E-cadherin–independent mechanism of mucosal invasion. Whether innate immune activation in the intestinal mucosa enhances the clearance of L. monocytogenes from the gut is not known.
Although the importance of TLR-mediated signaling in innate immune defense has been demonstrated after a variety of systemic infections, the role of TLRs in defense against intestinal infections is less clear. TLR expression in the intestine is highly regulated and restricted, perhaps because excessive signaling in response to commensal bacteria could result in detrimental inflammatory pathology in the gut (14). MyD88 deficiency increases susceptibility of mice to the intestinal toxicity of Dextran sulfate sodium, a defect attributed to impaired homeostasis of intestinal epithelial cells (15). Several studies have implicated signaling by inflammasome-associated cytosolic proteins in defense against intestinal bacterial infection. Mice deficient for Nod1, for example, have increased susceptibility to Helicobacter pylori infection (16), whereas mice lacking Nod2 have greater susceptibility to intestinal infection with L. monocytogenes (17).
The mammalian intestine expresses an array of antimicrobial molecules that can mediate direct bacterial killing (18). α-Defensins, called cryptdins in mice, are well-characterized antimicrobial peptides that kill bacteria by membrane disruption (19). Mice deficient for matrilysin-7, which is a metalloprotease that activates cryptdins, have increased susceptibility to intestinal Salmonella typhimurium infection (20). In the intestine, regulation of cryptdin expression remains largely undefined. Enteric cryptdins are constitutively secreted into intestinal crypts by Paneth cells, but secretion can also be induced by cholinergic agonists or microbial stimuli (19, 21). Nod2-mediated signals after L. monocytogenes infection are postulated to limit bacterial dissemination by enhancing cryptdin expression in the small intestine (17). TLR- mediated induction of defensins has also been shown in human epithelial cell lines (22, 23).
Recently, RegIIIγ, which is produced by intestinal Paneth cells, has been identified as a protein with potent in vitro bactericidal activity (24). In contrast to cryptdins, RegIIIγ acts exclusively against Gram-positive bacteria and mediates bacterial killing by binding to surface-exposed carbohydrate moieties of peptidoglycan. Commensal bacteria, such as Gram-negative Bacteroides thetaiotaomicron, induce RegIIIγ expression in the small bowel, whereas the Gram-positive probiotic bacterium Bifidobacterium longum down-regulates RegIIIγ expression (25). Infection of germ-free mice with L. monocytogenes, on the other hand, induces RegIIIγ expression, but whether this enhances in vivo bacterial clearance remains unclear (13). Furthermore, the innate immune signaling pathways inducing RegIIIγ expression in the gut remain undefined.
We report on our studies of intestinal L. monocytogenes infection of wt and MyD88-deficient mice. Although bacterial elimination from the stomach and proximal small intestine is rapid and similar in both mouse strains, clearance of L. monocytogenes is markedly impaired in the distal small intestine of MyD88-deficient mice. Quantitative analysis of antimicrobial proteins in the gut of infected mice demonstrated markedly diminished expression of RegIIIγ in the distal small intestine of MyD88-deficient mice, correlating with increased numbers of viable L. monocytogenes. Antibody-mediated in vivo depletion of RegIIIγ in wt mice resulted in diminished killing of L. monocytogenes in the lumen of the small intestine, implicating this antimicrobial lectin as a first-line defense against Gram-positive intestinal pathogens. Our studies indicate that MyD88-mediated signaling in intestinal epithelial cells enhances resistance to L. monocytogenes infection by increasing secretion of RegIIIγ into the intestinal lumen, thereby killing bacteria before they have a chance to invade the epithelium.
RESULTS
Early kinetics of oral L. monocytogenes infection in MyD88-deficient and wt mice
To determine the role of MyD88-mediated signals in the setting of intestinal infection, we measured bacterial transit through the gastrointestinal tract after intragastric inoculation with live, virulent L. monocytogenes. The number of L. monocytogenes CFUs was measured in the stomach, small intestine, and colon at different time points after infection. Transit of L. monocytogenes through the stomach and small intestine was rapid. As early as 2 h after inoculation, the majority of L. monocytogenes was detected in the colon. Bacterial transit through the stomach and small intestine was similar in MyD88-deficient and wt mice, with roughly equal numbers of L. monocytogenes in the colon 2 h post-infection (p.i.; Fig. 1). Bacterial killing in the stomach, which results in a 99% reduction of CFUs, was similar in MyD88-deficient and wt mice. 5 h after oral infection, however, cultures of the small intestinal lumen of MyD88-deficient mice revealed 100-fold higher numbers of L. monocytogenes compared with wt mice (Fig. 1). Separate cultures of luminal and intestinal wall CFUs revealed, at this early time point, that all live bacteria remained in the lumen and had not entered the mucosa. Although these results suggest that MyD88-mediated signals do not alter the rate at which bacteria move through the intestinal tract (i.e., from the stomach to the colon), clearance of L. monocytogenes from the small intestine, but not the stomach or colon, is impaired in the absence of MyD88.
Figure 1.

L. monocytogenes transit through the gastrointestinal tract after intragastric inoculation of MyD88+/+ and MyD88−/− mice. Quantitation of L. monocytogenes in stomach (wall + lumen), small intestine (wall + lumen), and colon (wall + lumen) in MyD88+/+ and MyD88−/− mice after oral infection with 109 L. monocytogenes, 15, 120, and 300 min p.i. Values are representative of two individual experiments with n = 3 in each group. **, P < 0.01. Error bars represent the SD.
MyD88-deficient mice are more susceptible to oral infection with L. monocytogenes
To determine the longer-term impact of diminished early L. monocytogenes growth inhibition in the small intestine of MyD88-deficient mice, we measured bacterial growth in the small intestinal wall and dissemination to the draining mesenteric lymph nodes (MLNs) at time points after infection. L. monocytogenes CFUs in the small intestinal mucosa were detected as early as 12 h after oral infection in MyD88-deficient mice, whereas no bacteria were detected in the intestinal mucosa of wt mice (Fig. 2 A). In contrast, CFUs in the colonic wall did not differ between wt and MyD88-deficient mice 12 h p.i. (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20070563/DC1). However, at all time points tested, the small intestine in MyD88−/− mice harbored 10–100-fold greater numbers of L. monocytogenes than in wt mice (Fig. 2 A). Draining MLNs in MyD88−/− mice also contained ∼100-fold higher numbers of bacteria than wt mice 24, 48, and 72 h after oral infection with 109 L. monocytogenes (Fig. 2 B). Similarly, differences were found in the spleen and liver of MyD88−/− and wt mice 24, 48, and 72 h p.i. (Fig. S1, B and C). Although increased numbers of L. monocytogenes at later time points might result from diminished recruitment or activation of inflammatory cells in the bowel wall of MyD88−/− mice, it is likely that diminished bacterial killing in the intestinal lumen of MyD88−/− mice at early time points (Fig. 1) contributes to the increased intestinal and MLN bacterial burden seen at later time points.
