

Abstract
Although cyclooxygenase (COX)-2 inhibitors (coxibs) are effective in controlling inflammation, pain, and tumorigenesis, their use is limited by the recent revelation of increased adverse cardiovascular events. The mechanistic basis of this side effect is not well understood. We show that the metabolism of endocannabinoids by the endothelial cell COX-2 coupled to the prostacyclin (PGI2) synthase (PGIS) activates the nuclear receptor peroxisomal proliferator–activated receptor (PPAR) δ, which negatively regulates the expression of tissue factor (TF), the primary initiator of blood coagulation. Coxibs suppress PPARδ activity and induce TF expression in vascular endothelium and elevate circulating TF activity in vivo. Importantly, PPARδ agonists suppress coxib-induced TF expression and decrease circulating TF activity. We provide evidence that COX-2–dependent attenuation of TF expression is abrogated by coxibs, which may explain the prothrombotic side-effects for this class of drugs. Furthermore, PPARδ agonists may be used therapeutically to suppress coxib-induced cardiovascular side effects.
The cyclooxygenase (COX) pathway in vascular endothelium plays important roles in thrombosis, atherosclerosis, and vascular inflammation (1). Vascular endothelial cells (ECs) constitutively express COX-1 and -2 isoenzymes, leading to the generation of prostacyclin (PGI2) and related molecules (2). PGI2, a well-known inhibitor of platelet aggregation and a vasodilator, activates the IP-subtype of G protein–coupled receptors on the plasma membrane of platelets and vascular smooth muscle cells (1, 3). In addition to activating cell surface receptors, PGI2 and related molecules are potent activators of nuclear peroxisomal proliferator–activated receptor (PPAR) δ (4–6). This mechanism was shown to be important for embryo implantation in mice (6) and in intestinal adenoma cell proliferation (7) and angiogenesis (8).
The role of COX-2 in the regulation of EC phenotype is not well understood. In small-vessel endothelium, COX-2 is induced by growth factors and cytokines during inflammation and angiogenesis (9). In large-vessel ECs, COX-2 is constitutively expressed as a laminar shear–inducible gene (10), which may be important for normal vascular homeostasis (11). This issue has recently received considerable attention because administration of COX-2–specific inhibitors (also known as the coxibs) leads to a small but substantial increase in prothrombotic side effects in humans, leading to the withdrawal of rofecoxib and valdecoxib from the market (12, 13). The mechanistic basis of these side effects is not clearly understood, even though PGI2-dependent platelet effects and thromboxane-dependent vascular pathology have been implicated (14, 15). In this study, we show that metabolism of endocannabinoids by the COX-2 pathway results in direct activation of the nuclear receptor PPARδ. We further show that this pathway suppresses the expression of tissue factor (TF), which is a primary regulator of blood coagulation. This description of the antithrombotic function of COX-2 may contribute to the mechanistic understanding of coxib-induced cardiovascular side effects seen in humans.
RESULTS AND DISCUSSION
The COX-2 isoenzyme has a larger active site pocket than COX-1 and therefore is capable of oxidizing many polyunsaturated fatty acids in addition to the common substrate arachidonic acid (AA) (16). We tested if metabolism of various substrates of COX-2 would lead to intracellular activation of PPARδ in ECs. Human umbilical vein ECs (HUVECs), which express COX-2 were transfected with a PPAR-responsive transcription reporter (pACO-Luc) (17), incubated with various fatty acid substrates and transcriptional reporter (luciferase) activity was measured. As shown in Fig. 1 A, endocannabinoids, 2-arachidonyl glycerol (2-AG), noladin ether (NE), and anandamide (AEA) stimulated PPAR-dependent transcription. In contrast, the effect of AA was modest, and neither n-3 fatty acids (docosahexaenoic acid or eicosapentaenoic acid) nor non–COX-2 substrates (palmitate or oleate) induced PPAR-dependent transcription. The concentration of endocannabinoids that induced transcription is significantly below the Km of 2-AG for COX-2, which is estimated to be ∼4 μM (16). NE, which is a nonhydrolyzable ether analogue of 2-AG, is more potent, suggesting that hydrolytic pathways are involved in attenuating the 2-AG effect. These data provide evidence that endocannabinoid ligands, which are alternative substrates for COX-2 but not COX-1, are capable of activating the endogenous PPAR system in ECs.
Figure 1.

Endocannabinoids induce PPARδ-dependent transcription in HUVECs. (A) HUVECs were transiently transfected with PPRE-luciferase reporter plasmid pACO-gLuc, and after 24 h cells were incubated with vehicle (DMSO), fatty acids (AA, DHA, and OA), or endocannabinoids (2G, AEA, and NE). Subsequently, luciferase activity was measured as described in Materials and methods. (*, P < 0.05; **, P < 0.01 compared with AA). Structures of the endocannabinoids are shown in Fig. S4. (B) Endocannabinoid activation of endogenous PPARδ-dependent transcription. A chimeric receptor containing the ligand-binding domain of PPARα,γ,δ fused with the DNA-binding domain of yeast Gal4 transcription factor was used. Transactivation was detected by cotransfection with reporter gene (Gal4 response element [UAS-tk-luc]) in the presence of 2-AG (*, P < 0.05; **, P < 0.01, compared with vehicle). (C) Mouse lung ECs were isolated from wild-type (yellow), PPARδ−/− (green), or CB1−/−/CB2−/− (blue) mice, and the Gal4-UAS system of transcriptional activation was assayed in the presence of vehicle (DMSO), 1 or 10 μM 2-AG, or 10 μM PPARδ agonist cPGI as control (*, P < 0.05; **, P < 0.01 compared with vehicle). Fig. S4 is available at http://www.jem.org/cgi/content/full/jem.20070828/DC1.
The Gal4-UAS–based transcription reporter system was used to distinguish between the three PPAR isoforms (7), all of which are expressed in vascular ECs (17). We observed that 2-AG primarily induces PPARδ−dependent transcriptional activation in a dose-dependent manner, without significantly influencing PPARα- or PPARγ-dependent transcription (Fig. 1 B). In addition, a dominant-negative PPARδ construct (7) blocked the endocannabinoid-induced reporter activation (unpublished data).
