

Abstract
CD8+ T cell responses directed against multiple pathogen-derived epitopes are characterized by defined immunodominance hierarchy patterns. A possible explanation for this phenomenon is that CD8+ T cells of different specificities compete for access to epitopes on antigen-presenting cells, and that the outcome of this so-called cross-competition reflects the number of induced T cells. In our study using a vaccinia virus infection model, we found that T cell cross-competition is highly relevant during boost vaccination, thereby shaping the immunodominance hierarchy in the recall. We demonstrate that competition was of no importance during priming and was unaffected by the applied route of immunization. It strongly depended on the timing of viral antigen expression in infected APCs, and it was characterized by poor proliferation of T cells recognizing epitopes derived from late viral proteins. To our knowledge, this is the first demonstration of the functional importance of T cell cross-competition during a viral infection. Our findings provide a basis for novel strategies for how boost vaccination to defined antigens can be selectively improved. They give important new insights into the design of more efficient poxviral vectors for immunotherapy.
CD8+ T cells play a pivotal role for the clearance of viruses and other intracellular pathogens. Although the immune system is confronted with a vast variety of pathogen-specific determinants, T cell responses to viral infections have been found to be directed against a rather small number of antigens. Regarding their overall size, T cell responses against different epitopes can be arranged in an immunodominance hierarchy. Fundamental conclusions have been drawn from experiments investigating influenza virus, lymphocytic choriomeningitis virus (LCMV), or Listeria monocytogenes in infection models or by using peptide-pulsed DCs and adoptively transferred TCR transgenic T cells (1), indicating that T cells can compete at the level of APCs. Although the competition of T cells of the same specificity has been clearly demonstrated, competition between T cells of different specificities (cross-competition) is still controversial (2). There are only a few reports in the literature that argue either for (HSV and influenza) or against (LCMV) a relevance of cross-competition during the primary induction of an antiviral immune response (3–5).
Several important features for the development of immunodominance hierarchies of T cells have been identified, as follows: (a) CD8+ T cells of the same specificity compete for access to APCs, and (b) T cell expansion depends on the precursor frequency, (c) is affected by T cell receptor affinity, and (d) can be controlled by APC killing (LCMV) or (e) in the absence of APC killing (influenza) by downmodulation of antigen from the APC or competition for antiapoptotic cytokines (6–8). Importantly, the immunodominance hierarchy of T cells can dramatically change between primary and secondary immune responses. In the case of influenza virus, this has been imputed to differential antigen presentation during primary and secondary immune responses (9).
Until now, most of the work on immunodominance has been done on viruses with relatively small genomes (10–20 kb). Large DNA viruses, like Herpes or Poxviruses (∼200 kb), represent a great challenge for the analysis of immunodominance. However, they also offer the possibility to identify crucial mechanisms of immunodominance because the immune system exceedingly shapes the immune response by trimming it down to reactivity against a few epitopes. We and others recently identified HLA-A2– and H2-Kb/Db–restricted poxvirus determinants, which were derived from a large variety of proteins (10–15). Usage of these epitopes should allow us to identify and analyze the underlying mechanisms of immunodominance in poxviral T cell responses. We used different strains of vaccina virus (VV; modified VV Ankara [MVA], chorioallantois VV Ankara [CVA], and Wyeth) for our infection experiments in C57BL/6 and HLA-A2 transgenic mice (HHD mice). In contrast to the replication-competent VV strains CVA and Wyeth, MVA is unable to replicate in mammalian cells. Therefore, the infection is abortive and is largely synchronized with the advantage to minimize an overlap of the phases of viral gene expression by cells that are infected at different time points during virus spreading. MVA is already used in clinical trials as a recombinant vector, and it is also considered as a next generation poxvirus vaccine. An interesting hallmark of poxviruses is the cascade-like course of antigen expression. There are three distinct phases of viral gene expression driven by distinct promoters with early, intermediate, and late activity. Previous work suggested that the immunogenicity of recombinant proteins produced by VV is influenced by the promoter driving the respective genes (16). Therefore, we presumed a specific impact of viral gene expression on the immunodominance hierarchy of early and late viral proteins.
Our experiments show that the induction and strength of primary CD8+ T cell responses against various VV-specific epitopes in a naive host is largely independent from simultaneous priming of T cells specific for other antigenic determinants delivered by the virus. However, during boost vaccinations, T cell cross-competition seems to be a major regulator of the expansion of virus-specific T cells. In particular, T cells recognizing determinants derived from late viral proteins had a clear disadvantage to proliferate during secondary responses.
However, the efficiency of boost vaccinations was strongly enhanced by using promoters that are active early during the viral life cycle. Our work has important implications for the use and the future design of viral vectors for immunotherapy.
RESULTS
Priming of T cells is independent of T cell competition
To investigate the role of T cell cross-competition on shaping the T cell response to VV, we compared the immune responses against several replication-competent and -deficient strains, as well as recombinant MVA viruses. MVA constructs either expressed or lacked antigens bearing strongly immunogenic epitopes compared with WT virus (MVA WT). We hypothesized that the quantity of induced T cells would be influenced by additional introduction or removal of immunodominant epitopes if T cell cross-competition was functionally important during priming. We compared the CD8+ T cell responses directed against a variety of virus-specific epitopes. Fig. 1 shows the relative numbers of T cells producing IFN-γ after in vitro restimulation with the respective peptides analyzed by intracellular cytokine staining of freshly isolated splenocytes. 8 d after infection of HLA-A2 transgenic (HHD) mice with MVA WT, T cells recognizing epitopes from late gene products (A6L6, H3L184, and I1L211) were dominating the response (Fig. 1 B). T cells recognizing epitopes derived from early proteins were subdominant (B22R79) or close to detection limit (<0.1% of CD8+: C7L74 and D12L251). Recombinant viruses additionally expressing human tyrosinase (MVA hTyr) or human Her-2/neu (MVA-Her-2/neu) induced a strong Tyr369- or moderate Her-2/neu435–specific response. However, the vector-specific response remained unchanged compared with MVA WT. To exclude a mouse strain–specific effect, we also analyzed C57BL/6 mice. In these mice, the cellular immune response against MVA WT is highly dominated by B8R20-specific T cells, which recognize a determinant derived from an early gene product, followed by A3L270- (late gene product), K3L6- (early gene product), and A8R189-specific (early gene product) T cells. Again, a recombinant virus expressing OVA (MVA P7.5 OVA) induced a strong additional OVA257-specific response in these mice without altering the frequencies of vector-specific T cells (Fig. 1 D). Tetramer staining showed similar results (unpublished data). We further constructed a mutant virus (MVA ΔB8R) with a deleted B8R gene (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20070849/DC1) to investigate if the induction of B8R20-specific T cells possibly interfered with the primary induction of T cells specific for other VV epitopes. In vitro experiments confirmed similar growth kinetics for MVA ΔB8R and MVA WT (Fig. S2), excluding an attenuation of the deletion variant. MVA ΔB8R did not induce a B8R20-specific T cell response, and other vector-specific T cells did not compensate for that loss (Fig. 1 D). This was also seen at low doses (105 infectious units [IU]; unpublished data). From these experiments, we conclude that the primary induction of vector-specific T cells using MVA is not influenced by the priming of other vector-induced T cells. The time of expression (early vs. late) of viral antigens did not correlate with their position within the immunodominance hierarchy.
Figure 1.