Figure 2.

MyD88-deficient mice are more susceptible to intestinal L. monocytogenes infection. MyD88+/+ and MyD88−/− mice were infected with 109 L. monocytogenes by gastric gavage and the number of bacteria in the small intestinal wall (A) and MLNs (B) was determined 12 (for small intestine), 24, 48, and 72 h after infection; n = 5–8 mice/group. *, P ≤ 0.05; **, P < 0.01; ND = not detectable. Error bars represent the SD.
Clearance of L. monocytogenes is diminished in the distal small intestine of MyD88-deficient mice
The small intestine extends from the distal stomach to the colon, and expression of bactericidal peptides and proteins increases in proximity to the colon (26). We therefore asked whether the increased number of L. monocytogenes in the small intestine of MyD88−/− mice was general or focal. MyD88-deficient and wt mice were orally infected with L. monocytogenes and, 24 h later, the small intestinal wall was divided into proximal and distal segments of equal length, and the number of L. monocytogenes was determined. The distal small intestine of MyD88-deficient mice contained higher numbers of L. monocytogenes compared with wt mice (Fig. 3 A). In contrast, CFUs in the proximal small intestine did not differ between MyD88-deficient and wt mice (Fig. 3 A).
Figure 3.

Diminished clearance of L. monocytogenes in the distal small intestine of MyD88-deficient mice. (A) MyD88+/+ and MyD88−/− mice were infected with 1010 L. monocytogenes by gavage, and CFUs in the proximal (pro) and distal part of the small intestinal wall were determined 24 h p.i.; n = 8 mice/group. (B) Segments from proximal (pro) or distal small intestine were cultured in media containing 2,000 L. monocytogenes. After 4 h, bacterial burden in the supernatant was determined. Values are representative of two individual experiments. (C) Schematic representation of the in vivo luminal killing assay. (D) Bioluminescence imaging of luminescent L. monocytogenes in ileal loops of MyD88+/+ and MyD88−/− mice. Luminescent L. monocytogenes (2.5 × 105) was injected into ileal loops of MyD88+/+ and MyD88−/− mice and imaged 30 min (top) and 2 h (bottom) after injection with an IVIS Imaging System. One representative mouse per group is shown. n = 5. (E) 1,000 L. monocytogenes were injected into ileal loops of wt, MyD88−/−, TNF−/−/IFN-γ−/−, and Rip2−/− mice. 2 h after injection, the isolated section of the intestine was harvested and CFUs in the luminal fluid were detected. n = 3 for MyD88−/−; n = 5–9 for all other groups. Bacterial numbers are expressed as the percentage of recovered L. monocytogenes. *, P ≤ 0.05, ***, P < 0.001. Error bars represent the SD.
To examine differences in the ability of small intestine to inhibit bacterial growth, an in vitro system was devised in which proximal and distal intestinal segments from wt or MyD88-deficient mice were cultured for 4 h with media containing 2,000 L. monocytogenes. After incubation, CFUs in the culture supernatant were determined. The number of bacteria obtained from cultures containing MyD88−/− distal small intes tine was greater than from cultures containing wt distal small intestine. In contrast, no difference in bacterial survival was seen in cultures supplemented with wt or MyD88-deficient proximal small intestines (Fig. 3 B). These results suggest that the distal small bowel of wt mice produces factors that restrict L. monocytogenes survival or replication, and that expression of this factor is reduced in MyD88-deficient mice.
To investigate in vivo luminal killing of L. monocytogenes in the gut of wt and MyD88-deficient mice, we used a ligated ileal loop model. A 3–4-cm loop of distal ileum, immediately adjacent to the ileocecal valve, was ligated (without interrupting the blood supply) in anesthetized mice and injected with L. monocytogenes. At defined time points, the ileal loop was harvested, and the number of surviving bacteria in the lumen and intestinal wall was determined (Fig. 3 C). As a first step to analyze luminal killing in wt and MyD88-deficient mice, IVIS imaging of bioluminescent L. monocytogenes was performed 30 min (Fig. 3 D, top) and 2 h (Fig. 3 D, bottom) after injection of bacteria into ligated ileal loops. Whereas bioluminescent bacteria were detected in wt and MyD88−/− loops 30 min after infection, by 2 h, luminescence in wt mice was markedly attenuated, although persisting in MyD88-deficient mice (Fig. 3 D). This result suggests that L. monocytogenes is killed more effectively in ileal loops of wt mice than in MyD88-deficient mice. To more accurately assess the extent of bacterial killing, ileal loops of MyD88-deficient or wt mice were injected with wt L. monocytogenes, and the percentage of surviving bacteria in luminal contents was determined 2 h later. Consistent with the bioluminescence data, ileal loops of MyD88-deficient mice were less effective at killing bacteria than wt loops (Fig. 3 E). Although 90% of the inoculated L. monocytogenes were killed in wt mice, only 55% were killed in MyD88-deficient mice (Fig. 3 E).
Recent studies have demonstrated that mice deficient in Nod2, which is a cytosolic innate immune receptor that responds to peptidoglycan fragments of bacterial cell walls (27), develop greater systemic infection after oral inoculation with L. monocytogenes (17). This defect has been attributed to decreased intestinal expression of cryptdin 4 and 10 (17). Rip2 is a signaling molecule downstream of Nod2, and in its absence Nod2 signaling is abrogated (28). Killing of L. monocytogenes in ligated ileal loops of Rip2−/− mice, however, was similar to that seen in wt mice (Fig. 3 E), suggesting that the effects of MyD88- and Rip2-mediated innate immune signals provide protection against L. monocytogenes at distinct steps during the pathogenesis of intestinal infection. Along similar lines, TNF and IFN-γ are essential for clearance of L. monocytogenes from spleen and liver after systemic infection, and mice lacking these cytokines are profoundly susceptible to L. monocytogenes infection (29–32). Remarkably, however, killing of L. monocytogenes in bowel loops of mice deficient for both IFN-γ and TNF was comparable to that seen in wt mice, indicating that these cytokines are not necessary for the induction of this intestinal microbicidal activity (Fig. 3 E). These results indicate that impaired killing of L. monocytogenes in the intestinal lumen of MyD88-deficient mice is not a general characteristic of immunocompromised mice with increased susceptibility to infection.