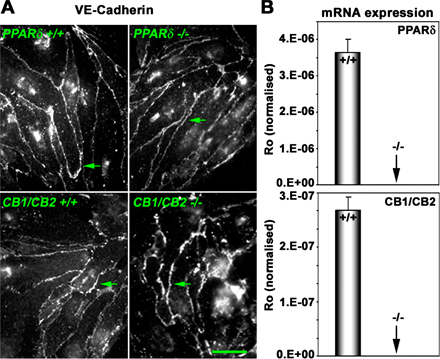
Endocannabinoids are known to activate their G protein–coupled receptors CB1 and CB2 (18). To determine whether the observed effects of endocannabinoids are mediated by CB receptors, we derived lung ECs from CB1/CB2 double knockout mice (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20070828/DC1) (19). We found that 2-AG stimulated PPAR-dependent transcription in these cells lacking both CB1 and CB2 to an equivalent level of wild-type cells, suggesting that endocannabinoids are not acting via CB receptors (Fig. 1 C). In contrast, when the PPARδ-null lung ECs (20) were used in a similar assay, 2-AG failed to induce transcriptional activation, suggesting an absolute requirement for this transcription factor.
We next tested if COX-2 enzyme activity is required for 2-AG–induced PPARδ transcription. Indeed, 2-AG–induced PPARδ transcription in ECs was completely inhibited by NS-398, a COX-2–selective inhibitor, but not by valeryl salicylate (VSA), a COX-1–specific inhibitor (Fig. 2 A). However, NS398 or VSA alone did not have any effect on PPARδ−dependent transcription. 2-AG–induced PPAR transcription was absent in HCT116 colon cancer cells, which lack COX-2 expression but express COX-1 endogenously (Fig. 2 B) (21). In contrast, direct activation of PPARδ by cPGI, which is a stable PGI2 analogue, induced transcription in HCT116 cells. More importantly, overexpression of COX-2 by adenoviral-mediated transduction into HUVECs strongly induced PPARδ transcription (∼25-fold), and this response was further potentiated (∼40-fold) by 2-AG (Fig. 2 C). This increase was blocked by NS-398. Although COX-2 expression with the adenovirus induced high basal activity of the PPAR-responsive reporter, this is probably caused by efficient transduction (>90% for HUVECs) and accumulation of COX-2 enzyme during the time course of the adenoviral expression. Similar arguments likely explain the incomplete inhibition by COX-2 inhibitor. Together, these results (overexpression, inhibitor sensitivity, and COX-2–null cell data) establish that COX-2 activity is required for 2-AG–induced PPARδ transcription.
Figure 2.

COX–2 activity is required for endocannabinoid-induced PPARδ transcription. (A) HUVECs were transiently transfected with pACO-gLuc as described in Fig. 1. Cells were pretreated with a COX-2–selective inhibitor (NS398; 10 μM) or a COX-1–selective inhibitor (VSA; 500 μM) for 30 min, followed by the addition of 2-AG or DMSO (vehicle) for 16 h (*, P < 0.05, compared with vehicle). (B) Transactivation of the PPRE reporter plasmid in HUVECs (blue), but not in HCT116 (gray), which is a COX-2–null cell line. (B, insets) mRNA levels of COX-1 and -2 in these cells by qRT-PCR (*, P < 0.05; **, P < 0.01, compared with vehicle). (C) Overexpression of COX-2 enhances 2-AG–induced PPARδ transactivation. HUVECs were transduced with either adeno-GFP (green) or adeno–COX-2 virus (blue) treated with 1 μM 2-AG as in Fig. 1, with or without 10 μM NS-398, and PPARδ transcriptional activity was assayed as in Fig. 1. (C, inset) COX-2 mRNA expression (**, P < 0.01, compared with control vector virus, inset *, P < 0.05; compared with vehicle).
Once fatty acid substrates are metabolized by COX-2 into endoperoxides, terminal synthases catalyze the formation of active prostanoids (1). To determine which of the prostanoids are involved in PPARδ transcription, we silenced the expression of genes encoding prostanoid synthases by siRNA in HUVECs and assayed the 2-AG–stimulated transcriptional response. As shown in Fig. 3 A, quantitative (q) RT-PCR analysis indicates that PGF2α synthase (PGFS) and PGI2 synthase (PGIS) mRNAs are highly expressed in HUVECs, although lower levels of cytosolic PGE2 synthase mRNA are also expressed. The inducible microsomal PGE2 synthase mRNA was not detected under basal conditions. This is consistent with our previous observation of abundant synthesis of 6-keto-PGF1α (a stable breakdown product of PGI2), PGF2α, and PGE2 in HUVECs (22). PGIS or PGFS siRNA significantly suppressed the expression of PGIS or PGFS mRNAs in HUVECs, respectively (Fig. 3 B). As shown in Fig. 3 C, 2-AG–induced PPARδ transcription was inhibited by the PGIS, but not by the PGFS siRNA. These data suggest that conversion of 2-AG by COX-2 and PGIS into PGI2 glycerol ester is critical for PPARδ activation in ECs. Attempts to purify the PGI2 glycerol ester or 6-keto-PGF1α glycerol ester from EC extracts were not successful, which is presumably caused by high hydrolytic conversion of prostaglandin glycerol esters by cytosolic esterases (23).
Figure 3.

Role of PG synthases in endocannabinoid-induced PPARδ transactivation. (A) Expression profile of PG synthases in HUVECs was determined by real-time PCR. Expression level (Ro) for each gene was normalized to glyceraldehyde-3-phosphate dehydrogenase. (B) Expression of PGIS or PGFS mRNA in HUVECs after treatment with 100 nM of PGFS or PGIS siRNA (*, P < 0.05, compared with control siRNA). (C) Effects of silencing PGFS or PGIS on PPARδ transactivation in response to 2-AG. Cells were transfected with PGFS or PGIS siRNA with Oligofectamine for 4 h, and were allowed to recover in complete growth medium. After 16 h, cells were transiently transfected with PPARδ-GAL4, UAS-tk-luc, and pβ-gal with Lipofectamine in OptiMEM, followed by addition of 2-AG. Cells were harvested for luciferase activity after 24 h (*, P < 0.05, compared with vehicle).