T cell cross-competition is absent during priming with MVA. (A) The applied gating strategy for analysis of viable (EMA-negative) CD8+- and CD62L-low IFN-γ–producing T cells (corresponding number is encircled). Numbers in the top left corner indicate the order of applied gates. (B) HHD mice were vaccinated i.p. (107 IU) with rec MVA or MVA WT. (C) Table shows early and late viral antigens and MHC-I restriction of the respective epitopes. (D) C57BL/6 mice were vaccinated i.p. (108 IU) with MVA WT, MVA P7.5 OVA, or MVA deletion mutant (MVA ΔB8R). 8 d after vaccination, tyrosinase-, her-2/neu–, OVA-, and vector-specific CD8+ T cell responses were analyzed by intracellular cytokine staining of splenocytes after a brief incubation with the A*0201-restricted tyrosinase peptide Tyr369, Her-2/neu435, MVA-specific peptides A6L6, B22R79, C7L74, D12L251, I1L211, H3L184, or H2b restricted OVA257 (SIINFEKL), MVA-specific peptides A3L270, A8R189, B8R20, K3L6, or an irrelevant peptide. Filled arrows indicate additional responses; open arrows represent missing responses compared with MVA WT. Results are representative of three independent experiments. n = 4.
Immunodominance hierarchy after secondary immunization correlates with viral gene expression
Next, we wanted to analyze whether the timing of antigen expression (early vs. late viral genes) was relevant for the reactivation of memory T cells. When boosting HHD mice with MVA WT, memory T cells recognizing epitopes derived from late gene products such as H3L184 and I1L211 did not expand (Fig. 2 A). A6L6-specific T cells were not amplified compared with the primary response. Interestingly, early gene product B22R79-specific T cells expanded vigorously during the secondary immunization and became the dominant T cell population among the tested epitope specificities. This severe switch is surprising because it contradicts the prediction that T cells dominating the primary response should also dominate the secondary response if memory precursor frequencies were merely the critical factor for the immunodominance hierarchy of recall responses. In C57BL/6 mice, the primary response is dominated by T cells specific for B8R20, which is derived from an early gene product. If early antigen expression supports recall expansion, boosting of C57BL/6 mice with MVA WT should still support the recall expansion of B8R20-specific T cells. Indeed, boosting with MVA WT led to a strong expansion of B8R20-specific T cells (Fig. 2 B). From these experiments, we hypothesized (a) that T cells specific for early viral proteins might be able to suppress the expansion of other virus-specific T cells, and (b) that T cells specific for late viral proteins should have a disadvantage to proliferate and expand during recall responses.
Figure 2.

Immunodominance hierarchy is changed after secondary immunization in HHD mice. (A) HHD or C57BL/6 mice were analyzed 8 d after prime (shaded bar) or boosted 35 d after prime and analyzed 6 d later (open bar). Mice were vaccinated i.p. with 108 IU. Only in HHD mice did B22R79-specific T cells increase in frequency; A6L6-, H3L184-, and I1L211-specific T cells do not proliferate during secondary immunizations. (B) In C57BL/6 mice, B8R20-specific T cells dominate the primary and secondary response. (C) Replication-competent virus CVA shows a shift similar to MVA in the immunodominance hierarchy. Results are representative of three independent experiments. n = 4.
Expansion of T cells specific for late gene products is broadly impaired
It has been shown that the route of vaccination can considerably change the outcome of immune responses toward vaccines and influences the immunodominance hierarchy (17). Therefore, we decided to include two clinically relevant application routes, intradermal and i.m. prime and boost immunizations, in our experiments. We found that irrespective of the administration route, the immunodominance hierarchy was conserved during priming, but again, T cells specific for late viral antigens had a disadvantage to expand during recall responses (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20070849/DC1). Nevertheless, the T cell frequencies varied between different vaccination routes. The strongest responses could be measured in the spleen after i.p. priming, followed by i.m. and intradermal immunizations.
Next, we wanted to analyze whether the poor proliferation of T cells specific for late viral antigens during recall responses was caused by the lack of replication of MVA in vivo. Comparative analysis of the immune response 8 d after prime using MVA or the replication-competent parental strain CVA revealed a similar immunodominance hierarchy for both viruses (Fig. 2, A and C). Also, in CVA-immunized mice, A6L6 was immunodominant, followed by I1L211- and H3L184-specific responses. Interestingly, we could detect a substantial response against B22R79 8 d after immunization with CVA. 6 d after boosting, we again found suppression of T cell responses against late viral epitopes using this replication-competent virus (Fig. 2 C). Similar results were obtained in experiments using the replication-competent VV strain Wyeth (unpublished data). To exclude that the lack of recall proliferation was caused by a general malfunction of T cells specific for epitopes derived from late gene products, we boosted MVA-primed mice with the respective peptides. As seen in Fig. S4 (available at http://www.jem.org/cgi/content/full/jem.20070849/DC1), H3L184-specific T cells could be readily amplified in vivo when mice were revaccinated with H3L184 peptide. Also, B22R79-specific T cells were expanded, when boosting with the respective peptide, yet to a lower extent. The frequency of C7L74-specific T cells did not exceed that of peptide-primed mice. In that respect, the expansion of MVA-primed T cells by single peptide revaccination resulted in a pattern that resembles the immunodominance hierarchy induced by MVA in the primary response (Fig. 1 B). This indicates that VV-specific T cells, which did not proliferate in recall responses to the virus, possibly lacked antigen-specific stimulation. Impairment of viral antigen processing or presentation might account for reduced or inhibited proliferation of T cells recognizing late gene product–derived epitopes during VV recall responses. To further analyze this issue, we decided to focus on the H3L late gene product.
Antigen presentation of late viral proteins is substantially delayed
As mentioned earlier, the viral life cycle of VV can be divided into three distinct phases: early, intermediate, and late viral gene expression. The interval between these phases is only about 1 h on the transcriptional level (18). To test if differences in viral transcription led to a relevant delay of the presentation of viral antigens, we established several T cell lines specific for VV early or late protein determinants. T cells recognizing early determinant B22R79 were able to efficiently lyse infected target cells 3 h after infection (Fig. 3 A). In contrast, T cells with specificity for late viral proteins, such as H3L184-specific T cells were not able to recognize infected cells before 15 h after infection. A6L6- and I1L211-specific T cell lines could not lyse infected target cells even at 15 h after infection (unpublished data). To gain more detailed information, we performed intracellular cytokine staining of T cells after coincubation with infected stimulator cells and performed detailed kinetic analyses. As shown in Fig. 3 C, at 2 h after infection the majority of B22R79-specific T cells (95%) were already stimulated. Again, A6L6- and I1L211-specific T cells did not get activated by infected cells, even at 8 h after infection, despite similar functional affinity for peptide-pulsed target cells (Fig. 3 B). Coincubation of T cells with stimulators that had been infected for a longer period (up to 14 h) did not lead to higher stimulation rates (unpublished data). H3L184-specific T cells were activated as early as 6 h after infection (20%), and ∼40% of the T cells produced IFN-γ at 8 h after infection. Notably, this T cell line has the highest functional affinity (Fig. 3 B). We concluded from these data that presentation of determinants derived from late viral gene products to CD8+ T cells is generally reduced and delayed for several hours compared with early viral gene products.
Figure 3.

Antigen presentation of late viral proteins is delayed. Specific 51Cr release of MVA WT–infected (MOI 10) A375 target cells is shown (E/T ratio = 10:1). (A) H3L184-specific T cells do not substantially lyse infected target cells before 15 h after infection. (B) Peptide titration shows similar affinity of T cell lines, except for H3L184-specific T cells showing a higher affinity. (C) Infected LCL (MOI 10) was used for a kinetic analysis to stimulate IFN-γ production in several V V-specific T cell lines. MVA WT–infected cells already stimulate B22R79-specific (early gene) T cells 2 h after infection. H3L184-specific (late gene) T cells get stimulated 6 h after infection. A6L6- and I1L211-specific T cells (both late) or control cell line are not stimulated by MVA WT–infected cells. (D) LCL infected by recombinant virus MVA ΔH3L P7.5 H3L expressing the H3L gene under an early/late promoter rapidly induce IFN-γ production in H3L184-specific T cells. Data are representative of three independent experiments.