RegIIIγ transcription and protein expression is markedly reduced in MyD88-deficient mice
The preceding results indicate that MyD88-mediated signals enhance microbial clearance from the distal intestinal lumen of infected mice. Secretion of antimicrobial peptides provides an important mucosal host defense mechanism against invasion by enteric pathogens. Therefore, we decided to quantify message levels of several cryptdins in the small intestines of MyD88-deficient and wt mice. No difference in Defensin-related cryptdin (Defcr)–related sequence (rs) 1, Defcr 4, Defcr 5, Defcr-rs 10 mRNA expression was detected between MyD88-deficient and wt mice (Fig. 4 A). The bactericidal lectin RegIIIγ was recently characterized as an antimicrobial molecule secreted by Paneth cells (24). In contrast to cryptdin expression, RegIIIγ mRNA levels were markedly decreased in the distal small intestine of MyD88-deficient mice (Fig. 4 B). As described by Cash et al. (24), we found increased RegIIIγ protein expression in the distal small bowel of wt mice. RegIIIγ protein expression, however, was profoundly diminished in uninfected MyD88-deficient mice (Fig. 4 C). To determine whether infection induces the expression of cryptdins or RegIIIγ in the distal small intestine of wt or MyD88- deficient mice, we orally infected mice with L. monocytogenes and measured mRNA expression levels 24 h later. We did not detect significant induction of cryptdins, and only a slight induction of RegIIIγ was measured 24 h after intestinal infection with L. monocytogenes (Fig. 4 D). These experiments demonstrate that MyD88-mediated signals induce the expression of RegIIIγ in the distal small intestine, providing a mechanism for the previous demonstrations that commensal bacteria are essential for mucosal expression of this protein (24, 25).
Figure 4.

RegIIIγ expression is dependent on MyD88-mediated signals. mRNA was extracted from the terminal ileum of MyD88+/+ and MyD88−/− mice. Defcr-rs 1, Defcr 4, Defcr 5, Defcr-rs 10 (A), and RegIIIγ (B) expression were examined by quantitative real-time PCR. Expression levels were normalized to GAPDH. Values are representative of two experiments with two mice per group. (C) Protein extracts from the proximal (pro) and distal (dis) small intestine of MyD88+/+ and MyD88−/− mice were analyzed by Western blotting with RegIIIγ-specific antiserum. Tubulin was used as a loading control. In D, mice were infected with 109 L. monocytogenes by gavage, and mRNA was prepared before and 24 h after infection from the terminal ileum of wt (left) and MyD88-deficient (right) mice. Expression was examined by quantitative real-time PCR and levels were normalized to GAPDH. Values are representative of two experiments with two mice per group. Error bars represent the SD.
Paneth cells in MyD88-deficient mice do not express RegIIIγ
Paneth cells are a major source of RegIIIγ in the small intestine. To determine whether Paneth cell development, morphology, or granule development is defective in MyD88-deficient mice, we performed conventional histologic studies of the small intestines of wt and MyD88−/− mice. Fig. 5 A demonstrates that Paneth cells in MyD88-deficient and wt mice are indistinguishable by conventional histologic examination. Immunohistochemistry using a polyclonal rabbit antiserum specific for RegIIIγ (33) demonstrated RegIIIγ expression in Paneth cells of the distal small intestine in wt mice and an absence of RegIIIγ in Paneth cells of MyD88−/− mice (Fig. 5 B). These results indicate that MyD88-mediated signals are not required for Paneth cell development or granule formation, but that induction of RegIIIγ expression in Paneth cells is MyD88 dependent.
Figure 5.
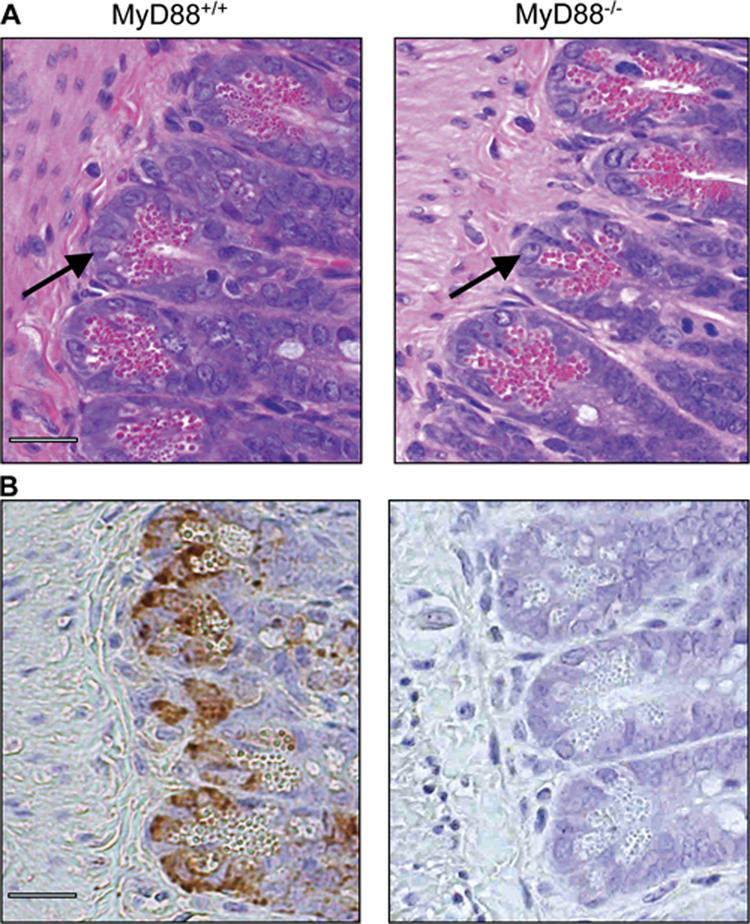
Histology of MyD88+/+ and MyD88-deficient mice. (A) Two representative small intestinal sections from the distal part of MyD88+/+ and MyD88−/− mice (hematoxylin and eosin–stained) are shown. Arrows indicate Paneth cells. (B) Immunohistochemical detection of RegIIIγ in paraffin-embedded distal small intestinal sections in MyD88+/+ and MyD88-deficient mice. Bars, 50 μm.