Because of the prothrombotic effects of coxibs observed in some patients (15, 24), we analyzed the effect of modulating the COX-2–PPARδ pathway on TF expression in ECs. TF is the primary cellular initiator of blood coagulation, and its expression within the vasculature is associated with thrombosis (25). We found that the COX-2 inhibitors celecoxib and NS398 induced TF mRNA expression, as determined by qRT-PCR (Fig. 4 A). In contrast, activation of the COX-2–PPARδ pathway with either 2-AG or GW501516 decreased basal TF mRNA expression (Fig. 4 A). Furthermore, 2-AG reduced celecoxib induction of TF mRNA expression (Fig. 4 A). These data suggest that inhibition of COX-2 induces TF expression, whereas activation of the COX-2–PPARδ pathway suppresses TF expression. We isolated lung ECs from PPARδ −/− mice and tested the expression of TF by qRT-PCR (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20070828/DC1). Unexpectedly, TF expression was lower than the wild-type counterparts. This phenomenon is analogous to the regulation of inflammatory gene expression by PPARδ in macrophages (26). These authors showed that although PPARδ agonists suppressed inflammatory genes, PPARδ-null macrophages had low expression of such genes. They further showed that release of the transcriptional repressor Bcl6 from the apo-PPARδ is responsible for suppression of inflammatory gene expression. We believe that an analogous phenomenon is operative in our PPARδ-null ECs. Although molecular details of such mechanisms need to be further elucidated, our data suggest that COX-2–dependent PPARδ activation maintains the antithrombotic phenotype of the normal endothelium by suppressing the TF gene.
Figure 4.

COX-2 inhibitors enhance, whereas PPARδ activators suppress, TF gene expression in ECs. (A) qRT-PCR analysis of total RNA extracted from HUVECs upon treatment with 10 μM celecoxib, 10 μM NS398, 10 μM GW501516, 10 μM 2-AG, or DMSO (vehicle) for 3 h. Celecoxib and NS-398 were added 30 min before stimulation with 2-AG or GW501516 for 6 h. Human TF mRNA levels were normalized by the internal control gene GAPDH (*, P < 0.05; **, P <0.01, compared with vehicle). (B) Northern blot analysis of HUVECs treated with LPS for 3–6 h in the presence or absence of COX-2 inhibitors, GW501516, or 2-AG, as described in A. (C) Western blot analysis of TF expression in HUVECs. (D) TF expression in HUVEC extracts was assayed by Western blot analysis as described in Materials and methods. Cells were treated with indicated concentrations (micromolar) of various compounds (D and E) in the presence of 10 μg/ml LPS. (E) TF activity assay in LPS-stimulated HUVEC lysates treated as outlined above. TF concentration of test samples was directly calculated from the standard curve. *, P < 0.05; **, P < 0.01, compared with vehicle.
When HUVECs were treated with LPS, large induction of TF mRNA was seen; however, PPARδ expression was not altered (Fig. 4 B and not depicted). In fact, TF mRNA can be detected by Northern blot only after LPS treatment. COX-2 inhibitors NS398 and celecoxib enhanced LPS induction of TF mRNA expression (Fig. 4 B). Similarly, celecoxib enhanced LPS induction of TF protein expression (Fig. 4 C). In contrast, the PPARδ agonist GW501516 and 2-AG both decreased LPS induction of TF mRNA expression (Fig. 4 B). Concomitant regulation of TF polypeptide expression was observed as determined by an immunoblot assay (Fig. 4 C). Rofecoxib, another COX-2–selective inhibitor, also enhanced LPS induction of TF protein expression in HUVECs in a dose-dependent manner, similar to celecoxib (Fig. 4 D).
Next, we measured TF activity in HUVECs under similar experimental conditions. LPS induction of TF activity in HUVECs was enhanced by celecoxib in a dose-dependent manner (Fig. 4 E). Preincubation of the cells with either GW501516 or 2-AG blocked this effect, and these agents decreased LPS induction of TF activity (Fig. 4 E). In contrast to the results with HUVECs, celecoxib did not induce TF activity in peripheral blood mononuclear cells and did not enhance LPS induction of TF activity (unpublished data), which may be caused by the differences in the COX-2–PGIS–PPARδ pathway in these cells. These findings indicate that the COX-2–PPARδ pathway serves to attenuate LPS-induced TF expression in the vascular endothelium. In addition, our data show that inhibition of this pathway by COX-2 inhibitors leads to the induction of TF expression.
Our results are in apparent contradiction with a recent study that showed that celecoxib, but not rofecoxib, inhibited the induction of TF expression in human aortic ECs stimulated with TNF-α in vitro (27). The authors of that paper suggested that inhibition of JNK activity is critical for the modulation of TF expression. There could be several reasons for these discrepancies, e.g., differences in signaling by different inflammatory stimuli (LPS vs. TNF-α) in cell-specific manner. However, in our work, we clearly show that the conversion of endocannabinoid (2-AG) by the COX-2–PGIS pathway, followed by activation of PPARδ, is involved in the regulation of TF mRNA and protein expression, as well as activity in HUVECs. In addition, as shown in the following paragraph, both COX-2– and PPARδ-dependent regulation of TF occurs in vivo as well.
We next determined whether the COX-2–PPARδ pathway regulates TF expression in ECs in vivo. Immunohistochemical analysis showed that LPS and/or celecoxib induced TF expression in ECs of blood vessels of lung and heart (Fig. 5 A). Modulation of TF expression is seen in both microvessels and large vessels. GW501516 abolished this effect (Fig. 5 A). The observation is consistent with other reports showing that TF is expressed in the endothelium in other diseases associated with thrombosis (28).
Figure 5.