Timing of viral antigen expression regulates T cell expansion
Next, we wanted to analyze whether the delayed antigen presentation of late viral proteins is related to the impaired proliferation of T cells specific for these proteins during boost vaccinations. We constructed a recombinant MVA in which the natural H3L gene has been knocked out (Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20070849/DC1) and reinserted into the viral genome under the control of a promoter (P7.5) with early/late activity (MVA ΔH3L P7.5 H3L). Northern blot analysis revealed that both viruses MVA WT and MVA ΔH3L P7.5 H3L express the H3L gene at comparable levels; but, in contrast to MVA WT, MVA ΔH3L P7.5 H3L already produces the H3 protein early during the viral life cycle (Fig. S6). In vitro, MVA ΔH3L P7.5 H3L–infected target cells induced IFN-γ in H3L184-specific T cells very early after infection (Fig. 3 D). Importantly, H3L184-specific T cells were readily expanded in the secondary response when boosting with MVA ΔH3L P7.5 H3L (Fig. 4 A). Thereby, the absolute numbers of H3L184-specific T cells reached similar levels as B22R79-specific T cells (Fig. 4 B). Importantly, the responses against other epitopes (A6L6, I1L211, and B22R79) were comparable for both viruses (Fig. 4 and not depicted).
Figure 4.
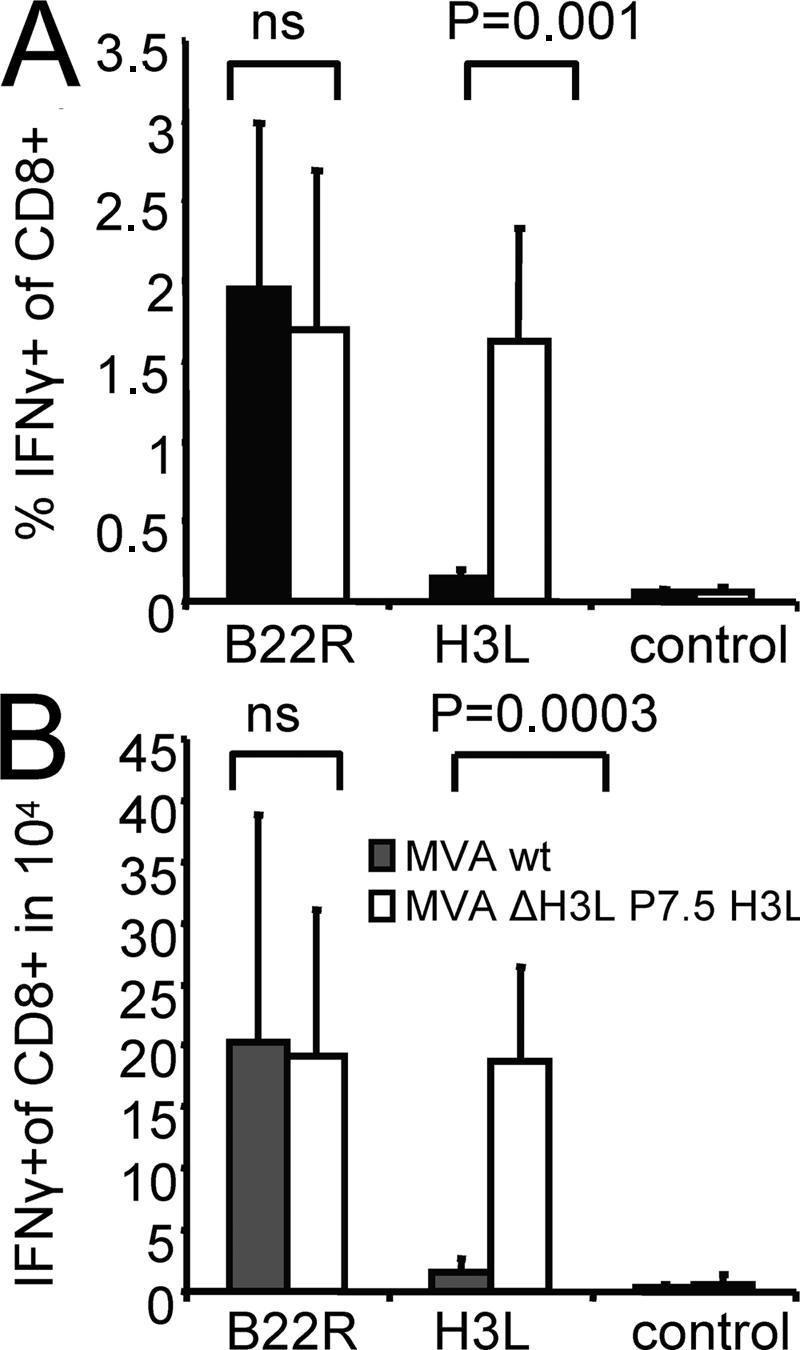
MVA ΔH3L P7.5 H3L amplifies H3L-specific T cell responses. HHD mice were primed i.p. with 108 IU of MVA WT and boosted with MVA WT (shaded bar) or MVA ΔH3L P7.5 H3L (open bar). MVA ΔH3L P7.5 H3L induces a significant expansion of H3L184-specific IFN-γ–producing T cells compared with MVA WT, without altering the T cell response against B22R79. Relative (A) and absolute numbers (B) of IFN-γ–producing, H3L184-specific T cells measured in the spleen compared with MVA WT. Data are representative of three independent experiments. n = 4.
Cross-competition of T cells regulates T cell expansion
Although epitopes derived from late viral proteins were presented with a strong delay on the surface of infected cells, one would expect that T cells recognizing these epitopes would eventually get stimulated and could proliferate. In vivo, however, H3L184- and I1L211-specific T cells were not amplified during recall responses (Fig. 2). Based on this observation, we speculated that in addition to the delayed presentation of viral late proteins, T cells with different specificities compete for infected APCs. In particular, T cells recognizing VV-epitopes from early proteins should have an advantage to cross-compete compared with T cells recognizing VV-epitopes from late proteins. Because peptide immunizations in HHD mice are very efficient (19), we tried to experimentally address this question by priming naive mice simultaneously with pairs of peptides. Cohorts of mice were immunized with H3L184 together with either B22R79 or C7L74, which are both derived from early proteins, or a control peptide derived from human tyrosinase (Tyr369; Fig. 5). 35 d after prime, T cell frequencies in the peripheral blood were determined by tetramer analysis. Thereafter, mice were boosted either with MVA WT or a recombinant virus expressing human tyrosinase under control of an early/late promoter (MVA hTyr). 5 d later, we again measured the specific T cell frequencies in the blood by tetramer staining. To compensate for variations of peptide-induced T cell frequencies within individual mice, we calculated the ratio of the frequencies before and after boosting with MVA vaccines as a relative measure of T cell proliferation. When H3L184-specific T cells were induced together with C7L74- or B22R79-specific T cells, they did not proliferate after boosting with MVA WT (Fig. 5). Importantly, when H3L184-specific T cells were accompanied by irrelevant Tyr369-specific T cells, they were readily amplified. However, when we boosted peptide-specific CD8+ T cells with MVA hTyr expressing human tyrosinase under the control of an early/late promoter, the H3L184-specific response was again suppressed. These data demonstrate that the proliferation of H3L184-specific T cells strongly depends on the ability of participating T cells of other specificity to cross-compete.
Figure 5.