RegIIIγ mediates in vivo killing of L. monocytogenes in the small intestinal lumen
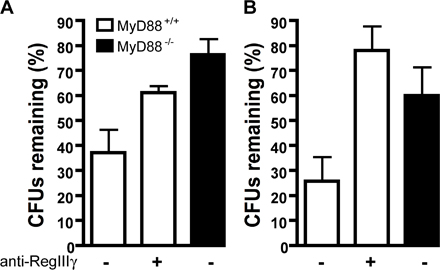
Because RegIIIγ expression in the distal small intestine correlates with regional inhibition of L. monocytogenes, we postulated that its absence might account for impaired bacterial clearance in the small intestine of MyD88−/− mice. To determine whether RegIIIγ mediates killing of L. monocytogenes, we injected polyclonal antiserum against RegIIIγ or preimmune serum into ligated ileal loops before injection of 1,000 L. monocytogenes, and the number of surviving bacteria was determined 2 h later. Polyclonal antiserum against RegIIIγ has been used before, and its generation and specificity is described by Cash et al. (24, 33). Whereas ∼90% of the inoculated bacteria were killed in ileal loops of mice treated with preimmune serum, only 40% of bacteria were killed in mice that received the RegIIIγ-specific antiserum (Fig. 6 A). Similar results were obtained with higher L. monocytogenes inoculum and different antibody concentrations (Fig. S2, A and B, available at http://www.jem.org/cgi/content/full/jem.20070563/DC1). Thus, antibody-mediated blockade of RegIIIγ significantly diminished luminal killing of L. monocytogenes. Luminal killing of L. monocytogenes in MyD88-deficient mice was similar, regardless of treatment with RegIIIγ antiserum (Fig. 6 A). To determine whether anti-RegIIIγ antiserum resulted in increased association of bacteria with intestinal tissues, we also quantified the number of bacteria associated with the small intestinal wall. Only a small proportion (∼2%) of inoculated bacteria gained entry into the intestinal wall, and no difference in bacterial numbers was detected between the experimental groups (unpublished data).
Figure 6.

RegIIIγ is responsible for diminished luminal killing in MyD88-deficient mice. (A) Antiserum against RegIIIγ or the preimmune serum was injected into ileal loops of MyD88+/+ and MyD88−/− mice before injection of 1,000 L. monocytogenes. After 2 h, bacterial growth in the luminal fluid of the isolated section of the small intestine was determined; n = 6 mice/group. (B) After reconstitution of MyD88-deficient mice with recombinant RegIIIγ or control protein (β2microglobulin), killing of 1,000 L. monocytogenes in ileal loops of MyD88-deficient mice was assessed; n = 5 each group. (A and B) Bacterial numbers are expressed as the percentage of recovered L. monocytogenes. **, P < 0.01; ***, P < 0.0001. Error bars represent the SD.
Although these results suggest that defective production of RegIIIγ in MyD88-deficient mice results in diminished luminal killing of L. monocytogenes in the distal small intestine, it is possible that other MyD88-dependent factors account for this defect. To address this issue, we injected recombinant RegIIIγ that was generated as described by Cash et al. (24, 33) into the lumen of MyD88-deficient ligated ileal loops before inoculation with L. monocytogenes and measured the impact on bacterial survival. Fig. 6 B demonstrates that administration of RegIIIγ markedly diminishes L. monocytogenes survival in the gut of MyD88-deficient mice, indicating that RegIIIγ is the major factor responsible for MyD88-mediated luminal killing of bacteria.
L. monocytogenes expresses cell surface and secreted proteins that enable attachment to host cells (internalin A and B mediate the binding to epithelial cells or hepatocytes, respectively), escape from the phagocytic vacuole (LLO), and locomotion in the cytosol (ActA) of the invaded cell (6). To investigate whether these virulence factors are necessary for efficient luminal killing in ligated ileal loops, L. monocytogenes strains deficient for LLO, ActA, internalin A, internalin B, and internalin A/internalin B, respectively, were examined. The percentage of recovered L. monocytogenes was similar for all of these strains (Table I). This indicates that bacterial adhesion, invasion, phagosomal escape, and locomotion within the host cells are not required for RegIIIγ-mediated killing of L. monocytogenes in the small intestinal lumen.
Table I.
Remaining CFUs 2 h after administering different L. monocytogenes strains into ileal loops of wt mice (n = 6 mice/group)
| L. monocytogenes strain | Mean ± SD CFUs remaining |
|---|---|
| % | |
| 10403s (wt) | 16.4 ± 21 |
| LLO−/− | 19 ± 17 |
| ActA−/− | 14.3 ± 14.1 |
| Internalin A−/− | 21 ± 14 |
| Internalin B−/− | 12 ± 3.7 |
| Internalin A−/−/Internalin B−/− | 17.4 ± 7.6 |
MyD88-mediated signaling in nonhematopoietic cells induces RegIIIγ expression and luminal killing of L. monocytogenes
Several recent studies demonstrated that dendritic cells extend processes into the intestinal lumen and contact luminal bacteria (34, 35). It is possible, therefore, that MyD88-mediated signals in intestinal dendritic cells are required for RegIIIγ induction in epithelial cells. Alternatively, MyD88-mediated signals may directly stimulate RegIIIγ production in intestinal epithelial cells. TLR signaling in epithelial cells influences the extent of dendritic cell sampling of intestinal contents (36), thereby demonstrating that innate immune signaling in epithelial cells is active. To distinguish between direct or indirect mechanisms of RegIIIγ induction in intestinal epithelial cells, we generated BM chimeric mice in which donor and/or recipients were either wt (CD45.1) or MyD88-deficient (CD45.2). Mice were studied 7 wk after transplantation and were found to be fully chimerized, as assessed by surface staining of BM and intestinal myeloid-derived cells with CD45.1- and CD45.2-specific anti bodies (Table S1 and S2, available at http://www.jem.org/cgi/content/full/jem.20070563/DC1). To determine the impact of MyD88-mediated signaling in hematopoietic or nonhematopoietic cells on RegIIIγ protein levels, Western blot analysis of intestines from chimeric mice was performed. Chimeric mice lacking MyD88 in hematopoietic cells expressed RegIIIγ levels similar to wt mice reconstituted with wt BM, demonstrating that MyD88-mediated signaling in hematopoietic cells does not induce RegIIIγ production (Fig. 7 A). In contrast, transplant recipients lacking epithelial MyD88 had markedly diminished RegIIIγ levels, regardless of whether the BM donor was wt or MyD88 deficient (Fig. 7 A). These findings indicate that RegIIIγ expression is induced by direct MyD88-mediated signals in intestinal epithelial cells. To determine whether RegIIIγ expression in BM chimeric mice influences luminal killing of L. monocytogenes, in vivo luminal killing assays were performed (Fig. 7 B). Luminal killing of L. monocytogenes correlated precisely with the level of RegIIIγ expression in the chimeric mice, further supporting the importance of this bactericidal lectin for in vivo microbial killing, but also demonstrating that MyD88-mediated signals in epithelial cells are essential for bacterial killing at this stage of infection.
Figure 7.