Regulation of TF expression in vivo by celecoxib and a PPARδ activator. C57BL/6J mice were treated with either vehicle (0.5% carboxymethyl cellulose), 25 mg/kg celecoxib, or 10 mg/kg GW501516 by gavage for 2 d. LPS (E. coli serotype 0111; B4; Sigma-Aldrich) was injected i.p. (100 μg/mouse). After 4–6 h, whole blood was collected from caudal vena cava into 10× acid citrate dextrose buffer to determine TF activity as previously described (see Materials and methods). Murine tissues were also harvested for immunohistochemistry. (A) Expression of TF protein in frozen tissue sections of lung and heart. Immunohistochemistry was conducted with monoclonal rat anti–mouse TF antibody and representative images are shown. The arrows and arrowheads indicate expression of TF protein in large vessels, as well as microvessels of both heart and lung. Bar, 20 μm. (B) TF activity was measured in PPP as described in Materials and methods. TF concentration was interpolated as ΔA405-490 from a standard curve. n = 10 mice/group. *, P < 0.05; **, P < 0.01.
Activated ECs release TF-positive microparticles into plasma (29). Therefore, we obtained platelet poor plasma (PPP) from mice orally administered with either celecoxib or GW501516, followed by intraperitoneal LPS administration. We observed that treatment of mice with either LPS or celecoxib increased plasma TF activity (Fig. 5 B). Treatment of mice with both LPS and celecoxib produced the highest levels of plasma TF, and such increase was abolished by pretreatment with the PPARδ activator GW501516 (Fig. 5 B). These data strongly suggest that the COX-2 pathway attenuates TF expression in vivo via the PPARδ pathway. Inhibition of COX-2 activity by coxibs results in the induction of TF expression in the endothelium, which is further enhanced under inflammatory conditions.
In summary, our results show the following novel findings. First, metabolism of endocannabinoids, particularly 2-AG, by the EC COX-2–PGIS system has important intracellular signaling fates. It is known that 2-AG is released upon cellular activation of platelets, monocytes, and ECs (30, 31). Recent work shows that metabolism of 2-AG by the COX-2 pathway of the vascular endothelium occurs under physiological conditions and limits the activation of the CB receptors (32). Although regulation of 2-AG production in human ECs need to be further examined, our data support the notion that 2-AG metabolism by the EC COX-2 pathway is biologically important. We speculate that the glycerol moiety of 2-AG may hinder its rapid secretion into the extracellular milieu, thereby facilitating the activation of intracellular receptors. Second, basal expression of COX-2 by EC, which is generally thought to be caused by physiological pulsatile shear forces (10), may be important for the tonic production of PPARδ activators by the PGIS pathway. Our data supports the notion that 2-AG is an endogenous mediator that regulates the thrombotic phenotype of the vascular endothelium. Thus, antagonism of this pathway by COX-2 inhibitors would be anticipated to disrupt this endogenous regulatory mechanism (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20070828/DC1). In this context, recent work shows that 15-epi-lipoxin A4, which is an endogenous antiinflammatory mediator derived from EC COX-2 is also inhibited by the coxib treatment (33), suggesting that multiple lipid mediators are perturbed by these drugs.
Third, we suggest that the PPARδ pathway attenuates prothrombotic TF gene expression, and thus keeps the endothelium in an antithrombotic state. Recent work has highlighted the prothrombotic side effects of COX-2 inhibitors (coxibs) in humans (34). Although the critical mechanisms involved remain to be elucidated, the importance of coxib inhibition of PGI2, the major vasoprotective prostanoid has been assumed to be the primary culprit (2, 11, 14). Indeed, studies in mice show that perturbation of the PGI2 receptor may control inflammatory vascular pathology (11,14). Our study provides an alternative mechanism in understanding the prothrombotic complications of COX-2 inhibitors. Elevated TF expression in the luminal surface of blood vessel and/or plasma may be one of the factors that contribute to the development of cardiovascular events in patients consuming COX-2 inhibitors (35). Recent reports show that the use of coxibs and nonsteroidal antiinflammatory drugs are associated with increased mortality risk in patients with a history of cardiovascular disease (36, 37). It is important to note that the side effects of coxibs only occur in a subset of patients, which is consistent with our in vitro data showing that coxibs induce TF expression modestly compared with LPS. Identification of the at-risk population would offer obvious advantages, as coxibs and nonsteroidal antiinflammatory drugs are some of the most widely consumed antiinflammatory drugs and have shown activity as anticancer and antiangiogenic agents. This study has provided a potential mechanism by which COX-2 inhibitors may contribute to thrombotic complications, whereas a PPARδ agonist would likely alleviate such adverse events.
MATERIALS AND METHODS
Cell culture, transfection, transduction, and luciferase assays.
HUVECs (p3–6; Glyco Tech Corporation or VEC TECHNOLOGIES, Inc.) were cultured as previously described (38) in complete growth medium (M199, 10% FBS, growth factor, and 1% antibiotic-antimicotic agent). Human colon carcinoma cells (HCT-116) were obtained from the American Type Culture Collection and were grown in Dulbecco's modified Eagle's medium or RPMI with 10% FBS and 1% antibiotic-antimicotic agent.
For transient transfection, HUVECs (7.5 × 104 cells) were seeded in 6-well plates, followed by transfection with a PPAR-responsive transcription reporter plasmid (pACO-gLUC), β-galactosidase plasmid (p-βgal), and Lipofectamine 2000 in OPTI-MEM (Invitrogen) for 4 h, followed by the addition of 10% FBS containing media. After 16–24 h, 0.01–0.05% vehicle (DMSO) or indicated compounds were added and cells were harvested after 24 h in 1× luciferase lysis buffer. Relative light intensity from firefly luciferase activity was determined by using a luciferase kit (Promega) and a luminometer (Turner Designs). Relative luciferase activity was normalized to β-galactosidase activity and protein content. All PPAR constructs were gifts from R. DuBois (Vanderbilt University Medical Center, Nashville, TN).