Cross-competition between T cells recognizing early and late determinants. HHD mice were simultaneously primed with pairs of peptides. 35 d after prime, the frequencies of respective tetramer-specific CD8+ T cells in the peripheral blood were determined. Mice were boosted i.p. with 108 IU of MVA WT or MVA hTyr. 5 d later, tetramer-specific CD8+ T cells were again measured in the blood. Numbers within dot plots indicate frequencies of H3L184-specific T cells before (left column) and after boosting (right column). Index shows relative increase of tetramer-specific T cells after boosting. Black line indicates that frequencies before and after boost are equal (index = 1). In contrast to control T cells (Tyr369), the presence of B22R79- or C7L74-specific T cells (both early genes) significantly suppresses the expansion of H3L184- (late gene) specific T cells after boosting with MVA WT. However, when MVA hTyr is used the expansion of H3L184-specific T cells is again suppressed, but Tyr369-specific T cells are readily amplified. Data are representative of three independent experiments. n = 5.
T cells cross-compete early after priming
Because we observed cross-competition between T cells only during boost vaccinations, we hypothesized that this phenomenon is linked to the presence of T cells with fully developed effector function. Naive T cells gain effector function early (∼3 d) during primary responses (20). Furthermore, by using L. monocytogenes, it has been previously demonstrated that a second infection during the priming phase can induce typical recall responses (21). We wanted to confirm our results by reimmunizing during the primary MVA response, and thereby define a time point after priming when T cells are able to execute cross-competition. For a detailed kinetic analysis, mice were primed and subsequently revaccinated once from 1 to 6 d after prime (Fig. 6 A). 6 d after revaccination, mice were killed, and the epitope dominance pattern was determined and compared with mice that had received MVA only once. Interestingly, when mice were reimmunized 1 or 2 d after prime, the epitope pattern did not change. When reimmunizing at 1 d after prime, A6L6-, I1L211-, and H3L184-specific CD8+ T cells even increased (Fig. 6 B and Fig. S7 B, available at http://www.jem.org/cgi/content/full/jem.20070849/DC1, for all epitopes). Reimmunization on day 2 did not alter the frequencies or absolute numbers of the different T cell populations. However, when boosting on day 3 or later, A6L6-, I1L211-, and H3L184-specific T cell frequencies were reduced, whereas B22R79-specific T cells expanded. From day 4 or later, B22R79-specific T cells were significantly amplified, as seen in secondary responses after 35 d. To confirm that T cells were able to cross-compete early after primary infection, we revaccinated MVA WT–primed mice with MVA WT or MVA ΔH3L P7.5 H3L. Indeed, similar to boosting at day 35 (Fig. 4), B22R79- and H3L184-specific T cells expanded when mice were boosted early (day 5) with MVA ΔH3L P7.5 H3L (Fig. 6 D and Fig. S7 D).
Figure 6.

Competition between T cells occurs early after priming. HHD mice were primed with MVA WT and boosted with the same virus (A and B) early at indicated days after priming, or at day 5 with MVA ΔH3L P7.5 H3L (C and D). (A) Schematic of prime/boost regimen. (B) Intracellular cytokine staining of splenocytes, comparing the VV-specific CD8+ T cell responses after priming (shaded bar) or 6 d after boosting (open bar). B22R79-specific T cells are significantly increased when boosting 4 d after prime or later. (C) Schematic of prime/boost regimen. (D) H3L184-specific T cell responses can be significantly amplified when using MVA ΔH3L P7.5 H3L (open bar) as compared with MVA WT (closed bar). Data are summary of three independent experiments. n = 6.
Cross-competition between T cells specific for early viral determinants
Thus far, we have shown that T cell cross-competition between T cells recognizing early or late viral products is functionally important for shaping the immunodominance hierarchy during recall responses. Next, we wanted to analyze whether T cells recognizing early epitopes also cross-compete with each other. In C57BL/6 mice, the B8R20-specific T cell response dominates both the primary and secondary response against VV. To analyze if B8R20-specific T cells were cross-competing with T cells of other specificities and impaired their expansion, we used MVA ΔB8R for prime/boost immunizations (Fig. S1). As anticipated, despite the absence of B8R20-specific T cells, the expansion of A3L270-specific T cells was suppressed in the recall response, presumably by cross-competing K3L6- and A8R189-specific T cells, which rapidly expanded. Again, we found a switch in the immunodominance hierarchy favoring the proliferation of T cells recognizing epitopes derived from early gene products (Fig. 7 A). In contrast, in a prime/boost regimen using MVA WT, K3L6- and A8R189-specific T cells were not amplified to the same level compared with MVA ΔB8R. To further characterize T cell cross-competition between T cells recognizing early viral products, we performed short interval prime/boost immunizations. We defined the K3L6- and A8R189-specific T cell response induced by MVA ΔB8R as the baseline and considered A3L270-specific T cell response as an internal control. We then compared the K3L6- and A8R189-specific T cell response induced by recombinant viruses, which contain an additional moderate (MVA OVA ΔB8R containing OVA257) or highly (MVA WT containing B8R20) immunogenic epitope or both (MVA OVA containing OVA257 and B8R20). As shown in Fig. 7 B, this stepwise additional induction of cross-competing T cells specific for early viral epitopes (OVA257/B8R20) leads to a gradually increased suppression of T cells specific for other early viral epitopes (K3L6/A8R189). Importantly, T cells specific for late viral epitopes (A3L270) remain fully suppressed. Hence, we conclude that cross-competition is also functional among T cells specific for early viral proteins during recall responses.
Figure 7.

Early virus-specific T cells suppress the expansion of early and late virus-specific T cells. (A) Intracellular cytokine staining of splenocytes comparing MVA ΔB8R (open bar) with MVA WT (shaded bar) 6 d after homologous boost (day 35). Immunodominance hierarchy is changing in MVA ΔB8R–immunized mice favoring the expansion of K3L6- and A8R189- (both early genes) specific T cells over A3L270- (late gene) specific T cells in the absence of B8R20-specific T cells. (B) Intracellular cytokine staining of splenocytes comparing MVA ΔB8R with MVA OVA P7.5 ΔB8R, MVA WT, and MVA OVA P7.5 6 d after homologous boost (day 5). The expansion of K3L6- and A8R189-specific (both early genes) T cells is successively suppressed by gradual appearance of cross-competing B8R20- and OVA257-specific (both early genes) T cells, wheras A3L270-specific (late gene) T cells remain fully suppressed. Data are representative of two independent experiments. n = 5.