Nonhematopoietic MyD88-mediated signaling is involved in RegIIIγ expression and RegIIIγ-mediated luminal killing of L. monocytogenes. (A) Protein extracts from distal small intestine of chimeric mice were analyzed by Western blotting with RegIIIγ-specific antiserum. Tubulin was used as a loading control. Shown are representative extracts from each genotype. n = 5 for wt→MyD88 and for MyD88→wt; n = 3 for wt →wt and for MyD88→MyD88. (B) 1,000 L. monocytogenes were injected into ileal loops of different chimeric mice. After 2 h, bacterial growth in the luminal fluid of the isolated section of the small intestine was determined; n = 5 for wt→MyD88 and for MyD88→wt; n = 3 for wt →wt and for MyD88→MyD88. Bacterial numbers are expressed as the percentage of recovered L. monocytogenes. **, P < 0.01. Error bars represent the SD.
DISCUSSION
The first, and arguably best, opportunity for the immune system to limit infection by orally acquired microbial pathogens is in the gastrointestinal tract. In this study, we demonstrate that mice deficient for MyD88 have a diminished capacity to kill L. monocytogenes in the lumen of the distal small intestine, a region that expresses high levels of antimicrobial peptides and proteins. MyD88-deficient mice express normal complements of cryptdins in the intestinal mucosa, but markedly diminished levels of RegIIIγ, a bactericidal lectin that selectively kills Gram-positive bacteria. We show that antibody- mediated depletion of RegIIIγ from the intestinal lumen of wt mice compromises killing of L. monocytogenes to an extent similar to that seen in MyD88−/− mice. Complementing MyD88-deficient mice with recombinant RegIIIγ, on the other hand, enhances intestinal killing of L. monocytogenes to nearly wt levels. These results strongly support the conclusion that RegIIIγ is a major effector molecule in defense against oral L. monocytogenes infection. Furthermore, these findings demonstrate, for the first time, that MyD88-mediated signals are essential for the induction of RegIIIγ, and that these innate immune signals mediate killing of micro organisms before invasion of the intestinal mucosa.
Several models can be proposed for MyD88-mediated defense against L. monocytogenes infection in the small intestine. The simplest model is that L. monocytogenes invades the intestinal epithelium, in the process triggering TLRs in the gut mucosa and inducing synthesis and secretion of RegIIIγ into the bowel lumen and subsequent microbial killing. We believe this model is unlikely for several reasons. First, we see high levels of RegIIIγ associated with the bowel mucosa in uninfected wt mice (Fig. 5 B and not depicted), suggesting that commensal bacteria have induced a high level of expression before infection with L. monocytogenes. Second, induction of RegIIIγ mRNA is minimal after intestinal infection with L. monocytogenes (Fig. 4 D). Third, killing of attenuated L. monocytogenes strains that are incapable of invading the intestinal epithelium and wt bacteria is similar, indicating that bacterial invasion is not required for the MyD88-mediated enhancement of L. monocytogenes killing. An alternative model, therefore, is that MyD88-mediated signals, presumably triggered by commensal organisms, induce RegIIIγ production in the distal small bowel, thereby conditioning the bowel and enhancing resistance to subsequent exposure to pathogenic bacteria. In support of this model, previous studies have demonstrated that commensal bacteria induce RegIIIγ in the distal ileum, and germ-free mice have markedly reduced expression of RegIIIγ (24). Not all intestinal bacteria are equivalent at inducing RegIIIγ, however. The Gram-negative bacterium B. thetaiotaomicron induces expression of RegIIIγ in intestinal epithelial cells, whereas the Gram-positive bacterium B. longum suppresses RegIIIγ expression (25). These findings suggest that resistance or susceptibility to enteric L. monocytogenes infection may be highly influenced by the composition of the commensal microbial flora.
MyD88- and RegIIIγ-mediated killing of pathogenic microbes in the intestinal lumen is likely to have beneficial consequences for the mammalian host. Indeed, clear correlations between the likelihood of systemic infection and the size of the bacterial inoculum have been demonstrated for most intestinal bacterial pathogens. Although it is conventional to think that microbes disseminating from the bowel transit through regional MLNs before entry into the blood stream, recent studies suggest that bacterial replication in the bowel lumen results in direct hematogenous spread of bacteria to spleen and liver (37). A mechanism that kills bacteria in the gut lumen, thus, could be predicted to significantly reduce the risk of bacterial dissemination. Although RegIIIγ likely provides such a mechanism, it is not the only protective mechanism. For example, the probiotic bacterium Lactobacillus salivarius UCC118 expresses a bacteriocin that kills L. monocytogenes in the gut lumen, thereby limiting systemic infection (38). Our finding that luminal clearance of L. monocytogenes was normal in mice lacking both TNF and IFN-γ, two cytokines that individually play essential roles in systemic immunity to this pathogen and are positively regulated by MyD88-mediated signals, illustrates how distinct mucosal and systemic innate immune defenses can be.
Nod2-deficient mice are more susceptible to intestinal L. monocytogenes infection, a phenotype that has been attributed to diminished cryptdin expression in the mouse intestine (17). Our results demonstrating that Rip2-deficient mice have normal clearance of L. monocytogenes from the lumen of the distal small intestine (Fig. 3 E) suggest that Nod2/Rip2-mediated antimicrobial activities are either temporally or anatomically distinct from those mediated by MyD88/RegIIIγ. Further studies will be required to precisely define the timing and localization of Nod2/Rip2-mediated inhibition of L. monocytogenes dissemination from the mouse intestine.
Several recent studies have demonstrated that intestinal dendritic cells can extend dendrites into the intestinal lumen and, in some circumstances, can also phagocytose bacteria and carry them to spleen and liver (34, 35). Because dendritic cells express a broad array of innate immune receptors, it is plausible that MyD88-mediated signaling in BM-derived intestinal DCs might contribute to the induction of RegIIIγ in the small intestine. Recent studies have also demonstrated a role for MyD88-mediated signaling in epithelial regeneration (15, 39, 40) after treatment with DSS. In this system, MyD88-mediated signals in macrophages underlying the epithelium (40) promote the repositioning of intestinal mesenchymal cells and their focal production of prostaglandin E2, which enhances the activity of colonic epithelial progenitor cells (41). Our experiments with BM chimeras, however, demonstrated that RegIIIγ induction in the small intestine is exclusively induced by MyD88-mediated signals in non–BM-derived cells, presumably intestinal epithelial cells. It remains unclear, however, whether MyD88-mediated induction of RegIIIγ occurs directly in Paneth cells, or whether innate immune signals initiated in intestinal epithelial cells near the lumen are transmitted to Paneth cells in intestinal crypts.