For adenoviral transduction, cells were infected with adenoviral vector containing human COX-2 cDNA or GFP cDNA as control for 48 h (multiplicity of infection, 500), and the level of COX-2 expression was measured by either Western blot analysis or qRT-PCR. For transactivation assays after transfection, cells were transduced with either vector virus or adeno–COX-2 virus for 16–24 h. Upon addition of COX-2 substrate, cells were harvested for luciferase activity as indicated above. Unless specified, all compounds (2-AG, AA, GW501516, NS398, NE, AEA, and cPGI) were purchased from Cayman Chemical.
qRT-PCR.
Total RNA was prepared using the RNA STAT-60 (Tel-test, Inc.) single-step isolation procedure according to the instructions provided by the manufacturer. 1 μg total RNA was treated with DNase I before reverse transcription to ensure removal of all contaminating DNA. Using SYBR Green I DNA-binding dye technology (QIAGEN) reactions for each sample were done using an ABI 7900HT instrument (Applied Biosystems). All reactions underwent a final dissociation curve determination to ensure a single PCR product at the correct melting temperature. Fluorescence data were exported and quantitated using a statistical model that corrects for PCR efficiency for each reaction. Results were expressed relative to the internal control gene glyceraldehyde-3-phosphate dehydrogenase for human or hypoxanthine phosphoribosyl-transferase I (HPRT) for mouse. Sequence of PCR primers were either obtained from the PrimerBank database (http://pga.mgh.harvard.edu/primerbank/) or designed with Primer Express software (Applied Biosystems) for optimal product length, GC content, and melting temperature.
Northern blot analysis.
Confluent cultures of HUVEC cells were rendered quiescent for 48 h, followed by pretreatment with either vehicle, celecoxib, NS398, 2-AG, or GW501516 in fresh HUVEC medium for 30 min. 10 μg/ml LPS (Escherichia coli 0111:B4; Calbiochem) was added, and cells were treated for an additional 3–6 h. Total RNA was isolated with the RNA STAT-60 reagent. 10 μg of total RNA, quantitated spectrophotometrically, was electrophoresed through 1% agarose gel containing 4% (vol/vol) formaldehyde, and the integrity of the RNA was monitored by ethidium bromide staining. RNA was transferred onto Zeta-Probe membrane (Bio-Rad Laboratories), and was UV cross-linked by Stratalinker (Stratagene). A 641-bp human TF cDNA fragment was used to detect the TF mRNA. Specific cDNA probes were radioactively labeled by using [32P]-α-dCTP (GE Healthcare) by a random primer labeling system (Roche). After cross-linking, the membranes were prehybridized in Church-Gilbert prehybridization buffer (7% SDS, 0.5 M phosphate buffer, 0.5% BSA, 40 mM EDTA, and 20% formamide) for 1 h. The membranes were hybridized with labeled probes for 18 h and washed following the Church-Gilbert protocol (38). The membranes were stripped in stripping buffer (0.1× SSC, 1% SDS, and 40 mM Tris-HCl, pH 7.6), rehybridized with different probes, and visualized by PhosphorImager (Molecular Dynamics). The bands were quantified using ImageQuant (Molecular Dynamics). The prehybridization, hybridization, and washing steps were all done at 65°C.
Small interfering RNA (siRNA)–mediated gene silencing.
Transfection of siRNA oligonucleotide duplexes to block PGIS and PGFS expression was done using Oligofectamine reagent (Invitrogen) according to the manufacturer's instructions. The siRNAs used were human PGIS (sense AGGCCAGGAUGAAACUGACdTdT and antisense GUCAGUUUCAUCCUGGCCUdTdT). Oligonucleotides were synthesized by Dharmacon, deprotected, and duplexed. For, silencing of PGFS, SMARTpool PGF synthase siRNA was obtained from Dharmacon. HUVECs were transfected with either 100 nM PGIS or PGFS siRNA complexed with Oligofectamine reagent (Invitrogen) and, 24 h after transfection, gene expression was examined by qRT-PCR analysis. E-cadherin (siRNA against epithelial cadherin was used as control).
Isolation of mouse lung ECs.
Lungs isolated from 21-d-old (C57/B6/129 mixed) mice were washed several times in Hanks' balanced salt solution and digested with 2 mg/ml collagenase A (Roche) in M199 medium, with occasional mixing at 37°C. After treatment with 1 μg/μl DNaseI, the suspension was passed through a cell strainer (40 μm nylon; BD Biosciences) to remove any undigested remains. After a brief spin, the supernatant was treated with rat anti–mouse CD31 monoclonal antibody (BD Biosciences) for 20 min at 4°C. The cell suspension was spun and washed twice in plain M199 medium. Resuspension of the pellet in M199 medium was followed by treatment with magnetic Dynabeads sheep anti–rat IgG (Dynal Biotech) at 4°C in a rotator. Bead-bound cells were washed and separated from the unbound cells by a magnetic bar. Cells were plated in fibronectin-coated tissue culture dish in M199, 10% FBS, growth factor, and antibiotic-antimicotic agents. After 24 h, the cells were immortalized with a retrovirus that expresses polyoma middle-T-antigen (39).
Immunofluorescence.
Cells grown on glass coverslips for 48 h were washed with PBS, fixed in 3.7% paraformaldehyde solution for 15 min at room temperature, and permeabilized with 0.2% Triton X-100 in PBS for 5 min at room temperature. After rinsing with PBS, cells were incubated with a goat polyclonal anti–VE-cadherin antibody (1:200; Santa Cruz Biotechnologies) for 1 h at room temperature. After washing with PBS few times, cells were further incubated with Alexa Fluor 488 donkey anti–goat IgG (1:1,000; Invitrogen) in 5% nonfat dry milk in PBS for 30 min at room temperature. After washing with PBS, cells were analyzed by a Zeiss Axiovert fluorescence microscope.
Western blot analysis.