Timing of viral antigen expression is crucial for vaccination strategies
To investigate the influence of viral antigen expression on T cell cross-competition more closely, we constructed two viruses expressing the model antigen OVA driven by a promoter with exclusive early (MVA K1L OVA) or late (MVA P11 OVA) activity. As shown by Western blot analysis, infection with MVA K1L OVA led to a rapid production of OVA, even in the presence of Ara-C, which is an effective inhibitor of late viral gene expression. In contrast, the synthesis of OVA in MVA P11 OVA–infected cells is initiated later during viral infection and abrogated upon Ara-C treatment (Fig. 8 A). After 24 h of infection, the overall amount of synthesized OVA is higher in MVA P11 OVA–infected cells (Fig. S8, available at http://www.jem.org/cgi/content/full/jem.20070849/DC1). Interestingly, when measuring the processing and presentation of OVA by specific staining of OVA257-loaded MHC-I complexes on RMA cells, we found a clear correlation with the time of expression, which depends on the promoter activity of the respective constructs (Fig. 8 B). 2 h after infection, we could already detect a substantial amount of H2-Kb OVA257 complexes on the surface of MVA K1L OVA–infected cells with a maximum at ∼12 h after infection. In contrast, we could not detect minimal amounts of OVA257 on the surface of MVA P11 OVA–infected RMA cells before 12 h after infection. In vivo, however, we found a comparable OVA-specific T cell response in the spleen of MVA P11 OVA– and MVA K1L OVA–immunized mice (Fig. 8 C). This implicates that during priming neither the amount of presented peptide/MHC-I complexes nor the time after infection at which viral antigen expression occurs determines the outcome of the T cell response. However, during secondary immunizations only MVA K1L OVA–immunized mice readily amplified OVA257-specific T cells, whereas MVA P11 OVA failed to expand OVA257-specific T cells (Fig. 8 C). Importantly, the T cell responses against virus-specific antigens were comparable for both viruses (Fig. S8). These data suggest that the time of viral antigen expression is of minor importance for T cell priming, but highly relevant for shaping the immunodominance hierarchy in secondary immunizations. This observation is independent from the route of vaccination or the ability of VV to replicate. The reduced proliferation of T cells specific for late viral proteins is founded on a delayed and impaired presentation of viral epitopes derived from these proteins. Additionally, these T cells are further inhibited to expand by other cross-competing T cells specific for early viral proteins. This has important consequences concerning the protection against lethal pathogens after vaccination. Only MVA constructs expressing OVA early during the viral life cycle were able to induce a protective immunity in mice when challenged with lethal doses of L. monocytogenes-OVA (Fig. 8 D). Mice immunized with MVA P11 OVA could reduce the bacterial load only slightly better than mice immunized with MVA WT.
Figure 8.

Timing of viral antigen expression is essential during secondary responses. (A) Western blot analysis of Hela cells infected with MVA K1L OVA or MVA P11 OVA in the presence (+) or absence (-) of Ara-C, which is a specific inhibitor of late viral gene expression. Cell lysates were harvested at 0, 5, or 12 h after infection. (B) For relative quantification of SIINFEKL-Kb complexes on infected cells, 25-D1.16 antibody was used. Mean fluorescence intensity of positive cells is shown. (C) Intracellular cytokine staining of splenocytes comparing MVA K1L OVA with MVA P11 OVA 8 d (shaded bar) after prime or 6 d (open bar) after boost, which was performed 35 d after priming with MVA P7.5 OVA, to allow for comparable memory T cell frequencies at the time of the second immunization. Mice were primed and boosted i.p. 5 d later with different MVA constructs. (D) 6 d after homologous boost, mice were challenged with different doses of L. monocytogenes-OVA (2 × 106 = shaded bar; 5 × 105 = open bar) i.v. and numbers of viable L. monocytogenes in spleen were determined 2 d later. nd = not detectable.
DISCUSSION
In our study, we identified T cell cross-competition to be responsible for the dramatic switch in the epitope dominance patterns of VV-specific CD8+ T cells by comparing primary with secondary infections. Changes in immunodominance hierarchies of T cells have also been reported for other pathogens, such as influenza, herpes viruses, or LCMV (22–24). In the LCMV infection model, these changes have been attributed primarily to T cell exhaustion. In contrast, upon influenza virus infection, the changing epitope pattern has been attributed to differential antigen expression, reflecting the capacity of memory T cells to respond to nondendritic cells (9). Yewdell's group, which works extensively on immunodominance in the influenza model, recently challenged this interpretation (8), and came to the conclusion that cross-presentation could also account for changes in dominance patterns. Furthermore, they demonstrated that immunodominance hierarchies were independent of perforin or Fas-mediated lysis in the secondary response, and are therefore not connected to APC killing or CD8+ T cell cross-competition. La Gruta et al. elegantly shifted epitopes within the same viral infection context to study immunodominance (25). They found that the epitope hierarchies were a result of antigen dose and the size of the preexisting T cell pool. Competitive interactions, as demonstrated in our study, seemed to have only little impact in their model. Removal or addition of immunogenic epitopes in influenza virus had no effect during priming, but led to compensation or suppression of other T cells during boosting, respectively (26, 27). The authors attributed their findings to effects of antibodies or limiting amounts of antigen. In our study, however, we could link the immunodominance pattern arising during secondary vaccinations to the presence of primed T cells. A reason for potential differences between VV and other infectious agents could be the high-level gene expression promoted by VV, leading to high antigen amounts. Hence, competition mediated by limited antigenic resources seems to be unlikely. Consistent with that view, a mathematical model of T cell competition recently predicted that a large set of different epitopes, such as that found for VV infection, should decrease T cell competition (28). However, under these conditions, high affinity of T cells and high expression levels of single epitopes would increase the chances for T cell competition. Hence, when immunizing with VV, the APC itself or resources of the APC other than the peptide–MHC complexes may become the limiting factor for competing T cells. Besides costimulatory or adhesion molecules, this could involve cytokines or access to APC. Another possibility is that memory T cells could be able to silence APCs after a certain number of interactions. Further work will be necessary to elucidate the cellular and molecular basis of T cell cross-competition.
Surprisingly, T cell cross-competition has been rarely documented so far. Marrack's group demonstrated T cell cross-competition by using peptide-pulsed DC and transferred TCR transgenic T cells (29). They found that the degree of competition depended on the affinity of responding T cells, but was much less efficient than competition among T cells with the same epitope specificity (30). In the LCMV model, T cell cross-competition is believed to be functionally unimportant (5). However, in this particular study, cross-competition was only analyzed during the priming phase. Our work demonstrates that cross-competition toward VV is active during the secondary response and depends on immediate T cell effector function (presence of primed T cells). This notion is supported by Kedl et al., who succeeded in demonstrating T cell cross-competition by using primed T cells (29). Additionally, we found that T cells were able to cross-compete 3 d after priming. This correlates with the development of cytotoxicity of naive T cells after exposure to APCs, which was detectable after 48 h and was fully present after 72 h (20). In a bacterial model, Wong and Pamer showed that CTL activity developed within 72 h, and that these T cells probably eliminated APCs, thereby regulating antigen presentation and, consequently, T cell priming and expansion (31). Interestingly, Belz et al. recently showed that during secondary influenza infections, T cells terminate the antigen presentation in a perforin-dependent manner (32). However, in the influenza model, perforin, granzyme B, or FAS–FASL interaction did not seem to influence the immunodominance hierarchy (7, 8). Other models showed that early onset of IFN-γ production by CD8+ T cells correlated with the immunodominant responses (33). Further work will be necessary to define which features of effector T cells are crucial to execute cross-competition.
The primary response to the replication-competent VV strain CVA was similar compared with MVA, apart from a substantial response against B22R79. Interestingly, we could also induce and detect an increased B22R79-specific response by performing short-interval prime/boost experiments with MVA. This, in a way, imitates replication caused by repeated infections during the priming phase. In that respect, the primary response to a replication-competent VV could be interpreted as a dynamic process of priming and boosting of T cells. This is in contrast to persistent chronic viral infections, where T cell exhaustion is a factor for changing immunodominance hierarchies. As recently shown, using LCMV as a model for a chronic viral infection, CD8+ T cells undergo extensive peptide-dependent division independent of IL-7 or -15. This suggests that these CD8+ T cells go through a fundamentally different pattern of differentiation compared with memory CD8+ T cells that develop after an acute infection (34). In addition to A*0201 transgenic mice, the suppression of secondary T cell responses against late viral epitopes by T cells recognizing early viral epitopes could also be confirmed in C57BL/6 mice, and seems to be a characteristic feature of VV infection. A similar connection between immunodominance and the kinetics of viral protein expression has been observed using LCMV (24, 35). Indeed, in an elegant model using hemisplenectomized mice and analysis of TCR-β motifs, Bousso et al. found that the timing of recruitment of individual T cell clones contributes more to the immune responses than their precursor frequency (36).