Our studies demonstrate an important, previously undescribed role for MyD88-mediated innate immune signaling in defense against infection by an intestinal pathogen. Although innate immune responses are typically considered to be rapid, at times even immediate, in this case, innate immune activation by commensal organisms occurs preemptively, before exposure to the pathogen. Importantly, the preemptive induction of RegIIIγ is not associated with overt evidence inflammation, but rather reflects a homeostatic, noninflammatory state mediated by commensal organisms. It is likely that MyD88-mediated expression of RegIIIγ influences the composition of the commensal flora, with downstream implications for mucosal homeostasis and susceptibility to Gram-positive intestinal pathogens.
MATERIALS AND METHODS
Mice, bacteria, infection.
MyD88−/− and Rip2−/− mice, which were backcrossed at least nine times onto the C57BL/6 background, were provided by S. Akira (Osaka University, Osaka, Japan) and R. Flavell (Yale University, New Haven, CT). TNF−/−/IFN-γ−/− mice were generated at Memorial Sloan-Kettering Research Animal Resource Center by crossing TNF−/− mice with IFN-γ−/− mice (The Jackson Laboratory). C57BL/6 and CD45 congenic C57BL/6 Ly5.1 mice (CD45.1) were purchased from The Jackson Laboratory. All mice were kept and bred at the Memorial Sloan-Kettering Research Animal Resource Center. For all experiments, mice were sex and age matched and maintained under specific pathogen–free conditions before infection. All animal procedures were approved and performed according to institutional guidelines for animal care. Mice were infected (via gavage) with 109 CFU of the streptomycin-resistant L. monocytogenes strain 10403s. 10403s, a wt L. monocytogenes strain and isogenic mutant strains, ActA-deficient L. monocytogenes strain (DP-L1942), LLO-deficient L. monocytogenes strain (DP-L2161), internalin A–deficient L. monocytogenes strain (DP-L4405), internalin B–deficient L. monocytogenes strain (DP-L4406), and internalin A/internalin B double-deficient L. monocytogenes strain (DP-L4404) were provided by D. Portnoy (University of California, Berkeley, Berkeley, CA). For bioluminescence imaging, luminescent L. monocytogenes strain Xen 32 (Xenogen Corporation) was used. Bacterial counts within the various organs were determined by homogenizing the tissues in PBS containing 0.05% Triton X-100 and plating on brain–heart infusion plates containing 100 μg/ml streptomycin and 50 μg/ml nalidixic acid to inhibit the growth of endogenous bacterial flora. Within the small intestine and the colon, differential counts of L. monocytogenes present in the gastrointestinal lumen vs. wall were performed; the luminal contents were flushed out with 2 × 5 ml of sterile PBS. L. monocytogenes colonies were identified by appearance and confirmed by Gram staining.
Generation of BM chimeric mice.
Recipient wt (CD45.1) or MyD88-deficient (CD45.2) mice were lethally irradiated with 950 rads using a 137Cs source and injected intravenously 2–3 h later with 5 × 106 BM cells derived from the tibia and femurs of the respective donors. 7 wk after engrafting, reconstitution was assessed by FACS analysis of BM and small intestinal lamina propria. After red blood cell lysis, BM and lamina propria cells were stained with FITC-labeled CD45.1 antibody and PerCPCy5.5–labeled CD45.2 antibody. Cell suspensions were analyzed on a BD LSR II (BD Biosciences).
Real-time PCR analysis.
The small intestine was divided into four parts, a 1.5-cm segment was excised from the distal portion, and total RNA was isolated using the Trizol reagent (Invitrogen). DNase-treated RNA underwent randomly primed cDNA synthesis and real-time PCR analysis. SYBR Green-based real-time PCR was performed using the DyNAmo SYBR Green qPCR kit (Finnzymes). Defcr-rs1-, Defcr5-, and RegIIIγ-specific primers were obtained from QIAGEN, and Defcr4- and Defcr-rs10–specific primers were purchased from SuperArray. Signals were normalized to GAPDH RNA (forward, 5′-ACCACAGTCCATGCCATCAC-3′; reverse, 5′-TCCACCACCCTGTTGCTGTA-3′). Normalized data were used to quantitate relative levels of Decr-rs1-, Defcr4-, Defcr5-, Defcr-rs10-, and RegIIIγ using ΔΔCt analysis.
Western blot analysis.
Small intestines were divided into four pieces and samples were taken from the first proximal part and from the fourth distal part and processed as described previously (24). Protein samples were analyzed by reducing 4–12% SDS-PAGE (Nupage Bis-Tris-Gel; Invitrogen) and detected with polyclonal RegIIIγ antiserum generated as described previously (33) or preimmune serum. As loading control, tubulin was detected with a monoclonal anti-tubulin antibody (Santa Cruz Biotechnology), followed by a horseradish peroxidase–conjugated anti–mouse IgG2 antibody (Santa Cruz Biotechnology). Bound antibody was detected by enhanced chemiluminescence (GE Healthcare).
Immunohistochemistry and hematoxylin-eosin staining.
Freshly isolated small intestine was divided into four parts. The fourth distal part was fixed in formalin and embedded in paraffin. Immunohistochemical staining for RegIIIγ was performed using the Envision System (DakoCytomation) for rabbit primary anti bodies according to the manufacturer's protocol. RegIIIγ polyclonal antiserum was diluted 1:1,000 in PBS containing 1% BSA. Control slides were stained with preimmune serum or rabbit IgG instead of the primary antibody, and did not show any positive staining. Hematoxylin-eosin staining was performed using a standard protocol.
In vitro luminal killing.
Small intestines were opened longitudinally, washed in PBS, and divided into fourths. Three 1.5-cm segments of the proximal (first fourth) and three 1.5-cm segments of the distal (last fourth) small intestine were cultured separately in a 24-well flat-bottom culture plate in RPMI 1640 medium containing 100 μg/ml streptomycin, 10% FCS, and 2,000 L. monocytogenes. After 4 h, supernatant was plated on BHI plates containing 100 μg/ml streptomycin and 50 μg/ml nalidixic acid.
In vivo luminal killing.