HUVECs were treated with indicated compounds, as described in Northern blot analysis. Cells were washed with ice-cold phosphate-buffered saline and harvested in radioimmunoprecipitation assay buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 0.25% sodium deoxycholate, 1% Nonidet P-40, 1 mM EDTA, 1 mM EGTA, and 1× protease inhibitor cocktail). Cells were lysed by three cycles of freeze (dry-ice/ethanol bath) and thaw (20°C), followed by centrifugation at 14,000 rpm for 20 min. Protein concentrations of supernatants were determined by BCA Protein Assay kit (Pierce Chemical Co.). Equal amounts of protein were separated on a 9% polyacrylamide gel and blotted to a nitrocellulose membrane for 1.5 h at 100 V. Immunoblot analysis was performed using 0.5 μg/ml goat anti–human TF antibody (a gift from W. Ruf, The Scripps Research Institute, La Jolla, CA) and detected by horseradish peroxidase–conjugated rabbit anti–goat secondary antibody. Blots were developed with the Western blot development kit from GE Healthcare.
TF activity assays.
Cell pellets from PBMCs or HUVECs were solubilized at 37°C for 15 min in 15 mM n-Octyl β-D-glucopyranoside (EMD Biosciences). TF activity in cell lysates was measured using a one-stage clotting assay using human pooled plasma (American Diagnostica Inc.) (40). Clotting was performed using a STart 4 clotting machine (Diagnostica Stago). Clotting times were converted to milliunits of TF activity using a standard curve established with purified human brain TF. Protein concentrations were measured using a DC protein assay kit (Bio-Rad Laboratories), and TF activity was normalized to protein concentration. Data were expressed as fold induction versus control for each experiment.
A chromogenic assay (Actichrome TF; American Diagnostica, Inc.) was used to analyze TF activity. In brief, HUVECs were lysed by repeated freeze-thaw in buffer containing 50 mM Tris-HCl, 100 mM NaCl, 0.1% Triton X-100, pH 7.4. TF activity was measured in cell supernatant according to the manufacturer's instructions (American Diagnostica, Inc.). Cell extracts were mixed with human factor VII and human factor X. The reagents were incubated at 37°C for complex formation to convert human factor X to factor Xa, and the amount of Xa generated was measured by its ability to cleave a chromogenic substrate Spectrozyme Xa. Absorbance was read at 405 nm with a microplate reader (Spectra Max 340; Molecular Devices). TF concentration was determined by interpolation of a standard curve obtained from different amounts of lipidated human TF standards. For TF activity measurement in mouse plasma, a mixture of 10% rat plasma/90% human plasma (Sigma-Aldrich) was added to the reaction mixture and ΔA405-490 was measured to calculate TF concentration.
In vivo studies.
All animal experiments were approved by the University of Connecticut Institutional Animal Care and Use Committee. C57/BL/6J (4–6 wk old; The Jackson Laboratory) mice were fed with either vehicle (0.5% carboxymethyl cellulose) or celecoxib (25 mg/kg body weight) or GW501516 (10 mg/kg body weight) by gavage for 2 d. 100 μg LPS (E. coli serotype 0111;B4; Sigma-Aldrich) was injected i.p. After 4–6 h, mice were anaesthetized by Avertin and whole blood was collected from caudal vena cava for TF activity assay, followed by isolation of murine tissues for immunohistochemistry.
Immunohistochemistry.
5–6-μm-thick frozen sections of mouse tissue were dried overnight at room temperature then fixed in acetone/ethanol (3:1 vol/vol) for 5 min at room temperature. All steps were performed in PBS buffer. Endogenous peroxidase was quenched with 3% hydrogen peroxide; nonspecific antibody binding was blocked with a 10% normal goat serum followed by blocking buffer. A rat anti–mouse TF antibody (41) (a gift from D. Kirchhofer, Genentech, Inc., San Francisco, CA) was biotinylated and used at 10 μg/ml in blocking buffer, which was applied at room temperature for 1 h. Primary antibodies were detected with 2.5 μg/ml rabbit anti–rat IgG, Vectastain ABC Elite (Vector Laboratories), and metal-enhanced DAB (Thermo Fischer Scientific). Sections were counterstained with Mayer's hematoxylin.
Isolation of blood and plasma.
Whole blood was collected in acid-citrate dextrose (20 mM citric acid, 110 mM sodium citrate, and 5 mM dextrose). PPP was obtained as follows. In brief, the unclotted blood was centrifuged at 310 g for 15 min to obtain platelet-rich plasma, followed by a series of centrifugations, first at 1,000 g for 15 min and finally at 5,000 g for 30 min to obtain PPP.
Statistical analysis.
All data were expressed as the mean ± the SEM. Statistical differences between two groups were analyzed using Student's t test unless specified. P < 0.05 was considered significant. All experiments represent the mean of three or more independent experiments. For in vivo experiments, a significance outlier detection program using Grubb's test was performed for each experimental group.
Online supplemental material.
Fig. S1 shows the characterization of mouse lung endothelial cells. Fig. S2 shows TF expression in PPARδ+/+ and PPARδ −/− mouse lung endothelial cells. Fig. S3 shows a schematic of COX-2 regulation of TF expression in endothelial cells. Fig. S4 shows structures of 2-AG, NE, AA, and AEA. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20070828/DC1.
Supplemental Material
Acknowledgments
We acknowledge the technical assistance of Todd Holcher. We also thank Drs. Daniel Kirchhofer and Wolfrum Ruf for their kind gifts of TF antibodies, Dr. James Morrissey for advice on TF activity measurements, and Dr. Raymond DuBois for the gift of PPAR constructs.
This work is supported by National Institutes of Health grants (HL49094, CA77839 to T. Hla), (DA006668 to S.K. Dey), and HL48872 to N. Mackman.
The authors have no conflicting financial interests.
Abbreviations used: AA, arachidonic acid; AEA, anandamide; AG, arachidonyl glycerol; COX, cyclooxygenase; EC, endothelial cell; HUVEC, human umbilical vein EC; NE, noladin ether; PGFS, PGF2α synthase; PGI2, prostacyclin; PGIS, PGI2 synthase; PPAR, peroxisomal proliferator–activated receptor; PPP, platelet poor plasma; q, quantitative; TF, tissue factor; VSA, valeryl salicylate.