In contrast to T cells specific for H3L184 or I1L211, which do not proliferate during secondary responses, the expansion of T cells specific for A6L6 is not fully suppressed when the boost is performed late in the memory phase (day 35). This might be explained by comparatively higher T cell numbers against A6L6, which cannot be controlled as easily by competing T cells recognizing early epitopes. After short-interval prime/boost experiments (day 5), the A6L6-specific T cell response is fully suppressed, possibly because during priming, the T cell size is increased compared with the memory phase (day 35), and therefore competition is enhanced. Nevertheless, we assume that the amount of B22R79-specific T cells induced after priming is probably too low to completely outcompete A6L6-, H3L184-, and I1L211-specific T cells. Additionally, ∼2% B22R79-specific T cells (Fig. 2) cannot fully account for the total VV-specific response after secondary immunization, which is ∼30–40% of all CD8+ T cells (11). Therefore, it is intriguing to speculate that one or more yet unidentified HLA-A*0201-restricted epitopes might exist.
It can be expected that T cells recognizing epitopes derived from early viral proteins confer better protection against infection. Because VV replication is completed at ∼8 h after infection, T cells specific for late viral proteins are likely to be activated too late to confer any protection because of delayed antigen presentation in infected cells. In this context, T cell cross-competition, and the subsequent preferential expansion of T cells specific for early proteins, reflects the host's ambition to rapidly clear viral infections. We speculate that this finding might also apply to other large DNA viruses, such as herpes viruses. Interestingly, a recent study analyzing T cells in CMV-infected humans described that the number of immediate early protein 1–specific (IE-1), and not pp65-specific (a late viral protein), T cells correlated with protection from disease (37).
In summary, we have demonstrated that the expansion of virus-specific CD8+ T cells was regulated by T cell cross-competition favoring T cells that are able to rapidly detect infected cells. Therefore, the outcome of this competition was heavily influenced by the timing of antigen expression, but independent of the route of vaccination or the ability of a virus to replicate. In immunotherapy using recombinant viral vectors, a successful expansion of the desired T cell response can be strongly impaired by cross-competing, vector-specific T cells. We show that this impairment can be compensated by expressing target antigens early during the viral life cycle, allowing antigen-specific T cells to successfully cross-compete. Thereby, we are able to improve the T cell responses against target antigens and simultaneously decrease the vector-specific response. The identification of other factors determining the outcome of T cell cross-competition and, ultimately, the elucidation of the molecular and cellular mechanism behind T cell cross-competition will allow for designing improved vaccines.
MATERIALS AND METHODS
Mice and vaccination.
HLA-A*0201-transgenic, H2Db −/−, β2m −/−, HHD mice (38) or C57BL/6 mice were derived from in-house breeding under specific pathogen-free conditions following institutional guidelines. Only female mice between 8 and 12 wk of age were used. Mice were vaccinated with 107 or 108 IU MVA. For peptide vaccination, mice were immunized s.c. with 0.1 mg peptide and 10 ng of synthetic CpG1668. Mice were killed on the indicated days after vaccination, and spleens were harvested to be analyzed by ICS.
Cell lines.
RMA cells were provided by F.A. Lemonnier (Institute Pasteur, Paris, France). Lymphoblastoid B cell line RL-LCL (HLA-A*0201 positive) was established in our laboratory, A375 human melanoma cells (CRL-1619) were purchased from American Type Culture Collection. Cell lines were cultured with RPMI 1640 supplemented with 10% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Viruses.
The VV strains New York City Board of Health (Wyeth) and Western Reserve (WR) were provided by B. Moss (National Institutes of Health, Bethesda, MD). CVA at second passage on chicken embryo fibroblasts (CEFs) or MVA (cloned isolate F6) at 582nd passage on CEF were used for this study. Replication-competent VV and MVA were routinely propagated and titered following standard methodology (39).
Quantification of antigen-specific CD8+ T cell responses.
Splenocytes from vaccinated HHD-mice were stimulated with either the HLA-A*0201 restricted human tyrosinase peptide Tyr369, Her-2/neu435, the vaccinia-specific peptides derived from H3L184, A6L6, I1L211, D12L251, C7L74, B22R79 (13), or a control peptide (Flu M158) for 5 h. C57BL/6 mice were stimulated with either H-2Kb– or H-2Db–restricted VV-specific peptides derived from A3L270, A8R189, B8R20, K3L6, OVA257 (13), or a control peptide (β-Gal96) for 5 h. 1 mg/ml Brefeldin A (Sigma-Aldrich) was present throughout stimulation. Cells were live/dead-stained with ethidium monoazide bromide (Invitrogen) and blocked with anti-CD16/CD32-Fc-Block (BD Biosciences). Surface markers were stained with APC-conjugated anti-CD8α and PE-conjugated anti-CD62L (Caltag Laboratories). Intracellular cytokine staining for IFN-γ production was performed with FITC anti–IFN-γ (XMG1.2) using the Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer's recommendations. Data were acquired by FACS analysis on a FACSCanto (BD Biosciences) and analyzed with FlowJo software (Tree Star, Inc.).
Northern blot analysis.
Cells were infected with MVA WT or MVA ΔH3L P7.5 H3L at multiplicity of infection (MOI) 10. Total RNA was isolated with guanidinium hydrochloride and ultracentrifugation over a CsCl cushion and separated by electrophoresis in 1% agarose glyoxal gels. Subsequently, RNA was transferred onto positively charged nylon membranes (Zeta-Probe; Bio-Rad Laboratories). Riboprobe for detection of MVA-encoded mRNA 093L (H3L) was obtained by excision of H3L DNA from the transfer plasmid (pIII-ΔHR-P7.5) using PmeI and BamH1 restriction enzymes. In vitro RNA labeling, hybridization, and signal detection were performed according to the manufacturer's instructions (Rediprime II Random Prime Labeling System; GE Healthcare), applying 68°C for hybridization and high-stringency wash in 1× SSC containing 0.1% SDS buffer.
Western blot analysis.
Expression of OVA was confirmed by Western blot analysis of cell lysates from infected cells. For some experiments, infections were performed in the presence of cytosine β-D-arabinofuranoside (Ara-C; Sigma-Aldrich) at 40 μg/ml to restrict the VV-specific gene expression to the early phase of the viral replication cycle. Proteins were resolved by electrophoresis on a SDS 10% polyacrylamide gel and electroblotted onto nitrocellulose for 20 min in a buffer containing 48 mM Tris, 39 mM glycine, 0.037% SDS, and 20% ethanol, pH 8.3. The blots were blocked overnight on ice in a tris-buffered saline blocking buffer containing 1% bovine serum albumin, 0.1% Tween-20, and 0.02% NaN3, and incubated for 1 h at room temperature with an OVA-specific antibody (rabbit polyclonal to OVA; Abcam) diluted 10,000-fold in blocking buffer. As a control, blots were incubated at similar dilutions with mouse anti–β-actin antibody (AC-15; Sigma-Aldrich). Blots were washed with 0.1% Tween-20 in tris-buffered saline, and incubated for 1 h at RT with POD-conjugated goat polyclonal anti–rabbit IgG (Dianova GmbH) diluted 15,000-fold, washed, incubated with chemiluminescent substrate (Lumi-Light; Roche), and exposed to a photographic film (BioMax; Kodak). For β-actin detection, POD-conjugated goat polyclonal anti–mouse IgG+IgM (Dianova) was used at the same dilution.
Chromium release assays.