After anesthesia, a midline laparotomy incision was made. The intestine was occluded with a vascular clip 1 cm proximal to the ileocecal junction and 3 cm proximal to this point. Care was taken to avoid disrupting the mesenteric vascular arcades. The length of intestine between the two clips was injected with 250 μl PBS containing 1,000 L. monocytogenes. After 2 h, mice were killed by exposure to carbon dioxide and the luminal fluid of the isolated segment was harvested and plated on BHI plates. For bioluminescence imaging 2.5 × 105 luminescent L. monocytogenes were injected into ileal loops. Bioluminescence imaging was performed using an IVIS Imaging System (Xenogen) per instruction of the manufacturer. Mice were kept anaesthetized and bioluminescence was recorded for 5 min at a pixel binning of 10; 30 min, and 2 h after injection of luminescent L. monocytogenes. For RegIIIγ blocking experiments, 250 μl of a 1:10 dilution of RegIIIγ polyclonal antiserum or preimmune serum in PBS was injected into ileal loops of MyD88+/+ or MyD88−/− mice. After 20 min, 250 μl PBS containing 1,000 L. monocytogenes was injected. Alternatively, an inoculum of 10,000 or 20,000 L. monocytogenes was used. Because of the higher inoculum (20,000 L. monocytogenes), polyclonal antiserum was diluted 1:4 in PBS. For reconstitution experiments, 250 μl MES-Buffer (25 mM pH 6, 25 mM NaCl) containing 20 μM purified recombinant RegIIIγ was injected before adding 1,000 L. monocytogenes in 100 μl MES-Buffer into the ileal loop. RegIIIγ was purified as previously described (33). As a control, purified β2microglobulin was substituted for RegIIIγ. After 2 h, the animals were killed by exposure to carbon dioxide immediately before harvesting the isolated section of the small intestine.
Statistical analysis.
Statistical analysis was performed on Prism software with the unpaired Student's t test. All P-values ≤ 0.05 were considered significant. *, P ≤ 0.05; **, P < 0.01; ***, P < 0.0001. Error bars denote the SDs.
Online supplemental material.
Fig. S1 A shows CFUs in the colonic wall of MyD88+/+ and MyD88−/− mice 12 h after oral infection with 109 L. monocytogenes; n = 4 each group. In Fig. S1 (B and C), bacterial burden in spleens and livers of MyD88+/+ and MyD88−/− mice was determined 24, 48, and 72 h after oral infection with 109 L. monocytogenes; n = 5–7 each group. Fig. S2 (A and B) demonstrates impaired luminal killing in MyD88−/− mice using 10,000 (A) and 20,000 (B) L. monocytogenes. Blocking of RegIIIγ in wt mice was performed using RegIIIγ polyclonal antiserum in different dilutions (1:10 for A and 1:4 for B). Table S1 and S2 show the degree of chimerism in BM chimeras as assessed by measuring CD45.1 and CD45.2 in BM cells (Table S1) and small intestinal lamina propria cells (Table S2).The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20070563/DC1.
Supplemental Material
Acknowledgments
The authors thank L.V. Hooper (University of Texas Southwestern Medical Center) for providing polyclonal RegIIIγ antiserum and recombinant RegIIIγ, as well as W. Falk (University of Regensburg, Germany), T. Hohl, and all other members of the laboratory for helpful discussions. The authors thank J. Zakrzewski for help with the bioluminescence imaging.
This research was supported by the Alexander von Humboldt Foundation through a Feodor Lynen postdoctoral fellowship to KB and National Institutes of Health grant AI39031 and AI42135 to EGP.
The authors have no conflicting financial interests.
Abbreviations used: Defcr, Defensin-related cryptdin; LLO, listeriolysin O; MLN, mesenteric lymph node; p.i., post-infection; TLR, Toll-like receptor; rs, related sequence.
References
- 1.Gellin, B.G., and C.V. Broome. 1989. Listeriosis. JAMA. 261:1313–1320. [PubMed] [Google Scholar]
- 2.Southwick, F.S., and D.L. Purich. 1996. Intracellular pathogenesis of listeriosis. N. Engl. J. Med. 334:770–776. [DOI] [PubMed] [Google Scholar]
- 3.Lecuit, M., S. Vandormael-Pournin, J. Lefort, M. Huerre, P. Gounon, C. Dupuy, C. Babinet, and P. Cossart. 2001. A transgenic model for listeriosis: role of internalin in crossing the intestinal barrier. Science. 292:1722–1725. [DOI] [PubMed] [Google Scholar]
- 4.Mengaud, J., H. Ohayon, P. Gounon, R.M. Mege, and P. Cossart. 1996. E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell. 84:923–932. [DOI] [PubMed] [Google Scholar]
- 5.Cossart, P., and P.J. Sansonetti. 2004. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science. 304:242–248. [DOI] [PubMed] [Google Scholar]
- 6.Pamer, E.G. 2004. Immune responses to Listeria monocytogenes. Nat. Rev. Immunol. 4:812–823. [DOI] [PubMed] [Google Scholar]
- 7.Edelson, B.T., and E.R. Unanue. 2002. MyD88-dependent but Toll-like receptor 2-independent innate immunity to Listeria: no role for either in macrophage listericidal activity. J. Immunol. 169:3869–3875. [DOI] [PubMed] [Google Scholar]
- 8.Seki, E., H. Tsutsui, N.M. Tsuji, N. Hayashi, K. Adachi, H. Nakano, S. Futatsugi-Yumikura, O. Takeuchi, K. Hoshino, S. Akira, et al. 2002. Critical roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. J. Immunol. 169:3863–3868. [DOI] [PubMed] [Google Scholar]
- 9.Serbina, N.V., W. Kuziel, R. Flavell, S. Akira, B. Rollins, and E.G. Pamer.. 2003. Sequential MyD88-independent and -dependent activation of innate immune responses to intracellular bacterial infection. Immunity. 19:891–901. [DOI] [PubMed] [Google Scholar]
- 10.Torres, D., M. Barrier, F. Bihl, V.J. Quesniaux, I. Maillet, S. Akira, B. Ryffel, and F. Erard. 2004. Toll-like receptor 2 is required for optimal control of Listeria monocytogenes infection. Infect. Immun. 72:2131–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huleatt, J.W., I. Pilip, K. Kerksiek, and E.G. Pamer. 2001. Intestinal and splenic T cell responses to enteric Listeria monocytogenes infection: distinct repertoires of responding CD8 T lymphocytes. J. Immunol. 166:4065–4073. [DOI] [PubMed] [Google Scholar]
- 12.Pope, C., S.K. Kim, A. Marzo, D. Masopust, K. Williams, J. Jiang, H. Shen, and L. Lefrancois. 2001. Organ-specific regulation of the CD8 T cell response to Listeria monocytogenes infection. J. Immunol. 166:3402–3409. [DOI] [PubMed] [Google Scholar]