References
- 1.Simmons, D.L., R.M. Botting, and T. Hla. 2004. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 56:387–437. [DOI] [PubMed] [Google Scholar]
- 2.McAdam, B.F., F. Catella-Lawson, I.A. Mardini, S. Kapoor, J.A. Lawson, and G.A. FitzGerald. 1999. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc. Natl. Acad. Sci. USA. 96:272–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cipollone, F., B. Rocca, and C. Patrono. 2004. Cyclooxygenase-2 expression and inhibition in atherothrombosis. Arterioscler. Thromb. Vasc. Biol. 24:246–255. [DOI] [PubMed] [Google Scholar]
- 4.Forman, B.M., J. Chen, and R.M. Evans. 1996. The peroxisome proliferator-activated receptors: ligands and activators. Ann. N. Y. Acad. Sci. 804:266–275. [DOI] [PubMed] [Google Scholar]
- 5.Gupta, R.A., J. Tan, W.F. Krause, M.W. Geraci, T.M. Willson, S.K. Dey, and R.N. DuBois. 2000. Prostacyclin-mediated activation of peroxisome proliferator-activated receptor delta in colorectal cancer. Proc. Natl. Acad. Sci. USA. 97:13275–13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim, H., R.A. Gupta, W.G. Ma, B.C. Paria, D.E. Moller, J.D. Morrow, R.N. DuBois, J.M. Trzaskos, and S.K. Dey. 1999. Cyclo-oxygenase-2-derived prostacyclin mediates embryo implantation in the mouse via PPARdelta. Genes Dev. 13:1561–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gupta, R.A., D. Wang, S. Katkuri, H. Wang, S.K. Dey, and R.N. DuBois. 2004. Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-delta accelerates intestinal adenoma growth. Nat. Med. 10:245–247. [DOI] [PubMed] [Google Scholar]
- 8.Wang, D., H. Wang, Y. Guo, W. Ning, S. Katkuri, W. Wahli, B. Desvergne, S.K. Dey, and R.N. DuBois. 2006. Crosstalk between peroxisome proliferator-activated receptor delta and VEGF stimulates cancer progression. Proc. Natl. Acad. Sci. USA. 103:19069–19074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crofford, L.J., R.L. Wilder, A.P. Ristimaki, H. Sano, E.F. Remmers, H.R. Epps, and T. Hla. 1994. Cyclooxygenase-1 and -2 expression in rheumatoid synovial tissues. Effects of interleukin-1 beta, phorbol ester, and corticosteroids. J. Clin. Invest. 93:1095–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Topper, J.N., J. Cai, D. Falb, and M.A. Gimbrone Jr. 1996. Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: cyclooxygenase-2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up-regulated by steady laminar shear stress. Proc. Natl. Acad. Sci. USA. 93:10417–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egan, K.M., J.A. Lawson, S. Fries, B. Koller, D.J. Rader, E.M. Smyth, and G.A. Fitzgerald. 2004. COX-2-derived prostacyclin confers atheroprotection on female mice. Science. 306:1954–1957. [DOI] [PubMed] [Google Scholar]
- 12.Fitzgerald, G.A. 2004. Prostaglandins: modulators of inflammation and cardiovascular risk. J. Clin. Rheumatol. 10:S12–S17. [DOI] [PubMed] [Google Scholar]
- 13.Warner, T.D., and J.A. Mitchell. 2004. Cyclooxygenases: new forms, new inhibitors, and lessons from the clinic. FASEB J. 18:790–804. [DOI] [PubMed] [Google Scholar]
- 14.Francois, H., K. Athirakul, D. Howell, R. Dash, L. Mao, H.S. Kim, H.A. Rockman, G.A. Fitzgerald, B.H. Koller, and T.M. Coffman. 2005. Prostacyclin protects against elevated blood pressure and cardiac fibrosis. Cell Metab. 2:201–207. [DOI] [PubMed] [Google Scholar]
- 15.Grosser, T., S. Fries, and G.A. FitzGerald. 2006. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J. Clin. Invest. 116:4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozak, K.R., J.J. Prusakiewicz, S.W. Rowlinson, C. Schneider, and L.J. Marnett. 2001. Amino acid determinants in cyclooxygenase-2 oxygenation of the endocannabinoid 2-arachidonylglycerol. J. Biol. Chem. 276:30072–30077. [DOI] [PubMed] [Google Scholar]
- 17.Bishop-Bailey, D., and T. Hla. 1999. Endothelial cell apoptosis induced by the peroxisome proliferator-activated receptor (PPAR) ligand 15-deoxy-Delta12, 14-prostaglandin J2. J. Biol. Chem. 274:17042–17048. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez de Fonseca, F., I. Del Arco, F.J. Bermudez-Silva, A. Bilbao, A. Cippitelli, and M. Navarro. 2005. The endocannabinoid system: physiology and pharmacology. Alcohol Alcohol. 40:2–14. [DOI] [PubMed] [Google Scholar]
- 19.Wang, H., Y. Guo, D. Wang, P.J. Kingsley, L.J. Marnett, S.K. Das, R.N. DuBois, and S.K. Dey. 2004. Aberrant cannabinoid signaling impairs oviductal transport of embryos. Nat. Med. 10:1074–1080. [DOI] [PubMed] [Google Scholar]
- 20.Peters, J.M., S.S. Lee, W. Li, J.M. Ward, O. Gavrilova, C. Everett, M.L. Reitman, L.D. Hudson, and F.J. Gonzalez. 2000. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta). Mol. Cell. Biol. 20:5119–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agarwal, B., P. Swaroop, P. Protiva, S.V. Raj, H. Shirin, and P.R. Holt. 2003. Cox-2 is needed but not sufficient for apoptosis induced by Cox-2 selective inhibitors in colon cancer cells. Apoptosis. 8:649–654. [DOI] [PubMed] [Google Scholar]