Specific lysis by A*0201-restricted murine CTL reactive against human tyrosinase peptide 369–377 or against VV-specific peptide A6L6, B22R79, H3L184, or I1L211 was determined in a 6-h standard 51Cr release assay, as previously described (10). In brief, HLA-A*0201–positive A375 or RL-LCL cells were infected at MOI 10, washed, and labeled for 1 h at 37°C with 100 μCi Na51CrO4, and then washed four times. Labeled target cells were plated in U-bottomed 96-well plates at 104 cells/well and incubated for 6 h with effector cells at various E/T ratios. The specific 51Cr release was determined in supernatants.
Antigen presentation assays.
RL-LCL cells were infected for 30 min at MOI 10 on ice and shifted on 37°C. At indicated time points, cells were cocultured at different ratios with peptide-specific CTL lines in the presence of 1 mg/ml BFA (Sigma-Aldrich) for 5 h. Staining and analysis for intracellular IFN-γ-production was performed as described above. For OVA257 presentation, RMA cells were infected for 30 min at MOI 20 on ice and then shifted on 37°C. At indicated time points, cells were harvested and stained with anti-SIINFEKL-Kb antibody (25-D1.16).
Statistical analysis.
All statistical analysis was performed using Excel Software. Results are expressed as the mean ± the SD. Differences between groups were analyzed for statistical significance using two-tailed student t test.
Online supplemental material.
Fig. S1 shows the construction of B8R-deficient MVA. Fig. S2 shows the multistep viral growth curve on CEF cells. Fig. S3 shows that the immunodominance hierarchy is changed after secondary immunization in HHD mice. Fig. S4 shows IFN-γ production of VV-specific T cells from freshly isolated splenocytes of MVA WT (107 IU)-primed and peptide-revaccinated HHD mice. Fig. S5 shows construction of H3L-deficient MVA revertant for the H3L gene. Fig. S6 shows that MVA ΔH3L P7.5 H3L expresses the H3L gene early during the viral life cycle. Fig. S7 shows that competition between T cells occurs early after priming. Fig. S8 shows the total amount of OVA-expressed and vector-specific responses induced by different MVA constructs. Additional information is provided in a Supplemental materials and methods. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20070489/DC1.
Supplemental Material
Acknowledgments
We would like to thank A. Dick and S. Eckle for their outstanding performance during their bachelor thesis. We also thank K.M. Huster for critical reading of the manuscript and helpful discussion.
This work was supported by the Deutschen Forschungsgemeinschaft SFB 456 (B7).
The authors have no conflicting financial interests.
Abbreviations used: CEF, chicken embryo fibroblast; CVA, chorioallantois VV Ankara; IU, infectious unit; LCMV, lymphocytic choriomeningitis virus; MOI, multiplicity of infection; MVA, modified VV Ankara; VV, vaccinia virus.
W. Kastenmuller and G. Gasteiger contributed equally to this paper.
References
- 1.Yewdell, J.W., and J.R. Bennink. 1999. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol. 17:51–88. [DOI] [PubMed] [Google Scholar]
- 2.Kedl, R.M., J.W. Kappler, and P. Marrack. 2003. Epitope dominance, competition and T cell affinity maturation. Curr. Opin. Immunol. 15:120–127. [DOI] [PubMed] [Google Scholar]
- 3.Stock, A.T., C.M. Jones, W.R. Heath, and F.R. Carbone. 2006. CTL response compensation for the loss of an immunodominant class I-restricted HSV-1 determinant. Immunol. Cell Biol. 84:543–550. [DOI] [PubMed] [Google Scholar]
- 4.Thomas, P.G., S.A. Brown, R. Keating, W. Yue, M.Y. Morris, J. So, R.J. Webby, and P.C. Doherty. 2007. Hidden epitopes emerge in secondary influenza virus-specific CD8+ T cell responses. J. Immunol. 178:3091–3098. [DOI] [PubMed] [Google Scholar]
- 5.Probst, H.C., T. Dumrese, and M.F. van den Broek. 2002. Cutting edge: competition for APC by CTLs of different specificities is not functionally important during induction of antiviral responses. J. Immunol. 168:5387–5391. [DOI] [PubMed] [Google Scholar]
- 6.Yang, J., S.P. Huck, R.S. McHugh, I.F. Hermans, and F. Ronchese. 2006. Perforin-dependent elimination of dendritic cells regulates the expansion of antigen-specific CD8+ T cells in vivo. Proc. Natl. Acad. Sci. USA. 103:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, W., J.R. Bennink, P.A. Morton, and J.W. Yewdell. 2002. Mice deficient in perforin, CD4+ T cells, or CD28-mediated signaling maintain the typical immunodominance hierarchies of CD8+ T-cell responses to influenza virus. J. Virol. 76:10332–10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, W., K. Pang, K.A. Masterman, G. Kennedy, S. Basta, N. Dimopoulos, F. Hornung, M. Smyth, J.R. Bennink, and J.W. Yewdell. 2004. Reversal in the immunodominance hierarchy in secondary CD8+ T cell responses to influenza A virus: roles for cross-presentation and lysis-independent immunodomination. J. Immunol. 173:5021–5027. [DOI] [PubMed] [Google Scholar]
- 9.Crowe, S.R., S.J. Turner, S.C. Miller, A.D. Roberts, R.A. Rappolo, P.C. Doherty, K.H. Ely, and D.L. Woodland. 2003. Differential antigen presentation regulates the changing patterns of CD8+ T cell immunodominance in primary and secondary influenza virus infections. J. Exp. Med. 198:399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drexler, I., C. Staib, W. Kastenmuller, S. Stevanovic, B. Schmidt, F.A. Lemonnier, H.G. Rammensee, D.H. Busch, H. Bernhard, V. Erfle, and G. Sutter. 2003. Identification of vaccinia virus epitope-specific HLA-A*0201-restricted T cells and comparative analysis of smallpox vaccines. Proc. Natl. Acad. Sci. USA. 100:217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moutaftsi, M., B. Peters, V. Pasquetto, D.C. Tscharke, J. Sidney, H.H. Bui, H. Grey, and A. Sette. 2006. A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-cell responses to vaccinia virus. Nat. Biotechnol. 24:817–819. [DOI] [PubMed] [Google Scholar]
- 12.Oseroff, C., F. Kos, H.H. Bui, B. Peters, V. Pasquetto, J. Glenn, T. Palmore, J. Sidney, D.C. Tscharke, J.R. Bennink, et al. 2005. HLA class I-restricted responses to vaccinia recognize a broad array of proteins mainly involved in virulence and viral gene regulation. Proc. Natl. Acad. Sci. USA. 102:13980–13985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pasquetto, V., H.H. Bui, R. Giannino, C. Banh, F. Mirza, J. Sidney, C. Oseroff, D.C. Tscharke, K. Irvine, J.R. Bennink, et al. 2005. HLA-A*0201, HLA-A*1101, and HLA-B*0702 transgenic mice recognize numerous poxvirus determinants from a wide variety of viral gene products. J. Immunol. 175:5504–5515. [DOI] [PubMed] [Google Scholar]