- 13.Lecuit, M., J.L. Sonnenburg, P. Cossart, and J.I. Gordon. 2007. Functional genomic studies of the intestinal response to a foodborne enteropathogen in a humanized gnotobiotic mouse model. J. Biol. Chem. 282:15065–15072. [DOI] [PubMed] [Google Scholar]
- 14.Abreu, M.T., L.S. Thomas, E.T. Arnold, K. Lukasek, K.S. Michelsen, and M. Arditi. 2003. TLR signaling at the intestinal epithelial interface. J. Endotoxin Res. 9:322–330. [DOI] [PubMed] [Google Scholar]
- 15.Rakoff-Nahoum, S., J. Paglino, F. Eslami-Varzaneh, S. Edberg, and R. Medzhitov. 2004. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 118:229–241. [DOI] [PubMed] [Google Scholar]
- 16.Viala, J., C. Chaput, I.G. Boneca, A. Cardona, S.E. Girardin, A.P. Moran, R. Athman, S. Memet, M.R. Huerre, A.J. Coyle, et al. 2004. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 5:1166–1174. [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi, K.S., M. Chamaillard, Y. Ogura, O. Henegariu, N. Inohara, G. Nunez, and R.A. Flavell. 2005. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 307:731–734. [DOI] [PubMed] [Google Scholar]
- 18.Sansonetti, P.J. 2004. War and peace at mucosal surfaces. Nat. Rev. Immunol. 4:953–964. [DOI] [PubMed] [Google Scholar]
- 19.Selsted, M.E., and A.J. Ouellette. 2005. Mammalian defensins in the antimicrobial immune response. Nat. Immunol. 6:551–557. [DOI] [PubMed] [Google Scholar]
- 20.Wilson, C.L., A.J. Ouellette, D.P. Satchell, T. Ayabe, Y.S. Lopez-Boado, J.L. Stratman, S.J. Hultgren, L.M. Matrisian, and W.C. Parks. 1999. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science. 286:113–117. [DOI] [PubMed] [Google Scholar]
- 21.Ayabe, T., D.P. Satchell, C.L. Wilson, W.C. Parks, M.E. Selsted, and A.J. Ouellette. 2000. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat. Immunol. 1:113–118. [DOI] [PubMed] [Google Scholar]
- 22.Vora, P., A. Youdim, L.S. Thomas, M. Fukata, S.Y. Tesfay, K. Lukasek, K.S. Michelsen, A. Wada, T. Hirayama, M. Arditi, and M.T. Abreu. 2004. Beta-defensin-2 expression is regulated by TLR signaling in intestinal epithelial cells. J. Immunol. 173:5398–5405. [DOI] [PubMed] [Google Scholar]
- 23.Ogushi, K., A. Wada, T. Niidome, T. Okuda, R. Llanes, M. Nakayama, Y. Nishi, H. Kurazono, K.D. Smith, A. Aderem, et al. 2004. Gangliosides act as co-receptors for Salmonella enteritidis FliC and promote FliC induction of human beta-defensin-2 expression in Caco-2 cells. J. Biol. Chem. 279:12213–12219. [DOI] [PubMed] [Google Scholar]
- 24.Cash, H.L., C.V. Whitham, C.L. Behrendt, and L.V. Hooper. 2006. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 313:1126–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sonnenburg, J.L., C.T. Chen, and J.I. Gordon. 2006. Genomic and metabolic studies of the impact of probiotics on a model gut symbiont and host. PLoS Biol. 4:e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ganz, T. 1999. Defensins and host defense. Science. 286:420–421. [DOI] [PubMed] [Google Scholar]
- 27.Girardin, S.E., I.G. Boneca, J. Viala, M. Chamaillard, A. Labigne, G. Thomas, D.J. Philpott, and P.J. Sansonetti. 2003. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 278:8869–8872. [DOI] [PubMed] [Google Scholar]
- 28.Park, J.H., Y.G. Kim, C. McDonald, T.D. Kanneganti, M. Hasegawa, M. Body-Malapel, N. Inohara, and G. Nunez. 2007. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J. Immunol. 178:2380–2386. [DOI] [PubMed] [Google Scholar]
- 29.Buchmeier, N.A., and R.D. Schreiber. 1985. Requirement of endogenous interferon-gamma production for resolution of Listeria monocytogenes infection. Proc. Natl. Acad. Sci. USA. 82:7404–7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harty, J.T., and M.J. Bevan. 1995. Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity. 3:109–117. [DOI] [PubMed] [Google Scholar]
- 31.Pfeffer, K., T. Matsuyama, T.M. Kundig, A. Wakeham, K. Kishihara, A. Shahinian, K. Wiegmann, P.S. Ohashi, M. Kronke, and T.W. Mak. 1993. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 73:457–467. [DOI] [PubMed] [Google Scholar]
- 32.Rothe, J., W. Lesslauer, H. Lotscher, Y. Lang, P. Koebel, F. Kontgen, A. Althage, R. Zinkernagel, M. Steinmetz, and H. Bluethmann. 1993. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature. 364:798–802. [DOI] [PubMed] [Google Scholar]
- 33.Cash, H.L., C.V. Whitham, and L.V. Hooper. 2006. Refolding, purification, and characterization of human and murine RegIII proteins expressed in Escherichia coli. Protein Expr. Purif. 48:151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niess, J.H., S. Brand, X. Gu, L. Landsman, S. Jung, B.A. McCormick, J.M. Vyas, M. Boes, H.L. Ploegh, J.G. Fox, et al. 2005. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 307:254–258. [DOI] [PubMed] [Google Scholar]
- 35.Rescigno, M., M. Urbano, B. Valzasina, M. Francolini, G. Rotta, R. Bonasio, F. Granucci, J.P. Kraehenbuhl, and P. Ricciardi-Castagnoli. 2001. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2:361–367. [DOI] [PubMed] [Google Scholar]
- 36.Chieppa, M., M. Rescigno, A.Y. Huang, and R.N. Germain. 2006. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J. Exp. Med. 203:2841–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barnes, P.D., M.A. Bergman, J. Mecsas, and R.R. Isberg. 2006. Yersinia pseudotuberculosis disseminates directly from a replicating bacterial pool in the intestine. J. Exp. Med. 203:1591–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corr, S.C., Y. Li, C.U. Riedel, P.W. O'Toole, C. Hill, and C.G. Gahan. 2007. Bacteriocin production as a mechanism for the antiinfective activity of Lactobacillus salivarius UCC118. Proc. Natl. Acad. Sci. USA. 104:7617–7621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fukata, M., K.S. Michelsen, R. Eri, L.S. Thomas, B. Hu, K. Lukasek, C.C. Nast, J. Lechago, R. Xu, Y. Naiki, et al. 2005. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 288:G1055–G1065. [DOI] [PubMed] [Google Scholar]
- 40.Pull, S.L., J.M. Doherty, J.C. Mills, J.I. Gordon, and T.S. Stappenbeck. 2005. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc. Natl. Acad. Sci. USA. 102:99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown, S.L., T.E. Riehl, M.R. Walker, M.J. Geske, J.M. Doherty, W.F. Stenson, and T.S. Stappenbeck. 2007. Myd88-dependent positioning of Ptgs2-expressing stromal cells maintains colonic epithelial proliferation during injury. J. Clin. Invest. 117:258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}