- 22.Trifan, O.C., R.M. Smith, B.D. Thompson, and T. Hla. 1999. Overexpression of cyclooxygenase-2 induces cell cycle arrest. Evidence for a prostaglandin-independent mechanism. J. Biol. Chem. 274:34141–34147. [DOI] [PubMed] [Google Scholar]
- 23.Ueda, N. 2002. Endocannabinoid hydrolases. Prostaglandins Other Lipid Mediat. 68-69:521–534. [DOI] [PubMed] [Google Scholar]
- 24.Solomon, S.D., M.A. Pfeffer, J.J. McMurray, R. Fowler, P. Finn, B. Levin, C. Eagle, E. Hawk, M. Lechuga, A.G. Zauber, et al. 2006. Effect of celecoxib on cardiovascular events and blood pressure in two trials for the prevention of colorectal adenomas. Circulation. 114:1028–1035. [DOI] [PubMed] [Google Scholar]
- 25.Mackman, N. 2004. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler. Thromb. Vasc. Biol. 24:1015–1022. [DOI] [PubMed] [Google Scholar]
- 26.Lee, C.H., A. Chawla, N. Urbiztondo, D. Liao, W.A. Boisvert, R.M. Evans, and L.K. Curtiss. 2003. Transcriptional repression of atherogenic inflammation: modulation by PPARdelta. Science. 302:453–457. [DOI] [PubMed] [Google Scholar]
- 27.Steffel, J., M. Hermann, H. Greutert, S. Gay, T.F. Luscher, F. Ruschitzka, and F.C. Tanner. 2005. Celecoxib decreases endothelial tissue factor expression through inhibition of c-Jun terminal NH2 kinase phosphorylation. Circulation. 111:1685–1689. [DOI] [PubMed] [Google Scholar]
- 28.Solovey, A., R. Kollander, A. Shet, L.C. Milbauer, S. Choong, A. Panoskaltsis-Mortari, B.R. Blazar, R.J. Kelm Jr., and R.P. Hebbel. 2004. Endothelial cell expression of tissue factor in sickle mice is augmented by hypoxia/reoxygenation and inhibited by lovastatin. Blood. 104:840–846. [DOI] [PubMed] [Google Scholar]
- 29.Morel, O., F. Toti, B. Hugel, B. Bakouboula, L. Camoin-Jau, F. Dignat-George, and J.M. Freyssinet. 2006. Procoagulant microparticles: disrupting the vascular homeostasis equation? Arterioscler. Thromb. Vasc. Biol. 26:2594–2604. [DOI] [PubMed] [Google Scholar]
- 30.Gauthier, K.M., D.V. Baewer, S. Hittner, C.J. Hillard, K. Nithipatikom, D.S. Reddy, J.R. Falck, and W.B. Campbell. 2005. Endothelium-derived 2-arachidonylglycerol: an intermediate in vasodilatory eicosanoid release in bovine coronary arteries. Am. J. Physiol. Heart Circ. Physiol. 288:H1344–H1351. [DOI] [PubMed] [Google Scholar]
- 31.Rouzer, C.A., and L.J. Marnett. 2005. Glycerylprostaglandin synthesis by resident peritoneal macrophages in response to a zymosan stimulus. J. Biol. Chem. 280:26690–26700. [DOI] [PubMed] [Google Scholar]
- 32.Ho, W.S., and M.D. Randall. 2007. Endothelium-dependent metabolism by endocannabinoid hydrolases and cyclooxygenases limits vasorelaxation to anandamide and 2-arachidonoylglycerol. Br. J. Pharmacol. 150:641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Birnbaum, Y., Y. Ye, Y. Lin, S.Y. Freeberg, M.H. Huang, J.R. Perez-Polo, and B.F. Uretsky. 2007. Aspirin augments 15-epi-lipoxin A4 production by lipopolysaccharide, but blocks the pioglitazone and atorvastatin induction of 15-epi-lipoxin A4 in the rat heart. Prostaglandins Other Lipid Mediat. 83:89–98. [DOI] [PubMed] [Google Scholar]
- 34.Solomon, S.D., J.J. McMurray, M.A. Pfeffer, J. Wittes, R. Fowler, P. Finn, W.F. Anderson, A. Zauber, E. Hawk, and M. Bertagnolli. 2005. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N. Engl. J. Med. 352:1071–1080. [DOI] [PubMed] [Google Scholar]
- 35.Steffel, J., T.F. Luscher, and F.C. Tanner. 2006. Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation. 113:722–731. [DOI] [PubMed] [Google Scholar]
- 36.Andersohn, F., S. Suissa, and E. Garbe. 2006. Use of first- and second-generation cyclooxygenase-2-selective nonsteroidal antiinflammatory drugs and risk of acute myocardial infarction. Circulation. 113:1950–1957. [DOI] [PubMed] [Google Scholar]
- 37.Gislason, G.H., S. Jacobsen, J.N. Rasmussen, S. Rasmussen, P. Buch, J. Friberg, T.K. Schramm, S.Z. Abildstrom, L. Kober, M. Madsen, and C. Torp-Pedersen. 2006. Risk of death or reinfarction associated with the use of selective cyclooxygenase-2 inhibitors and nonselective nonsteroidal antiinflammatory drugs after acute myocardial infarction. Circulation. 113:2906–2913. [DOI] [PubMed] [Google Scholar]
- 38.Hla, T., and K. Neilson. 1992. Human cyclooxygenase-2 cDNA. Proc. Natl. Acad. Sci. USA. 89:7384–7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paik, J.H., A. Skoura, S.S. Chae, A.E. Cowan, D.K. Han, R.L. Proia, and T. Hla. 2004. Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes Dev. 18:2392–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morrissey, J.H., D.S. Fair, and T.S. Edgington. 1988. Monoclonal antibody analysis of purified and cell-associated tissue factor. Thromb. Res. 52:247–261. [DOI] [PubMed] [Google Scholar]
- 41.Kirchhofer, D., P. Moran, S. Bullens, F. Peale, and S. Bunting. 2005. A monoclonal antibody that inhibits mouse tissue factor function. J. Thromb. Haemost. 3:1098–1099. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}