- 14.Terajima, M., J. Cruz, G. Raines, E.D. Kilpatrick, J.S. Kennedy, A.L. Rothman, and F.A. Ennis. 2003. Quantitation of CD8+ T cell responses to newly identified HLA-A*0201-restricted T cell epitopes conserved among vaccinia and variola (smallpox) viruses. J. Exp. Med. 197:927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tscharke, D.C., G. Karupiah, J. Zhou, T. Palmore, K.R. Irvine, S.M. Haeryfar, S. Williams, J. Sidney, A. Sette, J.R. Bennink, and J.W. Yewdell. 2005. Identification of poxvirus CD8+ T cell determinants to enable rational design and characterization of smallpox vaccines. J. Exp. Med. 201:95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bronte, V., M.W. Carroll, T.J. Goletz, M. Wang, W.W. Overwijk, F. Marincola, S.A. Rosenberg, B. Moss, and N.P. Restifo. 1997. Antigen expression by dendritic cells correlates with the therapeutic effectiveness of a model recombinant poxvirus tumor vaccine. Proc. Natl. Acad. Sci. USA. 94:3183–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yewdell, J.W. 2006. Confronting complexity: real-world immunodominance in antiviral CD8+ T cell responses. Immunity. 25:533–543. [DOI] [PubMed] [Google Scholar]
- 18.Kastenmuller, W., I. Drexler, H. Ludwig, V. Erfle, C. Peschel, H. Bernhard, and G. Sutter. 2006. Infection of human dendritic cells with recombinant vaccinia virus MVA reveals general persistence of viral early transcription but distinct maturation-dependent cytopathogenicity. Virology. 350:276–288. [DOI] [PubMed] [Google Scholar]
- 19.Firat, H., F. Garcia-Pons, S. Tourdot, S. Pascolo, A. Scardino, Z. Garcia, M.L. Michel, R.W. Jack, G. Jung, K. Kosmatopoulos, et al. 1999. H-2 class I knockout, HLA-A2.1-transgenic mice: a versatile animal model for preclinical evaluation of antitumor immunotherapeutic strategies. Eur. J. Immunol. 29:3112–3121. [DOI] [PubMed] [Google Scholar]
- 20.van Stipdonk, M.J., E.E. Lemmens, and S.P. Schoenberger. 2001. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat. Immunol. 2:423–429. [DOI] [PubMed] [Google Scholar]
- 21.Busch, D.H., K.M. Kerksiek, and E.G.P. Am. 2000. Differing roles of inflammation and antigen in T cell proliferation and memory generation. J. Immunol. 164:4063–4070. [DOI] [PubMed] [Google Scholar]
- 22.Nugent, C.T., J.M. McNally, R. Chervenak, R.M. Wolcott, and S.R. Jennings. 1995. Differences in the recognition of CTL epitopes during primary and secondary responses to herpes simplex virus infection in vivo. Cell. Immunol. 165:55–64. [DOI] [PubMed] [Google Scholar]
- 23.Belz, G.T., W. Xie, J.D. Altman, and P.C. Doherty. 2000. A previously unrecognized H-2D(b)-restricted peptide prominent in the primary influenza A virus-specific CD8(+) T-cell response is much less apparent following secondary challenge. J. Virol. 74:3486–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Probst, H.C., K. Tschannen, A. Gallimore, M. Martinic, M. Basler, T. Dumrese, E. Jones, and M.F. van den Broek. 2003. Immunodominance of an antiviral cytotoxic T cell response is shaped by the kinetics of viral protein expression. J. Immunol. 171:5415–5422. [DOI] [PubMed] [Google Scholar]
- 25.La Gruta, N.L., K. Kedzierska, K. Pang, R. Webby, M. Davenport, W. Chen, S.J. Turner, and P.C. Doherty. 2006. A virus-specific CD8+ T cell immunodominance hierarchy determined by antigen dose and precursor frequencies. Proc. Natl. Acad. Sci. USA. 103:994–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jenkins, M.R., R. Webby, P.C. Doherty, and S.J. Turner. 2006. Addition of a prominent epitope affects influenza A virus-specific CD8+ T cell immunodominance hierarchies when antigen is limiting. J. Immunol. 177:2917–2925. [DOI] [PubMed] [Google Scholar]
- 27.Webby, R.J., S. Andreansky, J. Stambas, J.E. Rehg, R.G. Webster, P.C. Doherty, and S.J. Turner. 2003. Protection and compensation in the influenza virus-specific CD8+ T cell response. Proc. Natl. Acad. Sci. USA. 100:7235–7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scherer, A., M. Salathe, and S. Bonhoeffer. 2006. High epitope expression levels increase competition between T cells. PLoS Comput. Biol. 2:e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kedl, R.M., B.C. Schaefer, J.W. Kappler, and P. Marrack. 2002. T cells down-modulate peptide-MHC complexes on APCs in vivo. Nat. Immunol. 3:27–32. [DOI] [PubMed] [Google Scholar]
- 30.Willis, R.A., J.W. Kappler, and P.C. Marrack. 2006. CD8 T cell competition for dendritic cells in vivo is an early event in activation. Proc. Natl. Acad. Sci. USA. 103:12063–12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong, P., and E.G.P. Am. 2003. Feedback regulation of pathogen-specific T cell priming. Immunity. 18:499–511. [DOI] [PubMed] [Google Scholar]
- 32.Belz, G.T., L. Zhang, M.D. Lay, F. Kupresanin, and M.P. Davenport. 2007. Killer T cells regulate antigen presentation for early expansion of memory, but not naive, CD8+ T cell. Proc. Natl. Acad. Sci. USA. 104:6341–6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu, F., J.L. Whitton, and M.K. Slifka. 2004. The rapidity with which virus-specific CD8+ T cells initiate IFN-gamma synthesis increases markedly over the course of infection and correlates with immunodominance. J. Immunol. 173:456–462. [DOI] [PubMed] [Google Scholar]
- 34.Shin, H., S.D. Blackburn, J.N. Blattman, and E.J. Wherry. 2007. Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J. Exp. Med. 204:941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tebo, A.E., M.J. Fuller, D.E. Gaddis, K. Kojima, K. Rehani, and A.J. Zajac. 2005. Rapid recruitment of virus-specific CD8 T cells restructures immunodominance during protective secondary responses. J. Virol. 79:12703–12713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bousso, P., J.P. Levraud, P. Kourilsky, and J.P. Abastado. 1999. The composition of a primary T cell response is largely determined by the timing of recruitment of individual T cell clones. J. Exp. Med. 189:1591–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bunde, T., A. Kirchner, B. Hoffmeister, D. Habedank, R. Hetzer, G. Cherepnev, S. Proesch, P. Reinke, H.D. Volk, H. Lehmkuhl, and F. Kern. 2005. Protection from cytomegalovirus after transplantation is correlated with immediate early 1–specific CD8 T cells. J. Exp. Med. 201:1031–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pascolo, S., N. Bervas, J.M. Ure, A.G. Smith, F.A. Lemonnier, and B. Perarnau. 1997. HLA-A2.1-restricted education and cytolytic activity of CD8+ T lymphocytes from β2 microglobulin (β2m) HLA-A2.1 monochain transgenic H-2Db β2m double knockout mice. J. Exp. Med. 185:2043–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Staib, C., I. Drexler, and G. Sutter. 2004. Construction and isolation of recombinant MVA. Methods Mol. Biol. 269:77–100. [DOI] [PubMed] [Google Scholar]
- 40.Drexler, I., E. Antunes, M. Schmitz, T. Wolfel, C. Huber, V. Erfle, P. Rieber, M. Theobald, and G. Sutter. 1999. Modified vaccinia virus Ankara for delivery of human tyrosinase as melanoma-associated antigen: induction of tyrosinase- and melanoma-specific human leukocyte antigen A*0201-restricted cytotoxic T cells in vitro and in vivo. Cancer Res. 59:4955–4963. [PubMed] [Google Scholar]
- 41.Staib, C., I. Drexler, M. Ohlmann, S. Wintersperger, V. Erfle, and G. Sutter. 2000. Transient host range selection for genetic engineering of modified vaccinia virus Ankara. Biotechniques. 28:1137–1148. [DOI] [PubMed] [Google Scholar]
- 42.Busch, D.H., and E.G.P. Am. 1999. T lymphocyte dynamics during Listeria monocytogenes infection. Immunol. Lett. 65:93–98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}